

Abstract
Transferrin receptor 2 (TFR2) is a transmembrane glycoprotein expressed in the liver and in the erythroid compartment, mutated in a form of hereditary hemochromatosis. Hepatic TFR2, together with HFE, activates the transcription of the iron-regulator hepcidin, while erythroid TFR2 is a member of the erythropoietin receptor complex. The TMPRSS6 gene, encoding the liver-expressed serine protease matriptase-2, is the main inhibitor of hepcidin and inactivation of TMPRSS6 leads to iron deficiency with high hepcidin levels. Here we evaluate the phenotype resulting from the genetic loss of Tmprss6 in Tfr2 total (Tfr2−/−) and liver-specific (Tfr2LCKO) knockout mice. Tmprss6−/−Tfr2−/− and Tmprss6−/−Tfr2LCKO mice have increased hepcidin levels and show iron-deficiency anemia like Tmprss6−/−mice. However, while Tmprss6−/−Tfr2LCKO are phenotypically identical to Tmprss6−/− mice, Tmprss6−/−Tfr2−/− mice have increased red blood cell count and more severe microcytosis than Tmprss6−/− mice. In addition hepcidin expression in Tmprss6−/−Tfr2−/− mice is higher than in the wild-type animals, but lower than in Tmprss6−/− mice, suggesting partial inhibition of the hepcidin activating pathway. Our results prove that hepatic TFR2 acts upstream of TMPRSS6. In addition Tfr2 deletion causes a relative erythrocytosis in iron-deficient mice, which likely attenuates the effect of over-expression of hepcidin in Tmprss6−/− mice. Since liver-specific deletion of Tfr2 in Tmprss6−/− mice does not modify the erythrocyte count, we speculate that loss of Tfr2 in the erythroid compartment accounts for the hematologic phenotype of Tmprss6−/−Tfr2−/− mice. We propose that TFR2 is a limiting factor for erythropoiesis, particularly in conditions of iron restriction.
Introduction
Transferrin receptor 2 (TFR2) is a transmembrane protein homologous to transferrin receptor 1 (TFR1) which is mutated in hereditary hemochromatosis type 3.1,2 TFR2 is expressed in the liver and, to a lower extent, in erythroid cells.3,4 TFR2 protein is stabilized on cell surface by binding to its ligand, diferric transferrin (holo-TF),5 and, in a complex with the hemochromatosis protein HFE, is considered a sensor of circulating iron. In the current model in conditions of iron deficiency HFE associates with TFR1; inversely, when transferrin saturation increases, competitive binding of holo-TF displaces HFE from TFR1 and the HFE-TFR2 complex activates HAMP transcription.6,7 However, the phenotype of the HFE and TFR2-related disease is different8 and the association between the two proteins has recently been questioned.9
Hepcidin blocks dietary iron absorption and iron recycling from senescent erythrocytes by inducing the degradation of the iron exporter ferroportin on enterocytes and macrophages, respectively.10 The mechanism of HAMP activation by TFR2 and HFE is still unclear. Both proteins probably contribute to HAMP upregulation by bone morphogenetic proteins (BMP) in response to increased tissue iron.11,12 BMP6, using hemojuvelin as a co-receptor, signals through sons-of-mothers-against-decapenthaplegic 1/5/8 (SMAD1/5/8) proteins. In agreement, HFE and TFR2 in vitro may form a multi-protein complex with hemojuvelin.13 The role of hepatic TFR2 as a regulator of HAMP transcription is confirmed by the phenotype of the Tfr2 total (Tfr2−/−) and liver-specific (Tfr2LCKO) knockout mouse models. Both mice are characterized by iron overload and low Hamp levels relative to their high iron stores, with Tfr2LCKO having more severe liver iron accumulation than Tfr2−/− animals.14,15
Recently TFR2 has been identified as a component of the erythropoietin receptor (EPOR) complex. TFR2 and the EPOR are co-expressed during erythroid differentiation, TFR2 associates with EPOR in the endoplasmic reticulum and is required for the efficient transport of the EPOR to the cell surface. Moreover TFR2 knockdown in vitro delays the terminal differentiation of erythroid precursors16 indicating that TFR2 is required for efficient erythropoiesis.
The BMP6-hemojuvelin-HAMP pathway is inhibited by matriptase-2, a type II transmembrane serine protease encoded by the TMPRSS6 gene. By cleaving hemojuvelin,17 TMPRSS6 strongly impairs BMP-mediated HAMP activation in the liver. TMPRSS6 mutations both in humans18 and in mice19,20 cause excessive HAMP production and iron-refractory, iron-deficiency anemia (IRIDA).21 The important role of TMPRSS6 in erythropoiesis is also highlighted by genome-wide association studies: indeed, common TMPRSS6 genetic variants associate with iron and erythrocyte traits in different populations.22–27 By studying Tmprss6 haploinsufficient mice28 and hepcidin levels of normal individuals and the TMPRSS6 common single nucleotide polymorphism (rs855791)29 we demonstrated that even a partial inability to modulate hepcidin influences iron parameters and, indirectly, erythropoiesis.
The regulation of TMPRSS6 and its activity is incompletely understood: besides hypoxia,30 iron and BMP6, through the BMP-SMAD pathway, induce TMPRSS6 expression, likely as a negative feedback loop to limit excessive increases of HAMP.31 However, the regulation of TMPRSS6 in vivo according to iron needs remains to be clarified. A possible role of Tmprss6 in iron overload was demonstrated by Finberg et al.32 who showed that Hfe−/−mice with complete loss of Tmprss6 revert from a phenotype of iron overload to one of iron-deficiency anemia with high Hamp levels. These findings suggest that HFE acts genetically upstream of TMPRSS6 in the modulation of the BMP-SMAD pathway and of HAMP expression. In analogy with these results and given the role of TFR2 in erythropoiesis16 we wondered whether TFR2 is involved in the regulation of TMPRSS6. To answer this question, we back-crossed Tmprss6−/− mice with animals with a complete deletion of Tfr2 (Tfr2−/−) and analyzed the hematologic phenotype and the Bmp-Smad-Hamp pathway of the double mutant mice. Moreover, in order to discriminate between the hepatic and extra-hepatic functions of TFR2, we performed the same analysis in Tmprss6−/− mice lacking Tfr2 specifically in the liver (Tfr2LCKO).15
Methods
Mouse models
Mice were maintained in the animal facility of the Department of Clinical and Biological Sciences, University of Turin (Italy) in accordance with European Union guidelines. Each study was approved by the Institutional Animal Care and Use Committee (IACUC) of the same institution.
A Tmprss6−/− mouse model on a mixed C57BL/6-Sv129 background was kindly provided by Prof. C. Lopez-Otin (University of Oviedo, Spain) and maintained by brother-sister mating for more than ten generations. Tfr2−/− and Tfr2LCKO mice on a pure 129S2 background were generated as previously described.15 For the experimental work described we bred Tfr2−/− or Tfr2LCKO mice with Tmprss6+/− mice and then intercrossed the F1 progeny to generate various genotype combinations (F2: wild-type, Tmprss6+/−, Tmprss6−/−, Tfr2−/−, Tmprss6+/−Tfr2−/−, Tmprss6−/−Tfr2−/−, Tfr2LCKO, Tmprss6+/−Tfr2LCKO, Tmprss6−/−Tfr2LCKO). Mice were given a standard diet (480 mg iron/Kg) and only male mice were analyzed when 10 weeks old. Blood was collected for hematologic analyses, transferrin saturation and erythropoietin levels. After sacrifice livers and spleens were dissected, weighed, and snap-frozen immediately for RNA analysis or dried for tissue iron quantification.
Hematologic analyses
Blood was obtained by retro-orbital puncture from anesthetized mice. Red blood cell and white blood cell counts, hemoglobin concentration, hematocrit and erythrocyte indices (mean corpuscular volume, mean corpuscular hemoglobin) were measured using an ADVIA®120 Hematology System (Siemens Diagnostics).
Transferrin saturation was calculated as the ratio of serum iron and total iron binding capacity levels, using the Total Iron Binding Capacity kit (Randox Laboratories Ltd.), according to the manufacturer’s instructions. Serum erythropoietin levels were measured using a mouse Erythropoietin Quantikine set (R&D System), according to the manufacturer’s instructions.
Tissue iron content
To measure iron concentration, tissue samples were dried at 110°C overnight, weighed, and digested in 1 mL of 3M HCl, 0.6M trichloroacetic acid for 20 h at 65°C. The clear acid extract was added to 1 mL of working chromogen reagent (1 volume of 0.1% bathophenanthroline sulfate and 1% thioglycolic acid solution, 5 volumes of water, and 5 volumes of saturated sodium acetate). The solutions were then incubated for 30 min at room temperature until color development and the absorbance measured at 535 nm. A standard curve was plotted using an acid solution containing increasing amounts of iron diluted from a stock solution of Titrisol iron standard (Merck, Darmstadt, Germany).
Quantitative reverse transcriptase polymerase chain reaction
Total RNA was extracted from the liver and spleen using the guanidinium thiocyanate-phenol-chloroform method (Trizol Reagent), following the manufacturer’s (Invitrogen) recommendations. RNA (2 μg) was used for quantitative polymerase chain reaction (PCR) analysis for first-strand synthesis of cDNA with the High Capacity cDNA Reverse Transcription kit (Applied Biosystems), according to the manufacturer’s instructions.
For real-time PCR analysis, specific murine Assays-on-Demand products (20x) and TaqMan Master Mix (2x) from Applied Biosystems were used, and the reactions were run on a 7900HT Fast Real-Time PCR System (Applied Biosystems) in a final volume of 20 μL. Each cDNA sample was amplified in triplicate and the RNA level was normalized to the corresponding level of Hprt1 mRNA. Primers used for the quantitative reverse transcriptase PCR are listed in Online Supplementary Table S1.
Statistical analysis
Data are presented as mean ± standard deviation. Unpaired two-tailed Student t-tests were performed using GraphPad PRISM 5.0 and a P value less than 0.05 was considered statistically significant.
Results
Tmprss6−/−Tfr2−/− mice are anemic and have increased red cell numbers
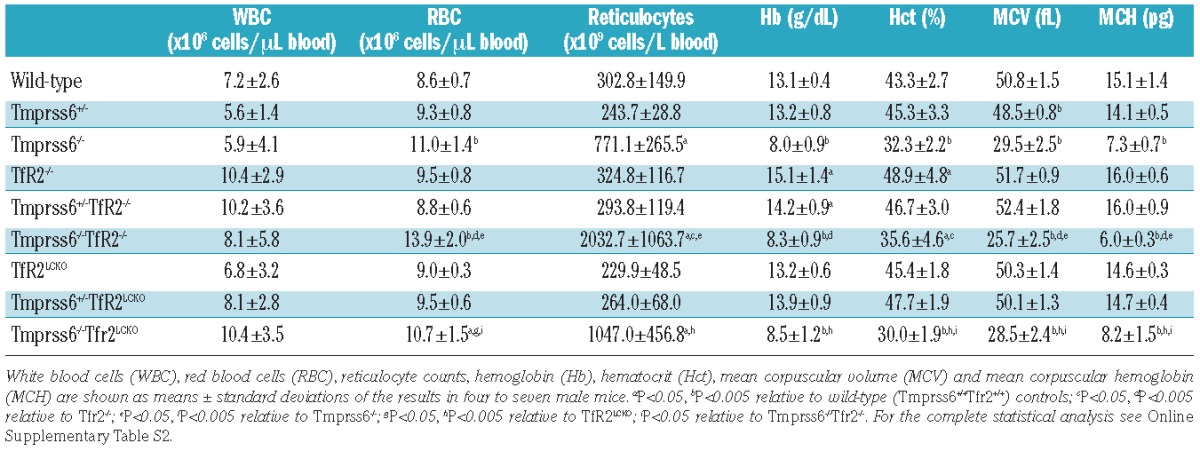
Ten-week old Tfr2−/− mice had higher hemoglobin levels than controls, while Tfr2LCKO mice had levels comparable to those in wild-type animals, as previously reported.15 Conversely Tmprss6−/− mice had the hematologic phenotype of microcytic anemia with increased red blood cell and reticulocyte counts accompanied by low levels of hemoglobin, hematocrit, mean corpuscular volume and mean corpuscular hemoglobin. The heterozygous loss of Tmprss6 in Tfr2−/− mice slightly reduced hemoglobin levels although the difference from the levels in Tfr2−/− mice was not statistically significant. On the contrary, Tmprss6+/−Tfr2LCKO mice had hemoglobin levels comparable to those of Tfr2LCKO animals. Both Tmprss6−/−Tfr2−/− and Tmprss6−/−Tfr2LCKO mice were anemic and had hemoglobin levels similar to those of Tmprss6−/− mice. However, the Tmprss6−/−Tfr2−/− mice had higher numbers of red blood cells than did Tmprss6−/− animals, resulting in more severe microcytosis, while this was not the case for Tmprss6−/− mice with specific liver deletion of Tfr2 (Table 1). In the absence of Tmprss6, reticulocytes were increased only in Tfr2−/− animals.
Table 1.
Hematologic data of all the genotype combinations analyzed.
Homozygous loss of Tmprss6 reduces systemic and tissue iron levels of Tfr2−/− and Tfr2LCKO mice
Transferrin saturation (Figure 1A) and liver iron content (LIC) (Figure 1B) were significantly lower in the iron-deficient Tmprss6−/− mice than in wild-type mice (defined as Tmprss6+/+Tfr2+/+ in Figures 1 and 2), while Tfr2−/− and Tfr2LCKO animals showed an important iron overload.15 Deletion of the Tmprss6 gene in both Tfr2−/− and Tfr2LCKO mice had a dose-dependent effect. The loss of a single allele slightly reduced transferrin saturation and LIC in both models, although the differences were statistically significant only for the Tfr2−/− animals. The homozygous inactivation of Tmprss6 lowered LIC of both Tfr2−/− and Tfr2LCKO animals to the levels of Tmprss6−/− mice. The difference in LIC of the various genotypes was confirmed by analysis of the Tfr1 mRNA levels, which are known to be inversely related to the cell iron content. Tfr1 mRNA levels were high in Tmprss6−/−, Tmprss6−/−Tfr2−/− and Tmprss6−/− Tfr2LCKO animals and reduced according to gene-dosage of Tmprss6 (Figure 1C). We observed no differences in the spleen iron content among all the genotypes analyzed (Figure 1D). In addition spleen and liver sizes were similar between Tmprss6−/−, Tmprss6−/−Tfr2−/− and Tmprss6−/−Tfr2LCKO animals (data not shown).
Figure 1.
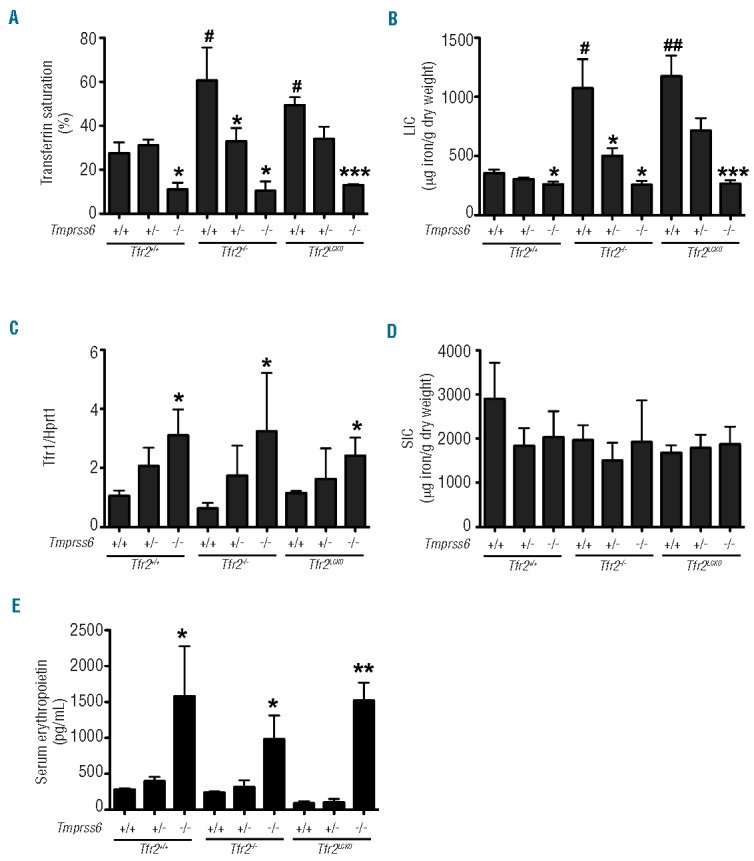
Effect of Tmprss6 deletion on iron parameters and serum erythropoietin levels of Tfr2−/− and Tfr2LCKO mice. The graphs show transferrin saturation (A); hepatic non-heme iron content (LIC) (B); liver mRNA expression of transferrin receptor 1 (Tfr1)(C); splenic non-heme iron content (SIC)(D); serum erythropoietin levels (E) in all genotype combinations analyzed. Mean values of three to six animals per genotype are shown and error bars indicate the standard deviation. Symbols refer to a statistically significant difference: *P<0.05, **P<0.01 and ***P<0.005 respective to the relative Tmprss6+/+ control of each Tfr2 genotype; #P<0.05 and ##P<0.01 respective to wild-type (Tmprss6+/+Tfr2+/+) controls. For the complete statistical analysis see Online Supplementary Table S2.
Figure 2.

Effect of Tmprss6 deletion on the Bmp-Smad pathway of Tfr2−/− and Tfr2LCKO mice. The graphs show liver mRNA expression of: bone morphogenetic protein 6 (Bmp6; A); hepcidin (Hamp; B); Hamp normalized on LIC (Hamp/LIC; C) and inhibitor of DNA binding 1 (Id1; D) in all genotype combinations analyzed. Mean values of three to six animals per genotype are shown and error bars indicate the standard deviation. Symbols refer to a statistically significant difference: *P<0.05, **P<0.01 and ***P<0.005 respective to the relative Tmprss6+/+ control of each Tfr2 genotype; #P<0.05 respective to wild-type (Tmprss6+/+Tfr2+/+) animals; §P<0.05 respective to Tmprss6−/− mice. For the complete statistical analysis see Online Supplementary Table S2.
Tfr2−/− and Tfr2LCKO mice had serum erythropoietin levels comparable to those of wild-type mice. As expected, anemic Tmprss6−/− mice had erythropoietin levels higher than those of wild-type mice and comparable to those of Tmprss6−/−Tfr2−/− and Tmprss6−/−Tfr2LCKO animals (Figure 1E).
Hamp levels are less inappropriately high in Tmprss6−/−Tfr2−/− mice than in Tmprss6−/− mice
The expression of Bmp6 reflected LIC in all the genotypes analyzed, being high in Tfr2−/− and Tfr2LCKO animals and low in Tmprss6−/− as compared to wild-type controls, although in the latter case the difference was not statistically significant. Bmp6 in Tmprss6 haploinsufficient Tfr2LCKO was indistinguishable from that in Tfr2LCKO animals (Figure 2A). The Bmp6/LIC ratio was comparable among all the genotypes analyzed proving that Bmp6 expression is adequate to the hepatic iron content (Online Supplementary Figure S1).
As expected, Hamp (Figure 2B) was over-expressed in Tmprss6−/− mice while comparable to wild-type levels in both Tfr2−/− and Tfr2LCKO mice. As a consequence the iron- deficient Tmprss6−/− mice had a Hamp/LIC ratio higher than that of wild-type animals, while both the iron-loaded Tfr2 knockout mice had a Hamp/LIC ratio lower than that of controls (Figure 2C).
In Tmprss6−/−Tfr2−/− mice Hamp expression was higher than in wild-type mice, but lower than in Tmprss6−/− animals, while levels in Tmprss6−/−Tfr2LCKO were comparable to those of Tmprss6−/− animals. This resulted in a Hamp/LIC ratio that was higher in Tmprss6−/−Tfr2−/− mice than in controls, but lower than in Tmprss6−/− animals, while the Hamp/LIC ratio of Tmprss6−/−Tfr2LCKO was comparable to that of Tmprss6−/− animals (Figure 2C). The mRNA levels of inhibitor of differentiation 1 (Id1), another target of the Bmp-Smad pathway, followed the same pattern as that of Hamp expression (Figure 2D), proving that in the double Tmprss6−/−Tfr2−/− mice the Bmp-Smad pathway is more active than in wild-type mice, but less active than in Tmprss6−/− mice.
Discussion
The analysis of animal models of Tfr2-hemochromatosis suggests that low hepcidin is due to an attenuated Bmp-Smad pathway. In theory TFR2 might promote BMP-SMAD signaling for hepcidin production by inhibiting the activity of TMPRSS6, as was hypothesized for HFE,32 by up-regulating BMP6 or through other unknown mechanisms. For this reason we compared the effect of Tmprss6 inactivation in mice with a total deletion of Tfr2 (Tfr2−/−) with that in mice with specific ablation of Tfr2 in the liver (Tfr2LCKO). Since the latter animals maintain Tfr2 function in other organs, the comparison of the phenotypes of the double knockout mice may provide clues to the extra-hepatic functions of TFR2.
We found that in adult Tfr2−/− mice the heterozygous loss of Tmprss6 slightly reduces the severity of hepatic iron overload and partially reverts the hematologic phenotype, reducing hemoglobin levels. In contrast, Tmprss6 haploin-sufficiency does not correct the iron-overload phenotype of Tfr2LCKO mice. This might be compatible with a more severe iron burden reported for the liver-specific knockout,14,15 although in the present study, in which only males were examined, LIC was similar in Tfr2−/− and Tfr2LCKO. Homozygous loss of Tmprss6 led to systemic iron deficiency and severe anemia in both genotypes with low levels of hemoglobin, transferrin saturation and LIC and enhanced hepatic Tfr1 expression, in analogy to what has been observed in Hfe knockout mice with deletion of Tmprss6.32 Similar results were also previously published for Tmprss6−/−Tfr2−/− mice,33 although differences of genetic backgrounds made the genotype comparison problematic.
The phenotype modification of Tfr2−/− and Tfr2LCKO from iron overload to iron deficiency in the absence of Tmprss6 demonstrates that TFR2 in the liver acts upstream of the serine protease and might control its activity, thus raising the possibility that pharmacological inhibition of TMPRSS6 is effective in limiting dietary iron absorption and redistributing iron to macrophages in TFR2-hemochromatosis, as shown for HFE-hemochromatosis.34,35
Loss of the protease activity of Tmprss6 leads to increased expression of hepcidin in Tfr2 iron-loaded animals which explains reduced iron absorption and iron deficiency. However, in Tmprss6−/− mice with complete loss of Tfr2 the hepatic mRNA levels of Hamp and Id1, although increased, do not reach the high levels observed in Tmprss6−/− mice. In contrast, the expression levels of Hamp and Id1 in Tmprss6−/−Tfr2LCKO are comparable to those in Tmprss6−/− mice. These differences are not mediated by an altered expression of Bmp6 since Bmp6 levels reflect the hepatic iron burden in all the genotypes analyzed. This appropriate regulation of Bmp6 in Tfr2−/− animals indicates that Tfr2 is not required for adequate Bmp6 response to increased tissue iron, a finding discordant from that in a recent report of Bmp6 being inappropriately low in Tfr2−/−mice.36 Based on our results we speculate that in Tmprss6−/−Tfr2−/− mice an inhibitory signal partially affects the efficiency of the Bmp-Smad pathway downstream of Bmp6 leading to a lower than expected hepcidin activation.
Since inhibition of hepcidin is largely dependent on erythropoietic signals we analyzed the hematologic phenotype of our models. The functional loss of both Tmprss6 and Tfr2 in the whole organism is associated with the same degree of iron deficiency as Tmprss6−/−. However, as compared to mice lacking Tmprss6 alone, Tmprss6−/−Tfr2−/−mice showed a consistent increase of red cell number and hematocrit, which was not observed when Tfr2 was specifically deleted in the liver. This observation supports the hypothesis that the hematologic phenotype of Tmprss6−/−Tfr2−/− is dependent on the lack of a still unknown extra-hepatic function of Tfr2.
We speculate that the loss of Tfr2 in the erythroid compartment accounts for the increased number of red cells observed in Tmprss6−/−Tfr2−/− mice and that the expansion of erythropoiesis is responsible for the partial inhibition of the Bmp-Smad pathway exclusively observed in these double mutant mice.
Iron-loaded Tfr2−/− mice are not characterized by increased red blood cell counts, but do have increased hemoglobin, as shown here and by others,15 as compared with Tfr2LCKO mice, indicating deregulated erythropoiesis. Indeed, the normal hemoglobin levels in the iron-loaded Tfr2LCKO mice indicate that the high hemoglobin levels observed in Tfr2−/− animals are not only due to their elevated iron burden, but to some other factors likely related to the absence of Tfr2 in the erythroid compartment.
In the attempt to verify whether the high red blood cell counts of Tmprss6−/−Tfr2−/− mice are due to increased erythropoietin levels, we measured serum erythropoietin in all the models. Since Tmprss6−/−, Tmprss6−/−Tfr2−/− and Tmprss6−/−Tfr2LCKO mice, which have the same degree of anemia, have comparable serum erythropoietin levels we conclude that erythroid precursors lacking Tfr2 might have enhanced sensitivity to erythropoietin stimulation. Our data seem discrepant with those reported by Foretnikova et al.,16 who found higher serum erythropoietin levels in Tfr2−/− mice than in Tfr2LCKO ones. However, the latter results were obtained in young animals (4-weeks old), while our data refer to adult, 10-weeks old mice. It is of interest that the same authors observed that TFR2-knockdown in human erythroid precursors led to a slight increase of total cell numbers after 12 days of cell culture.
In conclusion we propose that TFR2 is a modulator of erythropoiesis in keeping with its function as an EPOR partner. It is possible that TFR2, as an iron sensor, modulates the erythropoietin sensitivity of the erythroid precursors. The increased red cell numbers might be the result of this function in iron-deficient Tmprss6−/−Tfr2−/− animals. More specifically, since iron-loaded Tfr2−/− mice are not characterized by increased red blood cell counts, we propose that TFR2 is a limiting factor for erythropoiesis, which controls red cell numbers to avoid excessive production in conditions of iron-restriction. Further studies in mice with specific erythroid deletion of Tfr2 will clarify this possibility.
Acknowledgments
We acknowledge Prof. Carlos Lopez-Otin (Oviedo University, Spain) for releasing us the Tmprss6−/− mouse.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
Funding
This work was partially supported by Telethon Fondazione Onlus Rome (Grant GGP12025), MIUR PRIN 2010-2011 and the Italian Ministry of Health (Grant RF-2010-2312048) funds to CC and Progetti di Ateneo/CSP 2012 (TO_Call3_2012_0101) to GS.
References
- 1.Camaschella C, Roetto A, Cali A, De Gobbi M, Garozzo G, Carella M, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet 2000;25(1):14–5 [DOI] [PubMed] [Google Scholar]
- 2.Roetto A, Totaro A, Piperno A, Piga A, Longo F, Garozzo G, et al. New mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood 2001;97(9): 2555–60 [DOI] [PubMed] [Google Scholar]
- 3.Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem 1999;274(30):20826–32 [DOI] [PubMed] [Google Scholar]
- 4.Kawabata H, Germain RS, Ikezoe T, Tong X, Green EM, Gombart AF, et al. Regulation of expression of murine transferrin receptor 2. Blood 2001;98(6):1949–54 [DOI] [PubMed] [Google Scholar]
- 5.Johnson MB, Chen J, Murchison N, Green FA, Enns CA. Transferrin receptor 2: evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol Biol Cell 2007;18(3):743–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem 2006;281(39):28494–8 [DOI] [PubMed] [Google Scholar]
- 7.Gao J, Chen J, Kramer M, Tsukamoto H, Zhang AS, Enns CA. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab 2009;9(3):217–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood 2005;106(12):3710–7 [DOI] [PubMed] [Google Scholar]
- 9.Rishi G, Crampton EM, Wallace DF, Subramaniam VN. In situ proximity ligation assays indicate that hemochromatosis proteins Hfe and transferrin receptor 2 (Tfr2) do not interact. PLoS One 2013;8(10):e77267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306(5704):2090–3 [DOI] [PubMed] [Google Scholar]
- 11.Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet 2009; 41(4):478–81 [DOI] [PubMed] [Google Scholar]
- 12.Andriopoulos B, Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 2009; 41(4):482–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J Hepatol 2012; 57(5):1052–60 [DOI] [PubMed] [Google Scholar]
- 14.Wallace DF, Summerville L, Subramaniam VN. Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 2007; 132(1): 301–10 [DOI] [PubMed] [Google Scholar]
- 15.Roetto A, Di Cunto F, Pellegrino RM, Hirsch E, Azzolino O, Bondi A, et al. Comparison of 3 Tfr2-deficient murine models suggests distinct functions for Tfr2-alpha and Tfr2-beta isoforms in different tissues. Blood 2010;115(16):3382–9 [DOI] [PubMed] [Google Scholar]
- 16.Forejtnikova H, Vieillevoye M, Zermati Y, Lambert M, Pellegrino RM, Guihard S, et al. Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood 2010;116(24):5357–67 [DOI] [PubMed] [Google Scholar]
- 17.Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Met 2008;8(6):502–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finberg KE, Heeney MM, Campagna DR, Aydinok Y, Pearson HA, Hartman KR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet 2008;40(5):569–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du X, She E, Gelbart T, Truksa J, Lee P, Xia Y, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science 2008;320(5879):1088–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Folgueras AR, de Lara FM, Pendas AM, Garabaya C, Rodriguez F, Astudillo A, et al. Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 2008;112(6):2539–45 [DOI] [PubMed] [Google Scholar]
- 21.Hershko C, Camaschella C. How I treat unexplained refractory iron deficiency anemia. Blood 2014;123(3):326–33 [DOI] [PubMed] [Google Scholar]
- 22.Benyamin B, Ferreira MA, Willemsen G, Gordon S, Middelberg RP, McEvoy BP, et al. Common variants in TMPRSS6 are associated with iron status and erythrocyte volume. Nat Genet 2009;41(11):1173–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganesh SK, Zakai NA, van Rooij FJ, Soranzo N, Smith AV, Nalls MA, et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet 2009; 41(11):1191–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chambers JC, Zhang W, Li Y, Sehmi J, Wass MN, Zabaneh D, et al. Genome-wide association study identifies variants in TMPRSS6 associated with hemoglobin levels. Nat Genet 2009;41(11):1170–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soranzo N, Spector TD, Mangino M, Kuhnel B, Rendon A, Teumer A, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet 2009;41(11):1182–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka T, Roy CN, Yao W, Matteini A, Semba RD, Arking D, et al. A genome-wide association analysis of serum iron concentrations. Blood 2010;115(1):94–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traglia M, Girelli D, Biino G, Campostrini N, Corbella M, Sala C, et al. Association of HFE and TMPRSS6 genetic variants with iron and erythrocyte parameters is only in part dependent on serum hepcidin concentrations. J Med Genet 2011;48(9):629–34 [DOI] [PubMed] [Google Scholar]
- 28.Nai A, Pagani A, Silvestri L, Camaschella C. Increased susceptibility to iron deficiency of Tmprss6-haploinsufficient mice. Blood 2010;116(5):851–2 [DOI] [PubMed] [Google Scholar]
- 29.Nai A, Pagani A, Silvestri L, Campostrini N, Corbella M, Girelli D, et al. TMPRSS6 rs855791 modulates hepcidin transcription in vitro and serum hepcidin levels in normal individuals. Blood 2011;118(16):4459–62 [DOI] [PubMed] [Google Scholar]
- 30.Maurer E, Gutschow M, Stirnberg M. Matriptase-2 (TMPRSS6) is directly up-regulated by hypoxia inducible factor-1: identification of a hypoxia-responsive element in the TMPRSS6 promoter region. Biol Chem 2012;393(6):535–40 [DOI] [PubMed] [Google Scholar]
- 31.Meynard D, Vaja V, Sun CC, Corradini E, Chen S, Lopez-Otin C, et al. Regulation of TMPRSS6 by BMP6 and iron in human cells and mice. Blood 2011;118(3):747–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finberg KE, Whittlesey RL, Andrews NC. Tmprss6 is a genetic modifier of the Hfe-hemochromatosis phenotype in mice. Blood 2011;117(17):4590–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee P, Hsu MH, Welser-Alves J, Peng H. Severe microcytic anemia but increased erythropoiesis in mice lacking Hfe or Tfr2 and Tmprss6. Blood Cells Mol Dis 2012;48(3): 173–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt PJ, Toudjarska I, Sendamarai AK, Racie T, Milstein S, Bettencourt BR, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood 2013;121(7):1200–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo S, Casu C, Gardenghi S, Booten S, Aghajan M, Peralta R, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest 2013;123(4):1531–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald CJ, Wallace DF, Ostini L, Subramaniam VN. Parenteral vs oral iron: Influence on hepcidin signaling pathways though analysis of Hfe/Tfr2 null mice. Am J Physiol Gastrointestl Liver Physiol 2014; 306(2):G132–9 [DOI] [PubMed] [Google Scholar]