

Abstract
Erythropoiesis is the biological process that consumes the highest amount of body iron for heme synthesis. Heme synthesis in erythroid cells is finely coordinated with that of alpha (α) and beta (β)-globin, resulting in the production of hemoglobin, a tetramer of 2α- and 2β-globin chains, and heme as the prosthetic group. Heme is not only the structural component of hemoglobin, but it plays multiple regulatory roles during the differentiation of erythroid precursors since it controls its own synthesis and regulates the expression of several erythroid-specific genes. Heme is synthesized in developing erythroid progenitors by the stage of proerythroblast, through a series of eight enzymatic reactions divided between mitochondria and cytosol. Defects of heme synthesis in the erythroid lineage result in sideroblastic anemias, characterized by microcytic anemia associated to mitochondrial iron overload, or in erythropoietic porphyrias, characterized by porphyrin deposition in erythroid cells. Here, we focus on the heme biosynthetic pathway and on human erythroid disorders due to defective heme synthesis. The regulatory role of heme during erythroid differentiation is discussed as well as the heme-mediated regulatory mechanisms that allow the orchestration of the adaptive cell response to heme deficiency.
The heme biosynthetic pathway
Heme biosynthesis is a complex process that occurs in all cells through eight enzymatic reactions divided between mitochondria and cytosol. In the hemopoietic compartment, heme synthesis increases during differentiation of erythroid progenitors and it is tightly coordinated with iron acquisition and globin gene expression (Figure 1).
Figure 1.
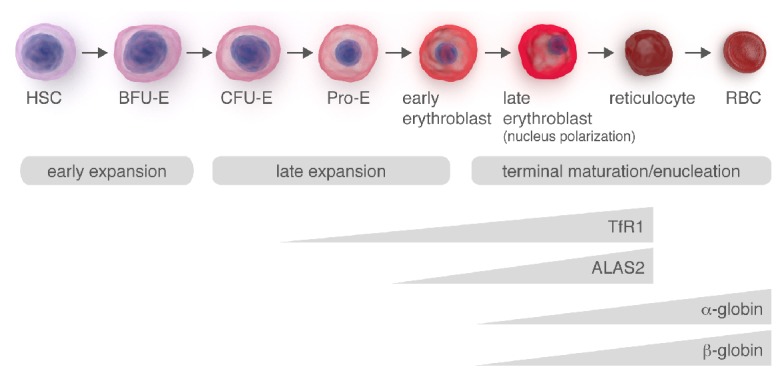
Schematic representation of erythroid differentiation. The timing of the processes leading to hemoglobin production during erythroid differentiation is illustrated. High amount of iron is required during erythroid differentiation to sustain heme biosynthesis. Pro-erythroblast increases iron uptake through the upregulation of TfR1. At the same time, the activity of ALAS2 also increases to provide the huge amounts of heme needed for hemoglobin production. Soon after its synthesis, heme activates the transcription and translation of globin chains, thus allowing hemoglobin synthesis.
Heme biosynthesis
The first step in heme biosynthesis is the condensation of succinyl-CoA and glycine to form δ-aminolevulinic acid (ALA) in the mitochondrial matrix (Figure 2). This reaction is catalyzed by ALA synthase (ALAS) and it is considered rate-limiting. There are two isoforms of ALAS, ALAS1 and ALAS2, which are encoded by separated genes. Alas1 gene is located on chromosome 3 and it is ubiquitously expressed. It plays an important housekeeping function in providing heme in non-erythroid tissues. Alas2 gene is located on the X chromosome and it is expressed exclusively in erythroid cells.1 Alas2 expression strongly increases during the late stages of erythroid differentiation and it is essential for the terminal maturation of red blood cells.2 The increased expression of Alas2 is a prerequisite for the full induction of the other genes of the heme biosynthetic pathway.3 The expression of Alas2 is regulated by erythroid-specific transcription factors, like GATA1.4,5 At the post-transcriptional level, Alas2 expression is regulated by iron. The Alas2 transcript contains a 5′ iron responsive element (IRE) that interacts with iron responsive proteins (IRPs), thus linking the regulation of heme biosynthesis in erythroid cells to the availability of iron. Under conditions of iron deficiency, the translation of Alas2 mRNA is inhibited by IRPs binding to the 5′IRE. On the other hand, when intracellular iron level increases, IRPs are degraded thus allowing the translation of Alas2 mRNA.6 Following its synthesis, ALA is exported to the cytosol where it is converted to coproporphyrinogen III (CPgenIII). All the remaining steps of heme biosynthesis take place inside mitochondria. CPgenIII is imported into the mitochondrial intermembrane space, where it is converted to protoporphyrinogen IX by coproporphyrinogen oxidase (CPOX). Then, protoporphyrinogen IX is oxidized to protoporphyrin IX (PPIX) by protoporphyrinogen oxidase (PPOX). Finally, ferrous iron is incorporated into PPIX to form heme in the mitochondrial matrix, a reaction catalyzed by ferrochelatase (FECH)7 (Figure 2). FECH is another rate-limiting enzyme of the heme biosynthetic pathway. FECH expression increases during erythroid differentiation and it is controlled by transcription factors Sp1, NF-E2 and GATA elements.8 FECH is an iron-sulfur cluster protein. At the post-transcriptional level, the expression of FECH is controlled by the availability of newly formed iron-sulfur clusters whose biogenesis is dependent on iron as well as on functional iron-sulfur cluster assembly machinery.9 Indeed, downregulation of FECH was observed during iron-deficient erythropoiesis in IRP2−/− mice, in iron-limited erythroid differentiation of MEL cells and in conditions of impaired iron-sulfur cluster biogenesis.9
Figure 2.

Heme biosynthesis. Schematic representation of the heme biosynthetic pathway. Heme synthesis starts with the condensation of Succynil-CoA and glycine to form ALA. ALA is then transported through the two mitochondrial membranes in the cytosol where it is converted to CPgenIII through a series of enzymatic reactions. Briefly, the aminolevulinate dehydratase (ALAD) catalyzes the condensation of two molecules of ALA to form one molecule of the monopyrrole porphobilinogen. Then, the hydroxymethylbilane synthase (HMBS) catalyzes the head-to-tail synthesis of four porphobilinogen molecules to form the linear tetrapyrrole hydroxymethylbilane which is converted to uroporphyrinogen III by uroporphyrinogen synthase (UROS). The last cytoplasmic step, the synthesis of CPgenIII, is catalyzed by uroporphyrinogen decarboxylase (UROD). CPOX is a homodimer weakly associated with the outside of the inner mitochondrial membrane and it converts CPgenIII to protoporphyrinogen IX. The following oxidation of protoporphyrinogen IX to PPIX is catalyzed by PPOX, located on the outer surface of the inner mitochondrial membrane. Finally, ferrous iron is incorporated into PPIX to form heme in the mitochondrial matrix, a reaction catalyzed by FECH. In hematopoietic tissue, iron is imported into mitochondria by MFRN1. FECH is localized in the inner mitochondrial membrane in association to MFRN1 and ABCB10. SLC25A38 and ABCB10 have been proposed as mitochondrial ALA exporters on the inner mitochondrial membrane. The ALA transporter located on the outer mitochondrial membrane has not been identified yet. ABCB6 has been proposed as a putative mitochondrial CPgenIII importer. However, this role is still controversial. Finally, several data suggest that FLVCR1b is a mitochondrial heme exporter.
Thus, heme biosynthesis is absolutely dependent on iron uptake by the developing erythroblast since iron is not only required for incorporation into the PPIX ring but it also controls the expression of Alas2 and FECH. Iron is acquired by differentiating erythroid progenitors via transferrin receptor 1 (TfR1)-mediated endocytosis and transferred to mitochondria for heme synthesis.10–12 Two mitochondrial iron importers localized on the inner mitochondrial membrane have been identified: mitoferrin1 (MFRN1) and mitoferrin2 (MFRN2), expressed in erythroid and non-erythroid tissues, respectively. They play an essential role in supplying iron for the biosynthesis of heme and iron-sulfur clusters.13,14
Due to the toxicity of intermediate products of the heme biosynthetic pathway, the product of each reaction has to be quickly delivered to the next enzyme in the pathway to avoid free-intermediates accumulation. It is still not completely understood how this is achieved and the existence of transient multi-enzyme complexes has been postulated.15 Recent evidence indicates that FECH is part of a complex in the inner mitochondrial membrane with MFRN1 and the ATP-binding cassette sub-family B, member 10 (ABCB10) transporter. The interaction between FECH and MFRN1 allows the coupling of iron import in mitochondria to its incorporation in the porphyrin ring. ABCB10 stabilizes MFRN1 expression16,17 (Figure 2).
Trafficking of heme and its precursors across mitochondrial membranes
While all the enzymatic steps leading to the production of heme are well characterized, it is still not completely understood how the compartmentalization of these reactions between mitochondria and cytosol occurs. How is ALA exported through the two mitochondrial membranes? How is CPgenIII imported into the mitochondrial intermembrane space? How is heme exported out of mitochondria? Only in the last decade these issues have began to be addressed and several novel players in the heme biosynthetic pathway have been identified.
Export of ALA from mitochondria
SLC25A38 (solute carrier family 25, member 38) and ABCB10 have been proposed to export ALA from mitochondria (Figure 2).
SLC25A38 is a member of the SLC25 family of inner mitochondrial membrane transporters, which promotes the exchange of one metabolite for another across the inner mitochondrial membrane.18 SLC25A38 is highly and preferentially expressed in erythroid cells. The knockdown of SLC25A38 in zebrafish causes anemia similar to Alas2 deficiency.19 Yeast lacking YDL119c, the ortholog of SLC25A38, shows a defect in the biosynthesis of ALA.19 Thus, it has been hypothesized that SLC25A38 could facilitate the production of ALA by importing glycine into mitochondria or by exchanging glycine for ALA across the mitochondrial inner membrane.19
ABCB10 is a member of the ATP-binding cassette family of transporters, which use the energy of ATP hydrolysis to transport diverse substrates across cellular membranes. ABCB10 is a mitochondrial inner membrane protein which homodimerizes to form a functional transporter.20 In addition to its role in the stabilization of MFRN1,17 ABCB10 plays an important role during erythroid differentiation. In fact, its overexpression enhances hemoglobin synthesis in differentiating MEL cells21 while its silencing impairs hemoglobinization of differentiating K562 cells.22 Abcb10−/− mice die in utero due to severe anemia.23 It was initially proposed that ABCB10 could export heme from mitochondria.16,21 However, ABCB10 silencing causes a decrease in cellular and mitochondrial heme levels associated to the reduced activity of several heme-containing enzymes. The administration of ALA fully restores heme levels in ABCB10-down-regulated cells whereas Alas2 overexpression fails to do this. Thus, it has been proposed that ABCB10 could facilitate mitochondrial ALA synthesis or its export from mitochondria.24
Both SLC25A38 and ABCB10 are located on the mitochondrial inner membrane. It remains to be understood how ALA is transported through the outer mitochondrial membrane.
Mitochondrial import of CPgenIII
ABCB6 (ATP-binding cassette, sub-family B, member 6) has been proposed to transport CPgenIII from the cytoplasm to the mitochondrial intermembrane space25 (Figure 2). ABCB6 is located in the outer mitochondrial membrane and its expression is positively regulated following the stimulation of erythroid differentiation of MEL and G1ER cells as well as by heme levels. ABCB6 binds porphyrins, included heme, and competition assays suggest that CPgenIII is the substrate.25
Nevertheless, the mitochondrial localization of ABCB6 as well as the role of ABCB6 in the translocation of porphyrins in mitochondria are still controversial. ABCB6 was found also on the plasma membrane, in the Golgi compartment and in lysosomes. Some studies even fail to detect ABCB6 in mitochondria26–28 In addition, ABCB6 has been associated to other functions unrelated to porphyrin homeostasis.29–31 Recently, it has been reported that Abcb6−/− mice completely lack mitochondrial ATP-driven import of CPgenIII whereas non-ATP-dependent CPgenIII uptake is unaffected.32 Indeed, loss of Abcb6 causes the upregulation of compensatory porphyrin and iron pathways. Abcb6−/− mice are phenotypically normal but increased mortality as well as reduced heme synthesis were observed following phenylhydrazine administration. Taken together, these data suggest that Abcb6 is dispensable for physiological heme biosynthesis, but it becomes essential during conditions of high porphyrin demand.32
Mitochondrial heme export
Recently, we provided several pieces of evidence that the mitochondrial isoform of the Flvcr1 (Feline Leukemia Virus subgroup C Receptor 1) gene could be a mitochondrial heme exporter33,34 (Figure 2). There are two different isoforms of FLVCR1: FLVCR1a and FLVCR1b, expressed on the plasma membrane and in mitochondria, respectively. FLVCR1a, a member of the major facilitator superfamily of transporters with 12 transmembrane domains, was initially identified as a heme exporter essential for erythropoiesis.35,36 FLVCR1b is a shorter protein33 with only 6 transmembrane domains translated from an mRNA that arises from an alternative transcription start site located in the first intron of the Flvcr1 gene (Figure 3). FLVCR1b is supposed to homo- or heterodimerize to form a functional transporter.33,37,38
Figure 3.
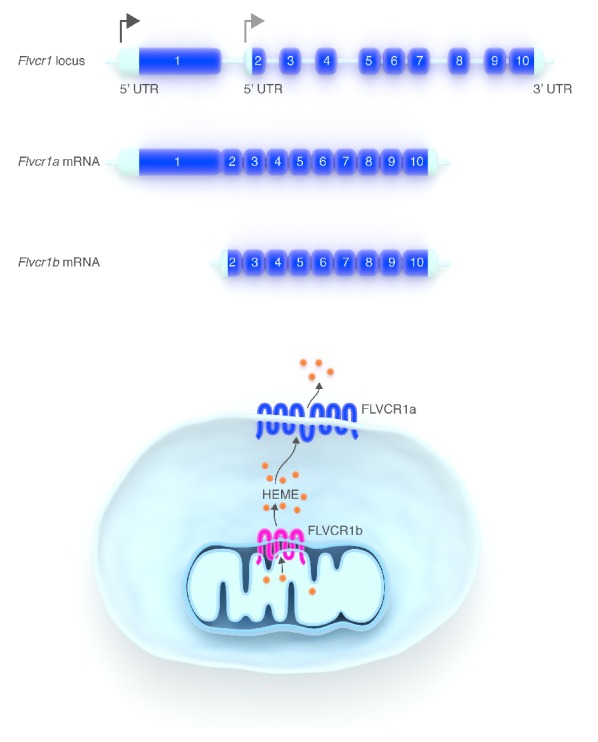
FLVCR1 isoforms. (A) Schematic representation of the Flvcr1 gene. Flvcr1a and Flvcr1b originate from two alternative transcription start sites (arrows). Flvcr1btranscript lacks the first exon of the Flvcr1gene. (B) Role of the two FLVCR1 isoforms. FLVCR1a, a 12-transmembrane domain protein, is a heme exporter localized at the plasma membrane. FLVCR1b has 6-transmembrane domains, it is supposed to homo/eterodimerize and it is expressed in mitochondria. There is much evidence to indicate that FLVCR1b is a mitochondrial heme exporter.
The role of FLVCR1b as a mitochondrial heme exporter is suggested by in vitro data indicating that Flvcr1b overexpression promotes heme synthesis whereas Flvcr1b silencing causes detrimental heme accumulation in mitochondria.33 Flvcr1b is essential for erythroid differentiation since Flvcr1b overexpression or silencing in K562 cells promotes or impairs, respectively, their differentiation.33 The comparison between different mouse models of Flvcr1 deficiency indicates that Flvcr1b controls fetal erythroid differentiation in vivo.33 Keel and collegues36 reported that mice lacking the Flvcr1 gene die in utero due to a complete block of erythroid differentiation. As the targeted disruption of the Flvcr1 gene was achieved by the deletion of the third exon, it is likely that both Flvcr1 isoforms have been deleted and thus the phenotype could result from the loss of Flvcr1a and/or Flvcr1b. Interestingly, Flvcr1a−/− mice have normal fetal erythropoiesis and die in utero due to severe hemorrhages and edema, indicating that Flvcr1b is able to support erythroid differentiation in the absence of Flvcr1a.33
FLVCR1b is the first mitochondrial heme exporter identified so far. We hypothesize that FLVCR1b function is finely coordinated with FLVCR1a-mediated heme export at the plasma membrane to ensure adequate heme content in cytosolic compartments.39 The aberrant expression of Flvcr1 isoforms may play a role in the pathogenesis of disorders characterized by heme deficiency and/or by an imbalance between heme and globin synthesis.
Regulatory roles of heme during erythroid differentiation
Heme is not only the prosthetic group of proteins involved in multiple cellular processes, but it also modulates gene expression through gene transactivation, translation, miRNA maturation and post-translational maturation. Heme regulates the transcription of several genes by binding to specific cis Heme Responsive Elements (HRE)40 and/or by controlling the activity of specific transcription factors. Moreover, heme controls translation through the heme-regulated eIF2α kinase (HRI)41 and it enhances the efficiency of pri-miRNA processing by binding and promoting the dimerization of the RNA-binding protein DiGeorge critical region-8 (DGCR8).42,43 Finally, heme controls the post-translational maturation of different proteins through a short cystein proline rich consensus sequence named Heme Regulatory Motif (HRM).44
Here we focus on the regulatory roles of heme during erythropoiesis. Recent data indicate that heme controls its own synthesis in differentiating erythroid cells. Moreover, heme controls the expression of α- and β-globin genes thus ensuring a balanced synthesis of all constituents of hemoglobin.
Heme regulates its own synthesis
In non-erythroid cells, heme synthesis is dependent on the activity of ALAS1, which is directly controlled by intracellular heme levels. Heme negatively regulates the transcription, translation and stability of Alas1 mRNA.45,46 The mitochondrial import of ALAS1 is also regulated by heme through the interaction with HRM.44 These represent crucial negative feedback mechanisms to maintain appropriate intracellular heme level in non-erythroid cells.
On the contrary, erythroid precursors undergoing differentiation need to strongly increase heme levels for hemoglobin synthesis. ALAS2 is not negatively regulated by heme.3,47 Recent data suggest that heme could enhance its own synthesis during erythroid differentiation by the regulation of ALAS2 expression through the IRE-IRP systems. The IRE binding activity of IRPs is regulated by cellular iron content through different mechanisms. The IRE binding activity of IRP1 is controlled by iron-sulfur clusters assembly while that of IRP2 by its oxidation, ubiquitination and degradation by the proteasome.6 This latter process is dependent on iron levels but also on heme. Heme interacts with an HRM consensus sequence in IRP248,49 and mediates the oxidation of IRP2 that triggers its ubiquitination and degradation. In addition, it has been reported that heme synthesis is positively regulated by the availability of iron within the range of iron concentrations in which IRP2 degradation is regulated.49 As Alas2 transcript contains a 5′IRE, the increase of heme level in erythroid precursors stimulates ALAS2 protein production due to the loss of IRP2-mediated translational repression.
Apart from Alas2, the expression of several other genes involved in iron metabolism is regulated by the IRE-IRP system. H-Ferritin, L-Ferritin, mAconitase and Ferroportin mRNAs contain a 5′IRE while DMT1 and TfR1 transcripts have a 3′IRE. The binding of IRPs to a 5′IRE prevents the initiation of translation while IRPs binding to the 3′IRE protects the mRNA from degradation.6 By controlling IRP2 oxidation/degradation, heme participates to the control of iron homeostasis in erythroid progenitors.
Heme controls the transcription of erythroid-specific genes
Heme directly regulates the transcription of several genes50–54 by binding the transcriptional repressor Btb And Cnc Homology 1 (BACH1).
BACH1 is a basic leucine zipper transcriptional repressor and a sensor of cellular heme levels. It antagonizes the activity of small Maf proteins (sMaf) that bind Maf recognition elements (MAREs) to activate the transcription of specific target genes.55 Heme binding to the BACH1 C-terminal domain inhibits its DNA binding activity, induces its dissociation from sMaf, triggers its export from the nucleus and induces its ubiquitination and degradation.55–57 In addition, heme stabilizes the nuclear factor erythroid-2-related factor 2 (NRF2) that binds to sMAF proteins to enhance gene transcription using specific Antioxidant Response Elements (ARE) which are specific forms of MAREs.58 If heme level is low, Kelch-like ECH-associated protein 1 (KEAP1) sequesters NRF2 in the cytoplasm by binding to its Neh2 domain.59 Oxidative stress inducers (sulforaphane or heme) react with specific cystein residues in Keap1 causing dissociation of the KEAP1-NRF2 complex and Nrf2 nuclear accumulation60 (Figure 4).
Figure 4.
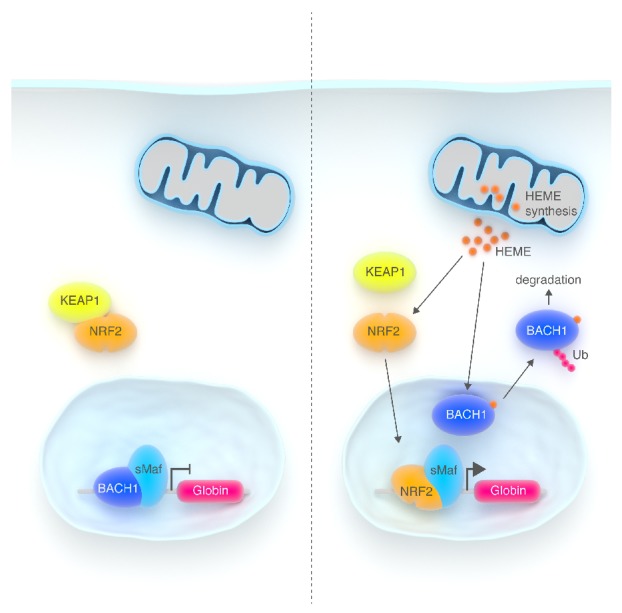
Heme controls the transcription of α- and β-globin genes in differentiating erythroid progenitors. (A) In early erythroid progenitors the transcription of α- and β-globin is inhibited by the transcriptional repressor BACH1 which antagonizes the activity of sMaf proteins that bind MAREs in the regulatory region of globin genes. (B) In late erythroid progenitors, when heme biosynthesis starts, heme binds to BACH1 in the nucleus and mediates its export in the cytosol. Finally, heme stabilizes the transcription factor NRF2 that accumulates in the nucleus. NFR2 associated with sMaf proteins activates the transcription of globin chains.
It has been reported that heme activates globin transcription by inhibiting the binding activity of BACH1 to the MARE sites in the locus control region of globin genes.52,53
In addition, heme also controls the transcription of other ubiquitously expressed genes, like the heme degrading enzyme heme-oxygenase-1,50 the iron storage proteins Hand L-ferritin51 and the iron exporter ferroportin.54 The role of heme-oxygenase1 during erythroid differentiation is still controversial. On the other hand, the transcriptional regulation of ferroportin, H- and L-ferritin by heme during erythroid differentiation has not been well investigated. However, this mechanism could contribute to the reorganization of iron metabolism in developing erythroblasts to ensure adequate iron supply to mitochondria for heme synthesis.
Heme regulates protein synthesis during erythroid differentiation
Protein synthesis in erythroid progenitors is mainly dependent on heme level that is sensed by HRI, a member of a family of protein kinases able to phosphorylate the α-subunit of the eukaryotic translation initiation factor (eIF2α). Once phosphorylated, eIF2α causes the inhibition of protein synthesis.41 HRI is predominantly expressed in the erythroid compartment61 and its kinase activity is directly regulated by heme level. Heme binding to the kinase domain of HRI inhibits its activity leading to decreased eIF2α phosphorylation and increased rate of translation, mainly of α- and β-globin mRNAs. On the other hand, during heme deficiency, the activation of HRI and subsequent phosphorylation of eIF2α cause the inhibition of protein synthesis. Thus, only following the initiation of heme biosynthesis in differentiating erythroid progenitors, the inhibition of HRI activity allows the synthesis of α- and β-globins. In this manner, HRI is fundamental to ensure that no globin is synthesized in excess of what can be assembled into hemoglobin tetramers (Figure 5).
Figure 5.

Heme controls the translation of α- and β-globin in differentiating erythroid progenitors. The translation of globin mRNAs is regulated by heme through the heme regulated kinase HRI. When heme binds HRI, HRI is inactivated and the translation initiation factor eIF2α is not phosphorylated allowing protein synthesis to occur.
Hri−/− mice show a mild hyperchromic, macrocytic anemia and altered adaptive response to iron deficiency. The normal adaptive response to iron deficiency is to shut down the synthesis of hemoglobin thus resulting in hypochromic, microcytic anemia. Hri−/− mice subjected to an iron-deficient diet develop a hyperchromic anemia with increased destruction of the late red cell precursors and compensatory erythroid hyperplasia. Excess globins resulting from HRI deficiency precipitate and form inclusions causing proteotoxicity, responsible for erythroid progenitor death.62 These data clearly demonstrate the essential role of HRI to shut off α- and β-globin synthesis in erythroid progenitors when iron supply is deficient, thus avoiding the detrimental precipitation of globin chains.62 Furthermore, the activation of HRI has been reported in a murine model of β-thalassemia intermedia as a consequence of the presence of denatured proteins and oxidative stress.63 HRI activation might represent an attempt to limit protein synthesis when α-globin-heme aggregates are formed as a consequence of reduced or absent β-globin synthesis. Consistently, the absence of HRI in β-thalassemic mice causes embryonic lethality due to severe anemia, thus indicating that HRI is required for the survival of differentiating erythroid progenitors.63
Recent data have started to elucidate the signaling pathway downstream HRI activation. It has been reported that HRI activates the Atf4 (Activating Transcriptional Factor 4) signaling pathway through the phosphorylation of eIF2α. The activation of the Atf4 signaling pathway is important to mitigate oxidative stress during chronic iron deficiency and is required for erythroid differentiation. Consistently, loss of Atf4 causes embryonic lethality due to severe fetal anemia.64 Moreover, an impairment of erythroid differentiation was observed in Hri−/− mice during chronic iron deficiency.65 The modulation of the HRI-eIF2α-Atf4 pathway, using salubrinal, a selective inhibitor of eIF2α dephosphorylation, has been proposed as a therapeutic strategy to ameliorate β-thalassemia. The increase in eIF2α phosphorylation, achieved by salubrinal treatment in β-thalassemic mice, results in the inhibition of protein synthesis and reduction of α-globin aggregates together with an enhancement of Atf4 signaling.65
Thus, by controlling HRI, heme regulates not only the synthesis of α- and β-globin under physiological conditions, but also the adaptive response of erythroid progenitors to stress conditions.
Erythroid disorders of heme biosynthesis
In the erythroid compartment, the alteration of heme biosynthesis rates causes pathological conditions like sideroblastic anemia or erythropoietic porphyria. The pathological alteration of the heme biosynthetic pathway may be due to different genetic abnormalities: mutation of genes coding for specific heme biosynthetic enzymes or for proteins involved in iron-sulfur cluster biogenesis.
Sideroblastic anemias
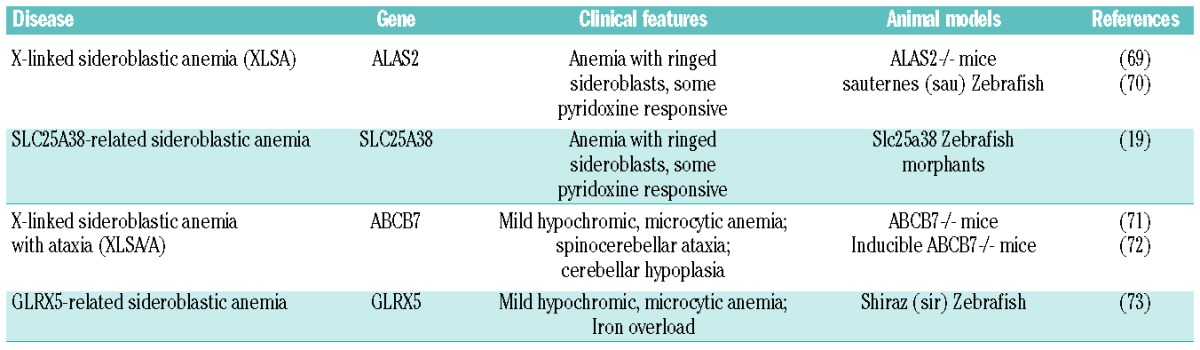
Sideroblastic anemias are genetically and clinically heterogeneous disorders characterized by the presence of bone marrow ‘ringed’ sideroblasts, iron-loaded mitochondria localized around the nucleus creating a ring-like appearance.66,67 Both congenital and acquired forms of sideroblastic anemias have been described. The inherited sideroblastic anemias are due to genes located on the X chromosome, genes on autosomal chromosomes or mitochondrial genes. Most of these conditions are characterized by decreased heme synthesis and mitochondrial iron overload.66,67 Acquired sideroblastic anemias are either primary, namely refractory anemia with ring sideroblasts (RARS) representing a type of myelodysplastic syndrome, or secondary due to some drugs, toxins, copper deficiency, or chronic neoplastic disease. Acquired sideroblastic anemias result from a defect in intracellular iron metabolism in erythroid cells.68 Here we focus on the inherited forms of sideroblastic anemia, highlighting how defects in the heme biosynthetic pathway or in iron-sulfur cluster biogenesis may lead to the pathological decrease of heme synthesis and to mitochondrial iron overload typical of this disorder (Table 1).69–82
Table 1.
Sideroblastic anemias.
X-linked sideroblastic anemia
X-linked sideroblastic anemia (XLSA) is the most common form of sideroblastic anemia, resulting from mutations in the gene coding for ALAS2.83–85 Recently, a novel condition of XLSA, due to mutation in an enhancer of the Alas2 gene, has been described. This mutation causes the disruption of a GATA binding site important for the transcriptional regulation of the gene, thus leading to a lower expression of Alas2 mRNA.4
Decreased activity of ALAS2 in bone marrow erythroblasts causes an impairment of heme biosynthesis and insufficient PPIX production to use all the available iron. Therefore, excess iron accumulates in mitochondria causing oxidative stress-induced cell damage. Patients show hypochromic, microcytic anemia of variable severity. Ineffective erythropoiesis, due to the increased ROS generation in erythroblasts, is also common in the most severe forms of XLSA.86 As in other conditions of ineffective erythropoiesis, increased intestinal iron absorption and systemic iron overload have been observed.86
Defects of ALAS2 cause a hypochromic, microcytic anemia in the sauternes (sau) zebrafish mutant. A delay in erythroid differentiation, abnormal globin gene expression and heme deficiency have been observed in sau mutants.70 The knockout of Alas2 in mice is embryonic lethal; Alas2−/− embryos develop a severe form of anemia characterized by a block of erythroid differentiation. In contrast to human patients, ring sideroblasts are not present and iron deposition occurs in the cytoplasm.69
SLC25A38-related sideroblastic anemia
Mutations in the gene coding for Slc25A38 have been identified in patients with an autosomal recessive form of sideroblastic anemia, similar to XLSA.19,87 It has been proposed that SLC25A38 is involved in the mitochondrial export of ALA;19 indeed, loss of Slc25A38 causes an impairment of heme synthesis leading to mitochondrial iron accumulation. The knockdown of SLC25A38 in zebrafish causes anemia similar to, although not as severe as, Alas2 deficiency.19
X-linked sideroblastic anemia with ataxia
X-linked sideroblastic anemia with ataxia (XLSA/A) is a rare form of sideroblastic anemia, characterized by a mild hypochromic, microcytic anemia associated with spin-ocerebellar ataxia and cerebellar hypoplasia. XLSA/A is due to mutations in the gene coding for the ATP-binding cassette sub-family B, member 7 (ABCB7) transporter,88 which is thought to be involved in the transfer of iron-sulfur clusters or their precursors from mitochondria to the cytosol.89 It has been proposed that ABCB7 deficiency could somehow diminish the availability of iron required for heme synthesis or, alternatively, cause the activation of IRP1 that interferes with the expression of Alas2.68 The knockout of Abcb7 gene is embryonic lethal in mice and the inducible deletion of Abcb7 in the bone marrow leads to bone marrow failure.71,72
GLRX5-related sideroblastic anemia
A homozygous mutation in the gene coding for glutaredoxin 5 (Glrx5) has been found in a patient with an autosomal recessive form of sideroblastic anemia. This patient is characterized by a hypochromic, microcytic anemia associated to iron overload.90 The same phenotype has been observed in the shiraz zebrafish model of GLRX5 deficiency.73,91 The molecular mechanism leading to sideroblastic anemia is similar to that observed in XLSA/A. GLRX5 is required for the assembly of iron-sulfur clusters, whose deficiency causes the activation of IRP1, mitochondria iron accumulation and cytosolic iron depletion that in turn activates IRP2. Thus, the activation of IRPs determines the translational repression of Alas2 resulting in a sideroblastic anemia.73,90,91
Therefore, a primary defect in iron-sulfur clusters biogenesis secondarily affects heme synthesis in erythroblast resulting in mitochondrial iron loading and the same pathophisiology of Alas2 deficiency.
Erythropoietic porphyrias
The inherited porphyrias are a group of genetic disorders resulting from mutations in specific genes coding for enzymes of the heme biosynthetic pathway. The porphyrias are characterized by the reduction of heme synthesis rates and accumulation of toxic heme precursors in different tissues, leading to hepatic and hematopoietic alterations, neurological and/or cutaneous symptoms. Depending on the primary site of porphyrin deposition, the porphyrias have been classified as hepatic or erythropoietic.92,93 Here, we focus on the erythropoietic forms of porphyria (Table 2).
Table 2.
Erythropoietic porphyrias.
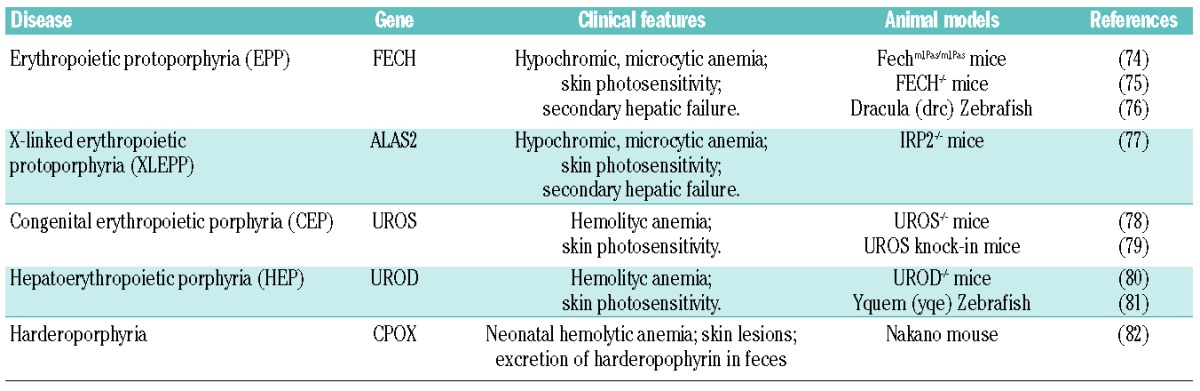
Erythropoietic protoporphyria
Erythropoietic protoporphyria (EPP) is an inherited disorder of heme biosynthesis due to a partial deficiency of FECH activity. In most patients, EPP results from the co-inheritance of a specific FECH mutation and a hypomorphic Fech allele common in the general population that reduces FECH activity below a critical threshold (residual activity lower than 35% of normal). In rare cases, EPP patients carry two trans mutations in the Fech gene.94,95
The reduced FECH activity leads to abnormally high levels of free PPIX in bone marrow, erythrocytes, plasma and liver. The major phenotype is porphyrin-induced photosensitivity as PPIX accumulation induces tissue damage through reactions with free radicals, triggered by light exposure. EPP may progress to severe hepatobiliary disease and hepatic failure.95 Hypochromic, microcytic anemia occurs in 20–60% of patients.96 Similar to what occurs in sideroblastic anemia, the presence of ring sideroblasts has been observed in EPP patients.97 It has been demonstrated that FECH deficiency leads to a steady state in which the decreased erythropoiesis is matched by reduced iron absorption and supply. This response is important to avoid the accumulation of toxic iron.96 The molecular mechanisms leading to decreased hemoglobin levels have been extensively studied in mouse models of the disease. Fechm1Pas/m1Pas mice, identified by a chemical mutagenesis screening, carry a loss of function mutation in the Fech gene. Fechm1Pas/m1Pas mice show a dramatic increase of PPIX in erythrocytes, plasma and liver resulting in microcytic anemia, skin-photosensitivity and hepatic failure.74 Another mouse model of EPP has been generated by the targeted disruption of Fech gene. Fech−/− mice die during embryonic development while Fech+/− mice show increased PPIX levels in erythrocytes and skin photosensitivity but no hepatic damage.75 The zebrafish model of Fech deficiency, Dracula, is characterized by PPIX accumulation, light-dependent hemolysis and liver disease76 The activation of HRI has been shown in Fechm1Pas/m1Pas mice as a consequence of heme deficiency and it is thought to prevent the accumulation of α- and β-globins. Consistently, in the absence of HRI, Fechm1Pas/m1Pas mice are more anemic and show a strong precipitation of globins in ‘inclusion bodies’. HRI deficiency in EPP also results in a dramatic increase in PPIX and more severe manifestation of liver pathology and skin photosensitivity.62 These data suggest a fundamental role for HRI in the adaptive response to low heme level in EPP patients.
X-linked erythropoietic protoporphyria
X-linked erythropoietic protoporphyria (XLEPP) is a clinically indistinguishable X-linked form of EPP, resulting from gain-of-function mutations of Alas2.98 In contrast to EPP, XLEPP patients show a high amount of zinc-protoporphyrin in erythrocytes. The overexpression of Alas2 in XLEPP patients increases PPIX production in spite of normal FECH activity. As iron becomes limiting, FECH uses its alternative metal substrate leading to the accumulation of Zn-protoporphyrin in erythrocytes.98 A phenotype similar to XLEPP has been described in Irp2−/− mice, which are characterized by a strong increase in ALAS2, resulting from the loss of IRP2-dependent translational repression. Irp2−/− mice develop erythroblast iron deficiency, microcytic anemia and erythropoietic protoporphyria.77 These data highlight the crucial role of IRPs in sensing iron levels and in coupling heme synthesis with iron availability in erythroid progenitors.
Congenital erythropoietic porphyria
Congenital erythropoietic porphyria (CEP) is an autosomal recessive disorder, resulting from mutations in the UROS gene. Reduced UROS activity causes the incomplete metabolism of hydroxymethylbilane and the accumulation of non-physiological porphyrin isomers in the bone marrow, erythrocytes, urine and other organs leading to hemolytic anemia, splenomegaly and cutaneous photosensitivity. Other clinical manifestations include erythrodontia and bone loss.78,93
The knockout of the Uros gene in mice is embryonic lethal. However, knock-in mouse models of CEP have been generated which mimic the disease phenotype, showing hemolytic anemia, hepatosplenomegaly, moderate photosensitivity and erythrodontia.78,79
Hepatoerythropoietic porphyria
Hepatoerythropoietic porphyria (HEP) is a very rare form of autosomic recessive erythropoietic porphyria due to mutation in the Urod gene. The reduced UROD activity leads to the accumulation of uroporphyrin, heptacarboxyl porphyrin and isocoproporphyrin in erythrocytes and liver. The phenotype of HEP is very close to that of CEP with hemolytic anemia associated to splenomegaly.93 The knockout of the Urod gene in mice is embryonic lethal and heterozygous mice do not completely recapitulate the disease.80 A zebrafish model of HEP has been generated which mimics the human disease.81
Harderoporphyria
Harderoporphyria is a rare autosomal recessive disorder due to homozygous or compound heterozygous mutations in the Cpox gene. It is characterized by neonatal hemolytic anemia, sometimes accompanied by skin lesions, and massive excretion of harderoporphyrin in feces. A mild residual anemia is chronically observed during childhood and adulthood.99 A hypomorphic mutation in the Cpox gene has been identified in the Nakano mouse, which is characterized by the excessive accumulation of CPgenIII in the lens causing hereditary cataract; unlike Nakano mice, human hereditary coproporphyria patients do not develop cataracts, likely reflecting species differences.82
Conclusions
Mutations in the Alas2 and Fech genes coding for the two rate-limiting enzymes of the heme biosynthetic pathway cause heme deficiency in erythroid compartment responsible for sideroblastic anemia and for the hypochromic, microcytic anemia observed in 20–60% of patients suffering from EPP,83–85 respectively. Similar anemic phenotypes result from mutations in genes coding for proteins involved in iron-sulfur cluster biogenesis highlighting the interplay between heme and iron metabolism.88,90 In addition, also the inability to absorb iron or alteration of iron transport, utilization and recycling reduce heme synthesis rates leading to iron deficiency anemia (IDA).67
As illustrated above, some studies suggested that heme, through HRI, may control the adaptive cell response to deficient erythropoiesis. It has been reported that HRI is required to prevent the phenotypic severity of IDA.62 A similar adaptive response to heme deficiency has been shown in the Fechm1Pas/m1Pas mouse model of EPP.63
Heme also regulates the transcription of many target genes by inhibiting BACH1. It has been demonstrated that in this way, heme controls the expression of globin genes in erythroid cells. In addition, heme might also control the expression of genes regulating iron uptake, storage and export as it has already been demonstrated in other cell types like macrophages, where heme-mediated BACH1 derepression orchestrates the response to heme overload.54
Thus, it is time to speculate that the adaptive cell response to heme deficiency may be manipulated pharmacologically to enhance cell performance ameliorating anemia. The successful treatment of thalassemic mice with salubrinal to potentiate HRI-mediated eIF2α phosphorylation highlighted the potential of these approaches.65 In the future, clarification of the HRI signaling pathway, along with the characterization of the complex regulatory network orchestrated by heme and iron, might open up the way to new therapeutic strategies.
Footnotes
Funding
This work was supported by Telethon Grant n. GGP12082 and by Regione Piemonte to E.T.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Bishop DF, Henderson AS, Astrin KH. Human delta-aminolevulinate synthase: assignment of the housekeeping gene to 3p21 and the erythroid-specific gene to the X chromosome. Genomics 1990;7(2):207–14 [DOI] [PubMed] [Google Scholar]
- 2.Harigae H, Suwabe N, Weinstock PH, Nagai M, Fujita H, Yamamoto M, et al. Deficient heme and globin synthesis in embryonic stem cells lacking the erythroid-specific delta-aminolevulinate synthase gene. Blood 1998;91(3):798–805 [PubMed] [Google Scholar]
- 3.Sadlon TJ, Dell’Oso T, Surinya KH, May BK. Regulation of erythroid 5-aminolevulinate synthase expression during erythropoiesis. Int J Biochem Cell Biol 1999;31(10):1153–67 [DOI] [PubMed] [Google Scholar]
- 4.Kaneko K, Furuyama K, Fujiwara T, Kobayashi R, Ishida H, Harigae H, et al. Identification of the novel erythroid-specific enhancer for ALAS2 gene and its loss-of-function mutation associated with congenital sideroblastic anemia. Haematologica 2014;99(2):252–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Surinya KH, Cox TC, May BK. Identification and characterization of a conserved erythroid-specific enhancer located in intron 8 of the human 5-aminolevulinate synthase 2 gene. J Biol Chem 1998;273(27): 16798–809 [DOI] [PubMed] [Google Scholar]
- 6.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 2004;117(3):285–97 [DOI] [PubMed] [Google Scholar]
- 7.Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta 2006;1763(7):723–36 [DOI] [PubMed] [Google Scholar]
- 8.Tugores A, Magness ST, Brenner DA. A single promoter directs both housekeeping and erythroid preferential expression of the human ferrochelatase gene. J Biol Chem 1994;269(49):30789–97 [PubMed] [Google Scholar]
- 9.Crooks DR, Ghosh MC, Haller RG, Tong WH, Rouault TA. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 2010;115(4):860–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008;320(5880): 1207–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaisman B, Fibach E, Konijn AM. Utilization of intracellular ferritin iron for hemoglobin synthesis in developing human erythroid precursors. Blood 1997;90(2):831–8 [PubMed] [Google Scholar]
- 12.Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 2007;110(1):125–32 [DOI] [PubMed] [Google Scholar]
- 13.Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol 2009;29(4):1007–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006;440(7080):96–100 [DOI] [PubMed] [Google Scholar]
- 15.Hamza I, Dailey HA. One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim Biophys Acta 2012;1823(9):1617–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W, Dailey HA, Paw BH. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010;116(4):628–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen W, Paradkar PN, Li L, Pierce EL, Langer NB, Takahashi-Makise N, et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc Natl Acad Sci USA 2009;106(38):16263–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch 2004;447(5): 689–709 [DOI] [PubMed] [Google Scholar]
- 19.Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet 2009;41(6):651–3 [DOI] [PubMed] [Google Scholar]
- 20.Graf SA, Haigh SE, Corson ED, Shirihai OS. Targeting, import, and dimerization of a mammalian mitochondrial ATP binding cassette (ABC) transporter, ABCB10 (ABC-me). J Biol Chem 2004;279(41):42954–63 [DOI] [PubMed] [Google Scholar]
- 21.Shirihai OS, Gregory T, Yu C, Orkin SH, Weiss MJ. ABC-me: a novel mitochondrial transporter induced by GATA-1 during erythroid differentiation. EMBO J 2000;19(11): 2492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang L, Bergevoet SM, Bakker-Verweij G, Harteveld CL, Giordano PC, Nijtmans L, et al. Human mitochondrial ATP-binding cassette transporter ABCB10 is required for efficient red blood cell development. Br J Haematol 2011;157(1):151–4 [DOI] [PubMed] [Google Scholar]
- 23.Hyde BB, Liesa M, Elorza AA, Qiu W, Haigh SE, Richey L, et al. The mitochondrial transporter ABC-me (ABCB10), a downstream target of GATA-1, is essential for erythropoiesis in vivo. Cell Death Differ 2012;19(7):1117–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bayeva M, Khechaduri A, Wu R, Burke MA, Wasserstrom JA, Singh N, et al. ABCB10 Regulates Early Steps of Heme Synthesis. Circ Res 2013;113(3):279–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, et al. Identification of a mammalian mitochondrial porphyrin transporter. Nature 2006;443(7111):586–9 [DOI] [PubMed] [Google Scholar]
- 26.Tsuchida M, Emi Y, Kida Y, Sakaguchi M. Human ABC transporter isoform B6 (ABCB6) localizes primarily in the Golgi apparatus. Biochem Biophys Res Commun 2008;369(2):369–75 [DOI] [PubMed] [Google Scholar]
- 27.Paterson JK, Shukla S, Black CM, Tachiwada T, Garfield S, Wincovitch S, et al. Human ABCB6 localizes to both the outer mitochondrial membrane and the plasma membrane. Biochemistry 2007;46(33):9443–52 [DOI] [PubMed] [Google Scholar]
- 28.Kiss K, Brozik A, Kucsma N, Toth A, Gera M, Berry L, et al. Shifting the paradigm: the putative mitochondrial protein ABCB6 resides in the lysosomes of cells and in the plasma membrane of erythrocytes. PLoS One 2012;7(5):e37378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelter G, Steinbach D, Konkimalla VB, Tahara T, Taketani S, Fiebig HH, et al. Role of transferrin receptor and the ABC transporters ABCB6 and ABCB7 for resistance and differentiation of tumor cells towards artesunate. PLoS One 2007;2(8):e798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helias V, Saison C, Ballif BA, Peyrard T, Takahashi J, Takahashi H, et al. ABCB6 is dispensable for erythropoiesis and specifies the new blood group system Langereis. Nat Genet 2012;44(2):170–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L, He F, Bu J, Zhen Y, Liu X, Du W, et al. ABCB6 mutations cause ocular coloboma. Am J Hum Genet 2012;90(1):40–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ulrich DL, Lynch J, Wang Y, Fukuda Y, Nachagari D, Du G, et al. ATP-dependent mitochondrial porphyrin importer ABCB6 protects against phenylhydrazine toxicity. J Biol Chem 2012;287(16):12679–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiabrando D, Marro S, Mercurio S, Giorgi C, Petrillo S, Vinchi F, et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J Clin Invest 2012; 122(12):4569–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fleming MD, Hamza I. Mitochondrial heme: an exit strategy at last. J Clin Invest 2012;122(12):4328–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, et al. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004;118(6):757–66 [DOI] [PubMed] [Google Scholar]
- 36.Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008;319(5864):825–8 [DOI] [PubMed] [Google Scholar]
- 37.Law CJ, Maloney PC, Wang DN. Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol 2008;62: 289–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pao SS, Paulsen IT, Saier MH., Jr Major facilitator superfamily. Microbiol Mol Biol Rev 1998;62(1):1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vinchi F, Ingoglia G, Chiabrando D, Mercurio S, Turco E, Silengo L, et al. The Heme Exporter FLVCR1a Regulates Heme Synthesis and Degradation and Controls Activity of Cytochromes P450. Gastroenterology 2014;146(5):1325–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sinclair J, Hamza I. A novel heme-responsive element mediates transcriptional regulation in Caenorhabditis elegans. J Biol Chem 2010;285(50):39536–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood 2007;109(7):2693–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Heme is involved in microRNA processing. Nat Struct Mol Biol 2007;14(1):23–9 [DOI] [PubMed] [Google Scholar]
- 43.Barr I, Smith AT, Chen Y, Senturia R, Burstyn JN, Guo F. Ferric, not ferrous, heme activates RNA-binding protein DGCR8 for primary microRNA processing. Proc Natl Acad Sci USA 2012;109(6):1919–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munakata H, Sun JY, Yoshida K, Nakatani T, Honda E, Hayakawa S, et al. Role of the heme regulatory motif in the heme-mediated inhibition of mitochondrial import of 5-aminolevulinate synthase. J Biochem 2004;136(2):233–8 [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto M, Hayashi N, Kikuchi G. Evidence for the transcriptional inhibition by heme of the synthesis of delta-aminolevulinate synthase in rat liver. Biochem Biophys Res Commun 1982;105(3):985–90 [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto M, Hayashi N, Kikuchi G. Translational inhibition by heme of the synthesis of hepatic delta-aminolevulinate synthase in a cell-free system. Biochem Biophys Res Commun 1983;115(1):225–31 [DOI] [PubMed] [Google Scholar]
- 47.Kramer MF, Gunaratne P, Ferreira GC. Transcriptional regulation of the murine erythroid-specific 5-aminolevulinate synthase gene. Gene 2000;247(1–2):153–66 [DOI] [PubMed] [Google Scholar]
- 48.Yamanaka K, Ishikawa H, Megumi Y, Tokunaga F, Kanie M, Rouault TA, et al. Identification of the ubiquitin-protein ligase that recognizes oxidized IRP2. Nat Cell Biol 2003;5(4):336–40 [DOI] [PubMed] [Google Scholar]
- 49.Ishikawa H, Kato M, Hori H, Ishimori K, Kirisako T, Tokunaga F, et al. Involvement of heme regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol Cell 2005;19(2):171–81 [DOI] [PubMed] [Google Scholar]
- 50.Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res 2007;35(21):7074–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hintze KJ, Katoh Y, Igarashi K, Theil EC. Bach1 repression of ferritin and thioredoxin reductase1 is heme-sensitive in cells and in vitro and coordinates expression with heme oxygenase1, beta-globin, and NADP(H) quinone (oxido) reductase1. J Biol Chem 2007;282(47):34365–71 [DOI] [PubMed] [Google Scholar]
- 52.Tahara T, Sun J, Igarashi K, Taketani S. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun 2004;324(1):77–85 [DOI] [PubMed] [Google Scholar]
- 53.Tahara T, Sun J, Nakanishi K, Yamamoto M, Mori H, Saito T, et al. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J Biol Chem 2004;279(7):5480–7 [DOI] [PubMed] [Google Scholar]
- 54.Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 2010;95(8):1261–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J 2001;20(11):2835–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki H, Tashiro S, Hira S, Sun J, Yamazaki C, Zenke Y, et al. Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J 2004;23(13):2544–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, et al. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol 2007;27(19):6962–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic Biol Med 2004;37(4):433–41 [DOI] [PubMed] [Google Scholar]
- 59.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci USA 2005;102(29):10070–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997;236(2):313–22 [DOI] [PubMed] [Google Scholar]
- 61.Crosby JS, Lee K, London IM, Chen JJ. Erythroid expression of the heme-regulated eIF-2 alpha kinase. Mol Cell Biol 1994;14(6):3906–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J 2001;20(23): 6909–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han AP, Fleming MD, Chen JJ. Heme-regulated eIF2alpha kinase modifies the phenotypic severity of murine models of erythropoietic protoporphyria and beta-thalassemia. J Clin Invest 2005;115(6):1562–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Masuoka HC, Townes TM. Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood 2002;99(3):736–45 [DOI] [PubMed] [Google Scholar]
- 65.Suragani RN, Zachariah RS, Velazquez JG, Liu S, Sun CW, Townes TM, et al. Heme-regulated eIF2alpha kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood 2012;119(22):5276–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fleming MD. Congenital sideroblastic anemias: iron and heme lost in mitochondrial translation. Hematology Am Soc Hematol Educ Program 2011;2011:525–31 [DOI] [PubMed] [Google Scholar]
- 67.Camaschella C, Poggiali E. Inherited disorders of iron metabolism. Curr Opin Pediatr 2011;23(1):14–20 [DOI] [PubMed] [Google Scholar]
- 68.Sheftel AD, Richardson DR, Prchal J, Ponka P. Mitochondrial iron metabolism and sideroblastic anemia. Acta Haematol 2009;122(2–3):120–33 [DOI] [PubMed] [Google Scholar]
- 69.Nakajima O, Takahashi S, Harigae H, Furuyama K, Hayashi N, Sassa S, et al. Heme deficiency in erythroid lineage causes differentiation arrest and cytoplasmic iron overload. EMBO J 1999;18(22):6282–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brownlie A, Donovan A, Pratt SJ, Paw BH, Oates AC, Brugnara C, et al. Positional cloning of the zebrafish sauternes gene: a model for congenital sideroblastic anaemia. Nat Genet 1998;20(3):244–50 [DOI] [PubMed] [Google Scholar]
- 71.Pondarre C, Antiochos BB, Campagna DR, Clarke SL, Greer EL, Deck KM, et al. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum Mol Genet 2006;15(6):953–64 [DOI] [PubMed] [Google Scholar]
- 72.Clarke SL, Vasanthakumar A, Anderson SA, Pondarre C, Koh CM, Deck KM, et al. Iron-responsive degradation of iron-regulatory protein 1 does not require the Fe-S cluster. EMBO J 2006;25(3):544–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wingert RA, Galloway JL, Barut B, Foott H, Fraenkel P, Axe JL, et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 2005;436(7053):1035–9 [DOI] [PubMed] [Google Scholar]
- 74.Tutois S, Montagutelli X, Da Silva V, Jouault H, Rouyer-Fessard P, Leroy-Viard K, et al. Erythropoietic protoporphyria in the house mouse. A recessive inherited ferrochelatase deficiency with anemia, photosensitivity, and liver disease. J Clin Invest 1991;88(5): 1730–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Magness ST, Maeda N, Brenner DA. An exon 10 deletion in the mouse ferrochelatase gene has a dominant-negative effect and causes mild protoporphyria. Blood 2002;100(4):1470–7 [DOI] [PubMed] [Google Scholar]
- 76.Childs S, Weinstein BM, Mohideen MA, Donohue S, Bonkovsky H, Fishman MC. Zebrafish dracula encodes ferrochelatase and its mutation provides a model for erythropoietic protoporphyria. Curr Biol 2000;10(16):1001–4 [DOI] [PubMed] [Google Scholar]
- 77.Cooperman SS, Meyron-Holtz EG, Olivierre-Wilson H, Ghosh MC, McConnell JP, Rouault TA. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 2005;106(3): 1084–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bishop DF, Johansson A, Phelps R, Shady AA, Ramirez MC, Yasuda M, et al. Uroporphyrinogen III synthase knock-in mice have the human congenital erythropoietic porphyria phenotype, including the characteristic light-induced cutaneous lesions. Am J Hum Genet 2006;78(4):645–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ged C, Mendez M, Robert E, Lalanne M, Lamrissi-Garcia I, Costet P, et al. A knock-in mouse model of congenital erythropoietic porphyria. Genomics 2006;87(1):84–92 [DOI] [PubMed] [Google Scholar]
- 80.Phillips JD, Jackson LK, Bunting M, Franklin MR, Thomas KR, Levy JE, et al. A mouse model of familial porphyria cutanea tarda. Proc Natl Acad Sci USA 2001;98(1):259–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang H, Long Q, Marty SD, Sassa S, Lin S. A zebrafish model for hepatoerythropoietic porphyria. Nat Genet 1998;20(3):239–43 [DOI] [PubMed] [Google Scholar]
- 82.Mori M, Gotoh S, Taketani S, Hiai H, Higuchi K. Hereditary cataract of the Nakano mouse: Involvement of a hypomorphic mutation in the coproporphyrinogen oxidase gene. Exp Eye Res 2013;112:45–50 [DOI] [PubMed] [Google Scholar]
- 83.Ducamp S, Kannengiesser C, Touati M, Garcon L, Guerci-Bresler A, Guichard JF, et al. Sideroblastic anemia: molecular analysis of the ALAS2 gene in a series of 29 probands and functional studies of 10 missense mutations. Hum Mutat 2011;32(6):590–7 [DOI] [PubMed] [Google Scholar]
- 84.Bergmann AK, Campagna DR, McLoughlin EM, Agarwal S, Fleming MD, Bottomley SS, et al. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer 2010;54(2):273–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cotter PD, Baumann M, Bishop DF. Enzymatic defect in “X-linked” sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase deficiency. Proc Natl Acad Sci USA 1992;89(9):4028–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Camaschella C. Hereditary sideroblastic anemias: pathophysiology, diagnosis, and treatment. Semin Hematol 2009;46(4):371–7 [DOI] [PubMed] [Google Scholar]
- 87.Kannengiesser C, Sanchez M, Sweeney M, Hetet G, Kerr B, Moran E, et al. Missense SLC25A38 variations play an important role in autosomal recessive inherited sideroblastic anemia. Haematologica 2011;96(6):808–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet 1999;8(5):743–9 [DOI] [PubMed] [Google Scholar]
- 89.Lill R, Muhlenhoff U. Iron-sulfur protein biogenesis in eukaryotes: components and mechanisms. Annu Rev Cell Dev Biol 2006;22:457–86 [DOI] [PubMed] [Google Scholar]
- 90.Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 2007;110(4):1353–8 [DOI] [PubMed] [Google Scholar]
- 91.Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, et al. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest 2010;120(5):1749–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Balwani M, Desnick RJ. The porphyrias: advances in diagnosis and treatment. Blood 2012;120(23):4496–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sassa S, Kappas A. Molecular aspects of the inherited porphyrias. J Intern Med 2000;247(2):169–78 [DOI] [PubMed] [Google Scholar]
- 94.Gouya L, Puy H, Robreau AM, Bourgeois M, Lamoril J, Da Silva V, et al. The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat Genet 2002;30(1):27–8 [DOI] [PubMed] [Google Scholar]
- 95.Lecha M, Puy H, Deybach JC. Erythropoietic protoporphyria. Orphanet J Rare Dis 2009;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Holme SA, Worwood M, Anstey AV, Elder GH, Badminton MN. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood 2007;110(12):4108–10 [DOI] [PubMed] [Google Scholar]
- 97.Rademakers LH, Koningsberger JC, Sorber CW, Baart de la Faille H, Van Hattum J, Marx JJ. Accumulation of iron in erythroblasts of patients with erythropoietic protoporphyria. Eur J Clin Invest 1993;23(2):130–8 [DOI] [PubMed] [Google Scholar]
- 98.Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet 2008;83(3):408–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schmitt C, Gouya L, Malonova E, Lamoril J, Camadro JM, Flamme M, et al. Mutations in human CPO gene predict clinical expression of either hepatic hereditary coproporphyria or erythropoietic harderoporphyria. Hum Mol Genet 2005;14(20):3089–98 [DOI] [PubMed] [Google Scholar]