

The vast majority of acute myeloid leukemia (AML) patients harboring an FLT3-ITD mutation experience relapse within a short period of time after discontinuation of chemotherapy.1 Treatment options include experimental trials using FLT3-tyrosine kinase inhibitors (TKI) or allogeneic stem cell transplantation (alloSCT). Inhibitors that are currently being investigated in advanced clinical trials with promising clinical responses include midostaurin (PKC412) and quizartinib (AC220). Resistance-mediating mutations emerging upon long-term exposure to these inhibitors have confirmed mutated FLT3-kinase as a valid therapeutic target.2,3 Both compounds have proven the ability to salvage refractory FLT3-ITD-positive AML and thus allowed ‘bridging’ of patients towards alloSCT. The role of alloSCT in first complete remission for FLT3-ITD-mutated AML remains a topic of current debate. Patients undergoing alloSCT do show a survival benefit in retrospective analyses; however, the majority of patients will eventually experience relapse.4 The combination alloSCT and concomitant inhibition of mutated FLT3-kinase may facilitate development of leukemia-specific T-cell (graft-versus-leukemia, GvL) responses after discontinuation of immunosuppression with the malignant clone being held in check by the kinase inhibitor. GvL activity has been documented in AML patients in general5 and even ITD-specific T-cell responses have been described in vitro.6 Kinase inhibitors can, in general, impair T-cell function through inhibition of various signaling pathways. Inhibition of this protective GvL effect after alloSCT could eventually lead to reactivation of the malignant clone. Although inhibiting T-cell reactivity at higher concentrations,7 TKI such as imatinib can be administered safely after alloSCT8,9 without any increased risk of relapse. Other TKI such as nilotinib10 or dasatinib11 do interfere with T-cell reactivity even at low nanomolar concentrations. Based on these findings, we aimed to assess the effects of clinically relevant doses of PKC412 and AC220 on T-cell signaling, proliferation and reactivity.
Most 1st-generation FLT3-TKIs such as PKC412 target various kinases and therefore act in relatively unspecific way while AC220 acts in a far more FLT3-specific manner.12 To investigate the effects of both clinically relevant FLT3-TKI on T-cell receptor (TCR) signaling we used primary human T cells derived from healthy donors (HD-TC) and applied a dose range of 5–50 nM midostaurin and 10–50 nM quizartinib. These concentrations had been described as trough levels during inhibitor therapy in early clinical trials.2,12–14 Thus, all effects observed resemble the clinical situation in terms of dosing and pharmacokinetics. The SRC-kinase inhibitor dasatinib was included as a positive control (at doses beyond the clinically achievable levels).
With regard to T-cell receptor (TCR)-mediated signaling, we investigated bona fide signaling molecules downstream of the TCR. Besides SRC-kinases such as LCK, also ZAP70, PLCG1 and the MAPK/ERK pathway have been described as playing a pivotal role in T-cell activation. Treatment with clinically relevant doses of midostaurin (Figure 1A, left panel) and quizartinib (Figure 1A, right panel) (50 nM) did not inhibit activation of any investigated TCR-signaling pathway. Comparable to DMSO control, overall phosphorylation was induced almost immediately after 0.5% PHA-stimulation (Figure 1A). Dasatinib treatment led to reduction in global tyrosine phosphorylation (Figure 1A, top panels) using the 4G10 antibody. Likewise, activation of all downstream signaling pathways appeared to be inhibited, consistent with previously published reports.11
Figure 1.
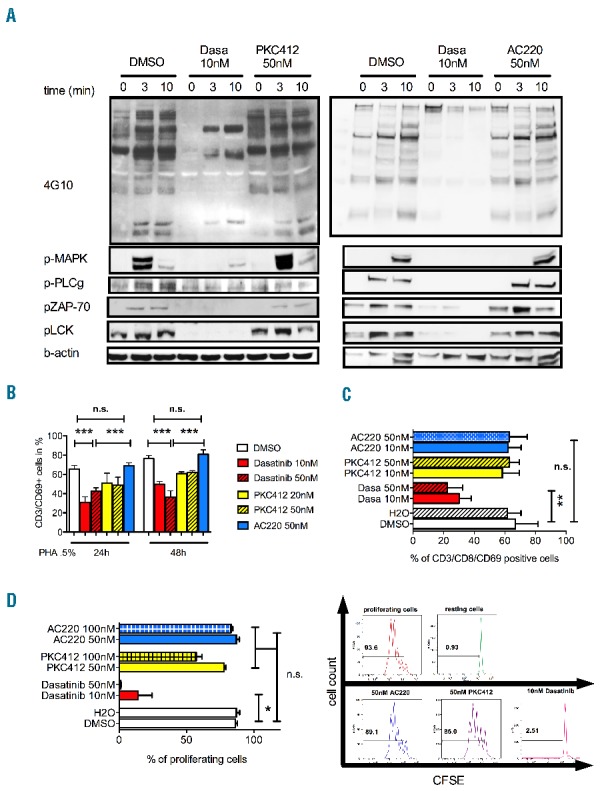
(A) Western blot analysis of T-cell receptor signaling upon inhibitor treatment. Cells were treated with the respective TKI for 48 h prior to short-term TCR stimulation. In detail, primary T cells from healthy donors (HD-TC) were treated with indicated concentrations of the respective kinase inhibitors (TKI) for 48 h and then stimulated with PHA (0.5%). Whole protein lysates were prepared immediately and after 3 and 10 min of PHA 0.5% stimulation. Midostaurin (PKC412, left panel) and quizartinib (AC220, right panel) did not reduce global tyrosine phosphorylation or activation of downstream signaling molecules (ZAP70, MAPK, LCK, PLCG1). Dasatinib treatment led to global reduction in global tyrosine phosphorylation and inhibition of downstream signaling pathways. Shown are 2 representative blots out of 6 healthy donors. (B, C) CD69 expression was determined by flow cytometry (n>5 per group). HD-TCs were stimulated with PHA 0.5% (B) or CD3/CD28-beads (for 48 h at a bead-to-cell ratio of 1:1; Dynabeads® Human T-Activator; Life Technologies) (C) and treated with indicated concentrations of the respective TKI. In both analyses all concentrations of midostaurin and quizartinib applied left CD69 expression unaffected. Dasatinib significantly reduced CD69 expression. (D) T-cell proliferation was assessed by CFSE labeling (n=4). HD-TCs stimulated for 24 h with 0.5% PHA/IL2 and co-incubated with kinase inhibitors dasatinib, midostaurin or quizartinib or DMSO. CFSE fluorescence was measured by flow cytometry on Day 5 post stimulation. 10nM dasatinib abrogated the proliferation activity (right). The FLT3-inhibitors midostaurin and quizartinib did not impair T-cell proliferation at concentrations applied.
Activation of primary T cells is a critical step in immune responses against viral and tumor antigens. Several surface markers such as CD69 have been described as indicators of T-cell activation. HD-TCs were stimulated using either PHA0.5% or CD3/CD28-beads. Applying clinically relevant doses of midostaurin or quizartinib had no impact on CD69 surface expression. CD69-expression on gated CD8+ cytotoxic T cells or (ungated) CD3+ cells was comparable to DMSO control, even in the presence of 50 nM midostaurin or quizartinib (Figure 1B and C). Dasatinib exposure significantly reduced CD69 expression on CD3+ T cells following TCR stimulation.
Reduction of the T-cell pool through decreased cell proliferation or induction of apoptosis could hamper immune responses against viral or tumor antigens. Therefore, we assessed T-cell proliferation by CFSE-labeling assays. Concentrations of up to 100 nM of either FLT3 inhibitor did not reveal any negative impact on proliferative capacity of previously stimulated T cells. Incubation with dasatinib almost abrogated proliferative activity (Figure 1D). Exposure of T cells to either inhibitor did not lead to any significant induction of apoptosis (data not shown).
Finally, we aimed to investigate the functional impact of FLT3-TKI on T-cell reactivity and function. To assess for T-cell reactivity directed against HLA-antigens, antigen-presenting cells (APC) derived from healthy donors were co-incubated with T cells derived from unmatched, unrelated donors in the presence of either FLT3 inhibitor or DMSO. Thymidine incorporation was used as the readout for T-cell reactivity. Neither FLT3-TKI affected T-cell reactivity when used at concentrations of up to 50 nM while dasatinib reduced T-cell reactivity to the maximum extent (Figure 2A). To confirm these findings we used an ELISPOT assay to assess for T-cell reactivity directed against viral peptides (CMVpp65) loaded on APC from healthy donors that were co-incubated with HD-TCs from the same individual. T-cell reactivity was preserved and comparable to DMSO control in the presence of 50 nM midostaurin or quizartinib (Figure 2B and C). Dasatinib abrogated the T-cell response. Similar effects could be confirmed in vivo using an intestinal graft-versus-host disease (GvHD) model. Mice were transplanted with unmatched T cells and treated with PKC412, dasatinib or vehicle control. Macroscopic analysis (soreness, Figure 2D) as well as histology of ileum and colon was performed to assess for GvHD development. Consistent with our previous findings, treatment with PKC412 or vehicle-control did not lead to any reduction of GvHD development (Figure 2E–G). Treatment with dasatinib significantly reduced the extent of GvHD.
Figure 2.
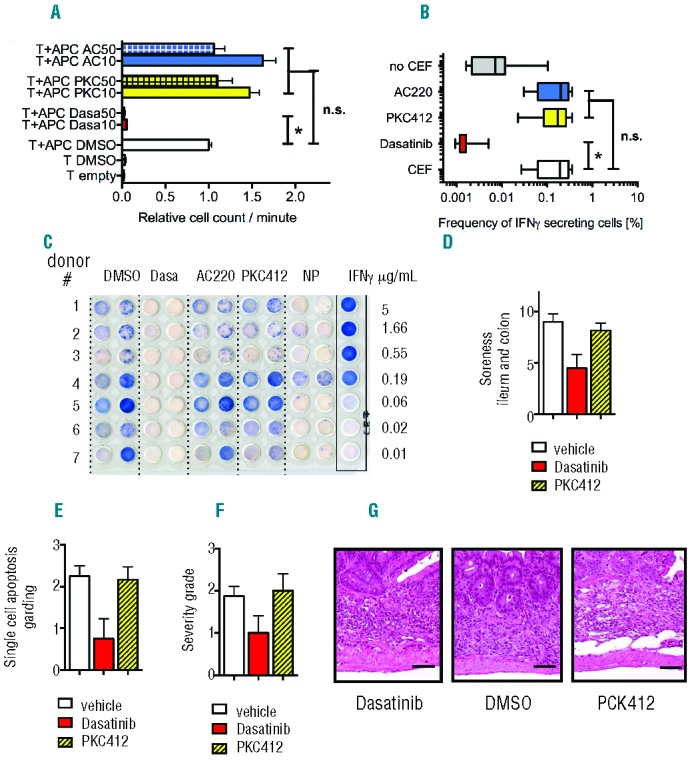
(A) Allogeneic (unmatched) T-cell reactivity was measured by 3H-thymidine incorporation. HD-TCs were co-incubated with antigen presenting cells (APC; T-cell depleted PBMC) from an unrelated healthy donor in the absence or presence of the respective TKI. 50nM of midostaurin and quizartinib did not have any negative impact on T-cell reactivity compared to DMSO control. Dasatinib impaired T-cell reactivity to less than 5% of control. (B, C) ELISPOT assay to assess for HD-TC-responses directed against viral antigens (n=10). Lytic T-cell activity was determined based on the frequency of IFN- secreting cells. Autologous APC loaded with a viral peptide pool (CMVpp65; PepMixTM HCMVA(pp65), JPT Inc., Berlin, Germany) were incubated with T cells (of the same individual) in the absence or presence of the respective TKI (as indicated). Midostaurin (50nM) and quizartinib (50nM) did not impair lytic activity. 10nM dasatinib significantly inhibited the virus-specific T-cell response. APC loaded with control peptides (CEF) or without peptides served as positive and negative control. These results could be recapitulated using human influenza virus derived peptides as an antigen. (D–G) Allogeneic (un-matched) GvHD (graft-versus-host-disease) assay in vivo: 0.5 × 106 CD3+ T cells derived from BL/6 mice were transplanted along with 2 × 106 BALB/c WBMC into lethally irradiated (13Gy) BALB/c recipient mice. Animals developed severe GvHD of the gut within 3–4 weeks after transplantation. Midostaurin treatment was performed after engraftment (Days 14–19; 100mg/kg body weight by gavage, every 24h). Ileum and colon fixed in formalin and embedded in paraffin were analyzed for apoptosis of crypt cells on Day 25. Midostaurin treatment (n=3) did not affect severity of GvHD in the mouse gut. Both, severity of GvH-reaction and induction of apoptosis in the ileum or colon were comparable to vehicle treated animals (n=2). Dasatinib (2.5mg/kg) treated animals (n=3) revealed decreased induction of apoptosis and reduced severity of GvHD in both, ileum and colon.
Taken together our results provide first evidence that clinically relevant doses of PKC412 and AC220 leave human T-cell signaling, proliferation and function unaffected. Although our study is limited by the use of HD-TC that may act differently to T cells derived from AML patients, our findings facilitate a pre-clinical assessment for the use of FLT3-TKI in the context of alloSCT. Without affecting T-cell function, midostaurin and quizartinib could be concomitantly used until discontinuation of immunosuppressive therapy and thereby prevent relapse prior to appearance of a sufficient GvL-response. Interestingly, differential effects of FLT3-TKI on dendritic cell function that may even stimulate GvHD cannot be excluded by our experiments and need to be considered.15
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
Funding: this work was supported by a grant from Novartis Inc. to F.H.H. and partially by a DFG-grant (FI405/5-1) to T.F. and F.H and the Collaborative Research Cluster (CRC854) to T.M.S., S.K., B.S., M.B.W., B.I., T.F. and F.H.H. All human samples were collected and stored in the Hematology Tissue-Bank Magdeburg (HTM) supported by a grant from the Jose-Carreras Foundation (SP12/04) to F.H.H. F.H.H. received research funding from Novartis Inc. All other authors have nothing to disclose.
References
- 1.Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008;111(5):2776–84 [DOI] [PubMed] [Google Scholar]
- 2.Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood 2006;107(1):293–300 [DOI] [PubMed] [Google Scholar]
- 3.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012;485(7397):260–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunet S, Labopin M, Esteve J, Cornelissen J, Socie G, Iori AP, et al. Impact of FLT3 internal tandem duplication on the outcome of related and unrelated hematopoietic transplantation for adult acute myeloid leukemia in first remission: a retrospective analysis. J Clin Oncol 2012;30(7):735–41 [DOI] [PubMed] [Google Scholar]
- 5.Van Driessche A, Gao L, Stauss HJ, Ponsaerts P, Van Bockstaele DR, Berneman ZN, et al. Antigen-specific cellular immunotherapy of leukemia. Leukemia 2005;19(11):1863–71 [DOI] [PubMed] [Google Scholar]
- 6.Graf C, Heidel F, Tenzer S, Radsak MP, Solem FK, Britten CM, et al. A neoepitope generated by an FLT3 internal tandem duplication (FLT3-ITD) is recognized by leukemia-reactive autologous CD8+ T cells. Blood 2007;109(7):2985–8 [DOI] [PubMed] [Google Scholar]
- 7.Seggewiss R, Lore K, Greiner E, Magnusson MK, Price DA, Douek DC, et al. Imatinib inhibits T-cell receptor-mediated T-cell proliferation and activation in a dose-dependent manner. Blood 2005;105(6):2473–9 [DOI] [PubMed] [Google Scholar]
- 8.Hess G, Bunjes D, Siegert W, Schwerdtfeger R, Ledderose G, Wassmann B, et al. Sustained complete molecular remissions after treatment with imatinib-mesylate in patients with failure after allogeneic stem cell transplantation for chronic myelogenous leukemia: results of a prospective phase II open-label multicenter study. J Clin Oncol 2005;23(30):7583–93 [DOI] [PubMed] [Google Scholar]
- 9.Pfeifer H, Wassmann B, Bethge W, Dengler J, Bornhauser M, Stadler M, et al. Randomized comparison of prophylactic and minimal residual disease-triggered imatinib after allogeneic stem cell transplantation for BCR-ABL1-positive acute lymphoblastic leukemia. Leukemia 2013;27(6):1254–62 [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Schmitt A, Chen B, Rojewski M, Rubeler V, Fei F, et al. Nilotinib hampers the proliferation and function of CD8+ T lymphocytes through inhibition of T cell receptor signalling. J Cell Mol Med 2008;12(5B):2107–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fei F, Yu Y, Schmitt A, Rojewski MT, Chen B, Greiner J, et al. Dasatinib exerts an immunosuppressive effect on CD8+ T cells specific for viral and leukemia antigens. Exp Hematol 2008; 36(10):1297–308 [DOI] [PubMed] [Google Scholar]
- 12.Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009;114(14):2984–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao Q, Sprankle KG, Grotzfeld RM, Lai AG, Carter TA, Velasco AM, et al. Identification of N-(5-tert-butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1, 3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor. Med Chem 2009;52(23):7808–16 [DOI] [PubMed] [Google Scholar]
- 14.Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer 2007; 7(5):345–56 [DOI] [PubMed] [Google Scholar]
- 15.Wolfl M, Schwinn S, Yoo YE, Ress ML, Braun M, Chopra M, et al. Src-kinase inhibitors sensitize human cells of myeloid origin to Toll-like-receptor-induced interleukin 12 synthesis. Blood 2013; 122(7):1203–13 [DOI] [PMC free article] [PubMed] [Google Scholar]