

Abstract
To help characterize the diversity in biological function of proteins emerging from the analysis of whole genomes, we present an operational definition of biological function that provides an explicit link between the functional classification of proteins and the effects of genetic variation or mutation on protein function. Using phylogenetic information, we establish definite criteria for functional relatedness among proteins and a companion procedure for predicting deleterious alleles or mutations. Applied to the functional classification of sequences similar to 13 human tumor suppressor proteins, our methods predict there are functional properties unique to mammals for three of them, BRCA1, BRCA2, and WT1. We examine protein variants caused by nonsynonymous single-nucleotide polymorphisms in a set of clinically important genes and estimate the magnitude of a disproportionate propensity for disruption of function among the nonsynomous single-nucleotide polymorphisms that are maintained at low frequency in the human population.
Although the idea that structural similarity between proteins can be anticipated from their sequences alone is well established, the notion that a signature of functional similarity exists in the comparison of sequences is much less well developed. In fact, the very definition of functional similarity is more elusive than that of structural similarity, which can be quantified (1–3), and pertains to relatively subtle aspects of proteins and their sequences. In proteins inferred to share a remote common ancestor, amino acids determined to be homologous from accurately aligned sequences may not share strictly analogous roles in function and stability, even though their relationship to an overall structural fold may be the same. This observation suggests an operational criterion for what it means that a set of proteins is functionally similar: corresponding amino acids at each residue position in functionally related proteins should serve analogous roles and should likely be interchangeable. From this perspective, the separate problems of functional classification of proteins and the prediction of functional consequences of amino acid substitutions are very closely related.
This study demonstrates how information in a multiple sequence alignment can provide an explicit link between protein functional classification and the tolerance of protein function to amino acid substitutions. In our analysis, we note that most multiple sequence alignments of a query and its homologues will contain too few sequences for the observed profile of amino acids at each residue position to reflect thorough sampling of all 20 amino acids by evolution. To overcome this paucity of empirical amino acid sampling, a key element of our analysis is the use of preexisting mixtures of Dirichlet prior distributions of amino acid frequencies (4) to infer which additional amino acids might be functionally consistent with the observed profiles. Using the Bayesian formalism associated with these distributions, we present a framework for the systematic functional classification of proteins and protein variants. In applications of our methodology, we examine both the functional properties of a group of human tumor suppressor proteins and the functional effects of nonsynonymous single-nucleotide polymorphisms (nsSNPs) in a set of clinically important genes.
Methods
Dirichlet Mixture Priors and Components. The mean posterior estimate of the probability of amino acid i in a residue position of a multiple sequence alignment, p̂i, is calculated from a Dirichlet mixture of priors as reported in equation 15 of Sjölander et al. (4):
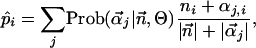 |
[1] |
where Θ refers to the entire set of parameters defining a prior, including the parameters 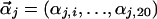 for each component j of the Dirichlet mixture, and
for each component j of the Dirichlet mixture, and 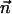 is the observed amino acid count vector. We find that different available Dirichlet mixtures (www.soe.ucsc.edu/research/compbio/dirichlets) give similar results in our analysis; the Blocks9 mixture of priors trained on the blocks database (5), however, performs best (data not shown).
is the observed amino acid count vector. We find that different available Dirichlet mixtures (www.soe.ucsc.edu/research/compbio/dirichlets) give similar results in our analysis; the Blocks9 mixture of priors trained on the blocks database (5), however, performs best (data not shown).
The sum of the contributions of Dirichlet components three and eight from the blocks9 mixture is calculated as follows:
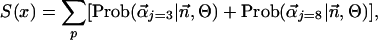 |
[2] |
where the summation is over all residue sequence positions for the subalignment defined by x, the level of sequence identity shared by the reference sequence and the most remote sequence in each subalignment (see Results and Discussion). An abrupt rise in S(x), when plotted against x, may be found by visual inspection of the curve. Alternatively, finding the rise may be automated with edge-detection algorithms, for example the Marr–Hildreth operator (6) (data not shown).
Predicting the Functional Consequences of Amino Acid Substitutions. At the two limits of either no observed amino acid counts or an abundance of observed counts of all amino acids, i.e., 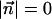 and αj,i << ni for all amino acids, Eq. 1 reduces to
and αj,i << ni for all amino acids, Eq. 1 reduces to 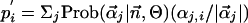 and
and 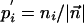 , respectively. We define a score for ranking the amino acids in a profile from high (rank 1) to low (rank 20) predicted exchangeability in the reference sequence as:
, respectively. We define a score for ranking the amino acids in a profile from high (rank 1) to low (rank 20) predicted exchangeability in the reference sequence as:
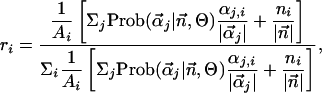 |
[3] |
where 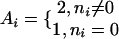 , and the ni are derived from the appropriately chosen subalignment. Alternative ranking schemes that use Eq. 1 directly were empirically found to give less good predictions in our experience.
, and the ni are derived from the appropriately chosen subalignment. Alternative ranking schemes that use Eq. 1 directly were empirically found to give less good predictions in our experience.
Amino acids with equal or similar ri (±0.003) are predicted to have the same degree of functional compatibility with the query amino acid and are grouped together. The highest-ranked group (i.e., the one with the smallest value of ri) that is predicted to include functionally tolerated amino acids is assigned as:
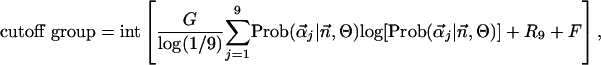 |
[4] |
where R9 is the rank of the ninth mixture component (1–9; 1 is highest) in terms of contribution relative to the other components; define r1,...,20 to represent the rank-ordered positions of amino acids in  , with r1 and r20 being the amino acids most and least likely, respectively, to substitute for the query amino acid; when R9 ≤ 2, G = number of groups within r1 to r7 and F = 0; and when R9 > 2, G = number of groups within r1 to r20 and F = 1. In our experience, these steps and parameters maximize overall prediction accuracy on the four mutation data sets. The primary determinant of accuracy appears to be the selection of the optimal subalignment. Other formulations based on the optimal subalignment are certainly possible, including approaches using machine learning.
, with r1 and r20 being the amino acids most and least likely, respectively, to substitute for the query amino acid; when R9 ≤ 2, G = number of groups within r1 to r7 and F = 0; and when R9 > 2, G = number of groups within r1 to r20 and F = 1. In our experience, these steps and parameters maximize overall prediction accuracy on the four mutation data sets. The primary determinant of accuracy appears to be the selection of the optimal subalignment. Other formulations based on the optimal subalignment are certainly possible, including approaches using machine learning.
Human Tumor Suppressor Protein Sequences. All human tumor suppressor protein sequences were obtained from Swiss-Prot (http://us.expasy.org/sprot) except for the ARF sequence, which was obtained from TrEMBL (7). The identifiers are APC_HUMAN, Q16360 (ARF); BRCA1_HUMAN; BRCA2_HUMAN; CDN2_HUMAN (INK4A); NF1_HUMAN; MERL_HUMAN (NF2); P53_HUMAN; PTC1_HUMAN; PTEN_HUMAN; RB_HUMAN; VHL_HUMAN; and WT1_HUMAN. We obtained sequences related to each tumor suppressor by querying the nonredundant database (posted August 2, 2002) (8) with the above sequences by using psi-blast (Ver. 2.2.3 with parameters -e 1.0, -h 0.001, -b 1000, -j 6, SEG filter) (9). An alternate approach would have been to retrieve sequences on a domain-by-domain basis as was done with the test sequences, but we chose to pursue here the simpler approach of querying with the entire sequence and to defer a possibly more accurate domain search method to a separate study.
Results and Discussion
We sought a general procedure for identifying proteins functionally related to a query protein through the application of Dirichlet mixtures to ordered sets of multiple sequence alignments. We suspected (and later confirmed; see Fig. 1C) that amino acids at each residue position in a multiple sequence alignment that are not functionally equivalent to the reference amino acid in the query protein are more likely to be found in proteins remotely related to the query by sequence. Accordingly, we tried to find a subset of sequences or “subalignment” that would provide the most extensive sampling possible of tolerated alternative amino acids but would exclude proteins that are functionally divergent from the query protein because their level of sequence similarity with the query was too low. To further guide our methodology with experimental data, we applied the Blocks9 Dirichlet priors to profiles from the subalignments for query protein domains that had been extensively mutagenized and functionally tested in standardized assays. The single domain polypeptides were the HIV protease (10), T4 lysozyme (11), the N-terminal DNA-binding domain of the lac repressor (Lac-N) (12, 13), and the C-terminal regulatory domain of the lac repressor (Lac-C) (12, 13). We extracted sequences related to these four query domains from the Pfam database (Ver. 6.5-A) (14) and formed the subalignments defined by successively smaller minimal fractions of amino acids shared with the reference sequence.
Fig. 1.

Shown are inferences of which protein sequences in a multiple sequence alignment are functionally related to a reference sequence. The plots correspond to the reference protein domains HIV protease (residues 4–99), T4 lysozyme (residues 24–148), Lac-N (residues 2–29), and Lac-C (residues 68–327). The abscissa of each plot is the minimal fraction of amino acids shared with the reference sequence for each subalignment of the full multiple sequence alignment extracted from the Pfam database. (A) The contribution from each of the nine Dirichlet components from the Blocks9 mixture. The numerical plot symbols refer to the contributing components. (B) The sum of the contributions from components 3 and 8 from A. (C) The ratio of posterior estimates of amino acids experimentally determined to be either functionally deleterious or functionally tolerated. The minimum of each curve suggests which subalignments optimally inform the functional predictions in D. (D) Overall prediction accuracy of tolerated vs. deleterious amino acid substitutions. The gray vertical line in each plot indicates the subalignment selected for representing each query sequence. The subalignment sequence identity cutoffs are 0.531 for HIV protease (although a larger rise occurs at ≈0.3, we interpret the smaller rise at ≈0.5 as indicating some level of functional divergence from the query sequence), 0.281 for T4 lysozyme, 0.545 for Lac-N, and 0.289 for Lac-C.
For each of the subalignments of these four domains ordered by the minimum fraction of amino acids shared with the query sequence, we examined the average contribution of each of the nine components in Blocks9 to the estimated profiles for all residue positions (see Methods). Notably, as the minimal fraction of shared amino acid identity with the query sequence decreased below a unique and different value for each of the four domains, there was an abrupt increase in the contribution of Blocks9 components 3 and 8 to the estimated amino acid profiles (Fig. 1A). Each component in the commonly used Dirichlet mixtures favors a different class of amino acids, implicitly reflecting their physiochemical properties, empirically determined exchangeability, and relative functional importance (4, 15). The contribution of each component to the estimated profile of amino acids at each residue position characterizes how well it matches the observed profile. Blocks9 components 3 and 8 are different from the others in that together they make relatively little distinction among the 20 amino acids (4). The abruptly increased contribution of these two components most likely signifies a loss of functional specificity of the amino acids observed at residue positions in the alignment caused by inclusion of sequences that are functionally remote from the reference sequence. The alternative explanation that this effect is due to sequence misalignment is unlikely because (i) the alignments are from the highly curated Pfam database, and (ii) for the cases of the HIV protease and the Lac-N, the abrupt increase occurs at levels of sequence identity well within the range of sequence similarity that can be accurately aligned in a structural sense by using sequence-based methods (e.g., psi-blast) (9, 16, 17).
We examined whether the direct experimental measurements of tolerated and deleterious mutations in the four domains were consistent with our functional classification of sequences based on Blocks9 components 3 and 8. For each profile in each subalignment, we compared the posterior estimates of amino acids (Eq. 1) that had been substituted and measured to be either functionally tolerated or deleterious. The ratio of these two values (p̂i of deleterious mutations/p̂i of tolerated mutations) is expected to suggest how well each subalignment reflects the query's tolerance to mutation, and its minimum is likely to estimate the subalignment that optimally represents the tolerated amino acid variability at each residue position. We found a remarkable correspondence between the subalignments minimizing the ratio and the abrupt increase in the summed contribution of Blocks9 components 3 and 8 that signifies a loss of functional character in the observed profiles (Fig. 1 B and C; see Methods). In general, there may be not one but several subalignments that optimally represent the query when the region around the rise is densely populated with subalignments. To borrow a term used in relating sequence to structural similarity, the fraction amino acid identity shared with the query around the abrupt rise may represent a “twilight zone” (17) for functional similarity.
Next, we devised an algorithm that uses the selected subalignments for the four domains to predict which amino acid substitutions at each query sequence position would have an effect on function. For each residue position, the algorithm ranks all 20 amino acids according to a modified posterior probability of being tolerated and then determines a cutoff probability value for separating amino acids predicted to be tolerated from those predicted to be deleterious (see Methods).
The accuracy of the predictive algorithm using only mutations that are unambiguously tolerated or deleterious in the four data sets was high, with overall values being 82% (216/263) for the HIV protease, 78% (587/750) for T4 lysozyme, 76% (205/268) for the N-terminal domain of the lac repressor, and 72% (2087/2905) for the C-terminal domain of the lac repressor (Table 1). In addition, the method is fairly balanced in its prediction accuracy between tolerated and deleterious mutations. The overall predictions are optimal using the subalignments selected as described above, reinforcing the approach of identifying the subalignment that optimally informs amino acid exchangeability through the summed contribution of Blocks9 components 3 and 8.
Table 1. Prediction accuracy of the effect of amino acid substitutions on function.
| Comparison with sift predictions
|
|||||||
|---|---|---|---|---|---|---|---|
| Protein | Predicted tolerated* | Predicted deleterious* | Total* | Rank-ordering accuracy† | Predicted tolerated | Predicted deleterious | Total |
| Hiv | 81% (85/105) | 83% (131/158) | 82% (216/263) | 95% (88/93) | 77% (81/105) | 79% (125/158) | 78% (206/263) |
| Lys | 79% (534/676) | 72% (53/74) | 78% (587/750) | 89% (132/149) | 66% (446/676) | 95% (70/74) | 69% (516/750) |
| Lac-N | 75% (86/115) | 78% (119/153) | 76% (205/268) | 88% (111/126) | 66% (76/115) | 76% (117/153) | 72% (193/268) |
| Lac-C | 72% (1,700/2373) | 73% (387/532) | 72% (2,087/2,905) | 85% (901/1066) | 73% (1,726/2373) | 79% (421/532) | 74% (2,147/2,905) |
Only nonintermediate phenotypes (either wild-type function or complete ablation of function as assayed) from the mutation studies of HIV protease, T4 lysozyme, and Escherichia coli lac repressor were included in the analysis, because they were regarded as being the most reliable.
‡Prediction results from sift Ver. 2 (http://blocks.fhcrc.org/~pauline/SIFT.html) (18) for comparison.
The overall prediction accuracy and the fraction of amino acid substitutions correctly predicted to be either functionally tolerated or functionally deleterious are listed for the four domains. The subalignments used to represent each query sequence are indicated in Fig. 1
The accuracy of the rank-ordering of amino acids in each residue profile as measured by their relative propensity to disrupt function. Only residue positions that contain both tolerated and deleterious substitutions were included in this analysis
To judge whether the rank ordering of the amino acids in each residue profile accurately reflects their relative propensity to disrupt function, we assigned a separate cutoff to each residue profile to maximize the distinction between tolerated and deleterious mutations. This exploratory procedure led to a very high classification accuracy that likely approaches the experimental accuracy in the analysis of the mutations (Table 1) and confirms that the rank ordering of amino acids in the residue profiles very accurately reflects their relative impact on protein function in these four test cases.
A previously reported method, sift (18, 19), also uses multiple sequence alignments and Dirichlet priors to generate predictions, but it uses very different ways of (i) choosing which sequences are included in a multiple sequence alignment (sift does not use Dirichlet priors in this step) and (ii) distinguishing between tolerated and deleterious amino acids. Predictions using sift (Ver. 2) are less accurate overall and less well balanced for false positive and false negative classifications [as previously noted (18); Table 1].
Using the behavior of Blocks9 components 3 and 8 for inferring functional relatedness among protein sequences, we examined proteins from the nonredundant database homologous to 13 well-studied human tumor suppressor proteins: APC, ARF (CDKN2A/P14), BRCA1, BRCA2, INK4A (CDKN2A), NF1, NF2, P53, PTC, PTEN, RB, VHL, and WT1. The summed contribution of components 3 and 8 to the estimated profiles in the successive subalignments for each protein family revealed distinct thresholds of amino acid similarity that we again interpret to reflect the limits on sequence similarity for functional similarity (Fig. 2). Barring a few short sequence fragments from nonmammalian species, only mammalian sequences were classified as having function similar to the human BRCA1, BRCA2, and WT1, which suggests a function unique to some mammals for these proteins (Table 2). For RB, our functional classification cutoff came at a very high level of sequence similarity (61%), but the sequences inferred to be functionally related to human RB are not all from mammals, and some mammalian sequences fall below the cutoff, which suggests diverse functions for RB-like proteins in diverse contexts as reported previously, e.g., the excluded human p107 [37% identity (residues 3–645) with RB] (20). Only mammalian sequences were obtained for ARF, so no distinctions could be made for this protein. For the other proteins, sequences inferred to be functionally related to the human query derive from both mammals and nonmammals.
Fig. 2.

The summed contribution from components 3 and 8 of the Blocks9 Dirichlet mixture for the multiple sequence subalignments corresponding to the 13 human tumor suppressor proteins. These plots are analogous to those in Fig. 1B (see Methods for details). Subalignments (gray vertical lines; sequence identities are listed in Table 2) were selected either by visual inspection or with an edge-detection algorithm (see Methods). No abrupt rise is seen for ARF (7), likely because all sequences are from mammals.
Table 2. Summary of functional classification of sequences related to 13 human tumor suppressor proteins.
| Protein (residues, subalignment identification, number of sequences in subalignment) | Examples of nonmammalian sequences (protein accession identification, residues, sequence identity to query) |
|---|---|
| APC (1—2843, 32%, 15) | Xenopus laevis (AAB41671, 1—2,829, 71%) |
| ARF (1—179, 44%, 11) | All mammals |
| BRCA1 (1—1863, 32%, 99) | X. laevis (AAL13037, 1—234, 41%), fragment |
| BRCA2 (1—3418, 37%, 24) | Arabidopsis thaliana (NP_191913, 106—204, 37%), fragment |
| INK4A (1—156, 33%, 19) | Xiphophorus helleri (AAD21313, 7—121, 57%) |
| NF1 (1—2839, 29%, 58) | Takifugu rubripes (AAD15839, 1—2,763, 88%) |
| Anopheles gambiae (EAA08440, 3—2,787, 60%) | |
| NF2 (1—595, 48%, 19) | A. gambiae (EAA07087, 56—635, 57%) |
| Drosophila melanogaster (AAM11326, 7—468, 54%) | |
| P53 (1—393, 53%, 118) | A. gambiae (P10361, 1—391, 79%) |
| X. laevis (P07193, 2—363, 56%) | |
| PTC (1—1447, 27%, 44) | X. laevis (AAK15463, 1—1,417, 80%) |
| D. melanogaster (A33468, 23—1,198, 41%) | |
| PTEN (1—403, 38%, 22) | X. laevis (AAD46165, 1—402, 89%) |
| T. rubripes (AAL08419, 1—412, 88%) | |
| RB (1—928, 61%, 25) | Gallus gallus (CAA51019, 1—921, 74%) |
| Notophthalmus viridescens (CAA70428, 2—899, 62%) | |
| X. laevis (A44879, 1—899, 61%) | |
| VHL (1—213, 33%, 9) | A. gambiae (EAA07955, 26—168, 33%) |
| WT1 (1—449, 31%, 110) | X. laevis (P18753, 591—760, 35%), fragment |
The only nonmammalian sequences included in the subalignments selected to represent BRCA1, BRCA2, and WT1 are short sequence fragments. The complete list of sequences is available upon request.
Using our algorithm for analyzing protein variants, we predicted effects on function for amino acid variants arising from nsSNPs in two published surveys of genetic variation in a selection of clinically important genes [Cargill et al. (21) and Halushka et al. (22); all predictions are listed in Table 3, which is published as supporting information on the PNAS web site]. For the multiple sequence alignments of protein families homologous to each of these proteins, Blocks9 components 3 and 8 revealed a different threshold of sequence similarity that was inferred to reveal the limits of functional similarity (Table 3). Overall, ≈30% of the polymorphisms [46/134 from Cargill et al. (21) and 15/51 from Halushka et al. (22)] were predicted to affect function, which is similar to the proportion found by others (19, 23–26), and the following were most strongly predicted to affect function: CETP-486VM, F5-817NT, F13A1-589LQ, FSHR-524SR, GH1-105SC, GHR-495PT, NTRK1-604HY, and TFPI-292VM. The variants in NTRK1 and TFPI had previously been reported to be associated with physiological effects (27, 28).
In addition, we found a statistically significant disproportionate representation of deleterious alleles at low frequency in the population in accordance with the expectation from genetic theory (26). In the data from Cargill et al. (21), 45% (37/83) of the nsSNPs at low frequency [<5%, classification from Cargill et al. (21)] were predicted to affect function compared with 27% (6/22) and 10% (3/29), respectively, of the nsSNPs occurring at medium frequency (5–15%; P = 0.03, χ2 test) and high frequency (15–50%; P = 7 × 10-4, χ2 test). When the predictions were distributed according to their group rank offset (Fig. 3 and Fig. 4, which is published as supporting information on the PNAS web site), we also found significantly more confident predictions of deleterious effects in the comparisons of (i) the low- to the high-frequency nsSNPs (P = 1 × 10-6, Mann–Whitney U test) and (ii) the low-frequency nsSNPs to the combined medium- and high-frequency nsSNPs (P = 1 × 10-5, Mann–Whitney U test). Insofar as they are consistent with genetic theory, the predictions are most directly interpreted as evidence for a significant effect on function for an appreciable fraction of human nsSNPS rather than a consequence of misclassification in the predictive algorithm, as has been suggested (19). Moreover, the consistency of our predictions with the genetic expectation may be taken as evidence of the essential validity of our approach. In the data from Halushka et al. (22), the most confident predictions of effect on function are seen almost exclusively in the low-frequency group, and none are seen in the high-frequency group, but these differences are not statistically significant [possibly because we used only confirmed polymorphisms, which made this data set less than half as large as the data set from Cargill et al. (21)].
Fig. 3.

Distributions of predicted effect on function for amino acid variants arising from nsSNPs at low (<5%), medium (5–15%), and high (>15%) minor allele frequency in the human population. Prediction confidence is measured by group rank offset (Fig. 4) from the cutoff, where offsets of zero (0) and above are variants predicted to affect function (highest confidence in right-most bin), and offsets below zero are variants predicted to be functionally tolerated (highest confidence in leftmost bin). The data from Cargill et al. (21) and Halushka et al. (22) are represented by the dark and light bars, respectively. Sequences related to each query were taken from Swiss-Prot, and subalignments were determined as described in Methods.
Conclusion
Previous studies had assigned functional classifications of reference proteins and their homologues by comparison of explicit functional information [e.g., Enzyme Commission (EC) numbers and Gene Ontology (GO) and Structural Classification of Proteins (SCOP)] and suggested that an average threshold of ≈40% minimal shared amino acid identity reflects functional relatedness (29–34). Our approach proposes an operational definition of functional similarity based on amino acid exchangeability in functionally related proteins that can be reduced to an algorithm by analysis of Blocks9 components 3 and 8 and suggests that the threshold for functional relatedness is a characteristic feature of each reference protein and its family of homologues. Still other approaches have considered phylogenetic tree analysis and use explicit functional information about some family members for classifying a new sequence of unknown function (35). Explicit knowledge of function is not required by our method's operational definition of functional similarity. Based on our findings for the four test domains, we argue that this definition of functional similarity can optimally inform predictions about the functional effects of mutations. We anticipate a wide-ranging application of our methods not only in the functional classification of proteins found in the growing number of fully sequenced genomes but also in predictions regarding the functional consequences of naturally occurring genetic variation.
Supplementary Material
Acknowledgments
We thank Gilbert Chu, Tyler Jacks, S. Roy Kimura, Alan Templeton, John Whittaker, Claudio Verzilli, Cursten Wiuf, and Gregory L. Verdine for insightful discussions and Ann E. Ferentz for helpful comments on the manuscript.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: nsSNP, nonsynomous single-nucleotide polymorphism; Lac-C, C-terminal DNA-binding domain of the lac repressor; Lac-N, N-terminal DNA-binding domain of the lac repressor.
References
- 1.Pearl, F. M., Bennett, C. F., Bray, J. E., Harrison, A. P., Martin, N., Shepherd, A., Sillitoe, I., Thornton, J. & Orengo, C. A. (2003) Nucleic Acids Res. 31, 452-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lo Conte, L., Brenner, S. E., Hubbard, T. J., Chothia, C. & Murzin, A. G. (2002) Nucleic Acids Res. 30, 264-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holm, L. & Sander, C. (1998) Nucleic Acids Res. 26, 316-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sjölander, K., Karplus, K., Brown, M., Hughey, R., Krogh, A., Mian, I. S. & Haussler, D. (1996) Comput. Appl. Biosci. 12, 327-345. [DOI] [PubMed] [Google Scholar]
- 5.Henikoff, J. G., Greene, E. A., Pietrokovski, S. & Henikoff, S. (2000) Nucleic Acids Res. 28, 228-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marr, D. & Hildreth, E. (1980) Proc. R. Soc. London Ser. B 207, 187-217. [DOI] [PubMed] [Google Scholar]
- 7.Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., Martin, M. J., Michoud, K., O'Donovan, C., Phan, I., et al. (2003) Nucleic Acids Res. 31, 365-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J. & Wheeler, D. L. (2003) Nucleic Acids Res. 31, 23-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loeb, D. D., Swanstrom, R., Everitt, L., Manchester, M., Stamper, S. E. & Hutchison, C. A., III (1989) Nature 340, 397-400. [DOI] [PubMed] [Google Scholar]
- 11.Rennell, D., Bouvier, S. E., Hardy, L. W. & Poteete, A. R. (1991) J. Mol. Biol. 222, 67-88. [DOI] [PubMed] [Google Scholar]
- 12.Suckow, J., Markiewicz, P., Kleina, L. G., Miller, J., Kisters-Woike, B. & Muller-Hill, B. (1996) J. Mol. Biol. 261, 509-523. [DOI] [PubMed] [Google Scholar]
- 13.Markiewicz, P., Kleina, L. G., Cruz, C., Ehret, S. & Miller, J. H. (1994) J. Mol. Biol. 240, 421-433. [DOI] [PubMed] [Google Scholar]
- 14.Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., Griffiths-Jones, S., Howe, K. L., Marshall, M. & Sonnhammer, E. L. (2002) Nucleic Acids Res. 30, 276-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ouzounis, C., Perez-Irratxeta, C., Sander, C. & Valencia, A. (1998) Pac. Symp. Biocomput. 3, 401-412. [PubMed] [Google Scholar]
- 16.Venclovas, C., Zemla, A., Fidelis, K. & Moult, J. (2001) Proteins Suppl., 163-170. [DOI] [PubMed]
- 17.Rost, B. (1999) Protein Eng. 12, 85-94. [DOI] [PubMed] [Google Scholar]
- 18.Ng, P. C. & Henikoff, S. (2001) Genome Res. 11, 863-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng, P. C. & Henikoff, S. (2002) Genome Res. 12, 436-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu, L., van den Heuvel, S., Helin, K., Fattaey, A., Ewen, M., Livingston, D., Dyson, N. & Harlow, E. (1993) Genes Dev. 7, 1111-1125. [DOI] [PubMed] [Google Scholar]
- 21.Cargill, M., Altshuler, D., Ireland, J., Sklar, P., Ardlie, K., Patil, N., Shaw, N., Lane, C. R., Lim, E. P., Kalyanaraman, N., et al. (1999) Nat. Genet. 22, 231-238. [DOI] [PubMed] [Google Scholar]
- 22.Halushka, M. K., Fan, J. B., Bentley, K., Hsie, L., Shen, N., Weder, A., Cooper, R., Lipshutz, R. & Chakravarti, A. (1999) Nat. Genet. 22, 239-247. [DOI] [PubMed] [Google Scholar]
- 23.Chasman, D. & Adams, R. M. (2001) J. Mol. Biol. 307, 683-706. [DOI] [PubMed] [Google Scholar]
- 24.Sunyaev, S., Ramensky, V., Koch, I., Lathe, W., III, Kondrashov, A. S. & Bork, P. (2001) Hum. Mol. Genet. 10, 591-597. [DOI] [PubMed] [Google Scholar]
- 25.Wang, Z. & Moult, J. (2001) Hum. Mutat. 17, 263-270. [DOI] [PubMed] [Google Scholar]
- 26.Fay, J. C., Wyckoff, G. J. & Wu, C. I. (2001) Genetics 158, 1227-1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gimm, O., Greco, A., Hoang-Vu, C., Dralle, H., Pierotti, M. A. & Eng, C. (1999) J. Clin. Endocrinol. Metab. 84, 2784-2787. [DOI] [PubMed] [Google Scholar]
- 28.Moatti, D., Seknadji, P., Galand, C., Poirier, O., Fumeron, F., Desprez, S., Garbarz, M., Dhermy, D., Arveiler, D., Evans, A., et al. (1999) Arterioscler. Thromb. Vasc. Biol. 19, 862-869. [DOI] [PubMed] [Google Scholar]
- 29.Parisi, G. & Echave, J. (2001) Mol. Biol. Evol. 18, 750-756. [DOI] [PubMed] [Google Scholar]
- 30.Wilson, C. A., Kreychman, J. & Gerstein, M. (2000) J. Mol. Biol. 297, 233-249. [DOI] [PubMed] [Google Scholar]
- 31.Andrade, M. A., Brown, N. P., Leroy, C., Hoersch, S., de Daruvar, A., Reich, C., Franchini, A., Tamames, J., Valencia, A., Ouzounis, C., et al. (1999) Bioinformatics 15, 391-412. [DOI] [PubMed] [Google Scholar]
- 32.Brenner, S. E. (1999) Trends Genet. 15, 132-133. [DOI] [PubMed] [Google Scholar]
- 33.Bork, P., Dandekar, T., Diaz-Lazcoz, Y., Eisenhaber, F., Huynen, M. & Yuan, Y. (1998) J. Mol. Biol. 283, 707-725. [DOI] [PubMed] [Google Scholar]
- 34.Tatusov, R. L., Koonin, E. V. & Lipman, D. J. (1997) Science 278, 631-637. [DOI] [PubMed] [Google Scholar]
- 35.Eisen, J. A. (1998) Genome Res. 8, 163-167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.