

Abstract
Novel therapies are needed for pediatric acute lymphoblastic leukemia resistant to conventional therapy. While emerging data suggest leukemias as possible targets of oncolytic attenuated measles virus, it is unknown whether measles virus can eradicate disseminated leukemia, in particular pediatric acute lymphoblastic leukemia. We evaluated the efficacy of attenuated measles virus against a large panel of pediatric xenografted and native primary acute lymphoblastic leukemias ex vivo, and against four different acute lymphoblastic leukemia xenografts of B-lineage in non-obese diabetic/severe combined immunodeficient mice. Ex vivo, attenuated measles virus readily spread among and effectively killed leukemia cells while sparing normal human blood cells and their progenitors. In immunodeficient mice with disseminated acute lymphoblastic leukemia a few intravenous injections of attenuated measles virus sufficed to eradicate leukemic blasts in the hematopoietic system and to control central nervous system disease resulting in long-term survival in three of the four xenografted B-lineage leukemias. Differential sensitivity of leukemia cells did not require increased expression of the measles entry receptors CD150 or CD46 nor absence of the anti-viral retinoic acid-inducible gene I/melanoma differentiation associated gene-5 /interferon pathway. Attenuated oncolytic measles virus is dramatically effective against pediatric B-lineage acute lymphoblastic leukemia in the pre-clinical setting warranting further investigations towards clinical translation.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common malignancy in childhood and has a guarded prognosis in the 20% of patients who relapse.1–3 Novel therapies are needed for poor prognosis ALL. Oncolytic viruses are an emerging therapeutic approach in solid tumors. Attenuated measles virus (MV), such as the Edmonston-B strain (MV-Edm) and its derivatives, is strongly oncolytic in solid tumor models.4–8 Its action is enhanced by chemotherapy,9 irradiation,10 radioactive nuclides11 and influx of neutrophils.12 The tropism of MV-Edm can be narrowed by genetic manipulations.13–17 MV-Edm and its derivatives have shown very few side-effects as vaccines in humans and when tested in susceptible and immunocompromised mouse models or non-human primates.6,9,18 Attenuated MV shows promise in clinical trials against solid tumors. Cutaneous T-cell lymphoma regressed after intratumoral injection of attenuated MV.19 MV-Edm administered intraperitoneally prolonged survival of patients with ovarian carcinoma, without dose-limiting toxicity.20,21 MV-Edm is also administered locally in patients with recurrent glioblastoma multiforme and intravenously into patients with multiple myeloma.22–24 While evidence for efficacy of attenuated MV in solid tumors is mounting, little is known about its effects in leukemias. Recent data suggest adult leukemias as possible targets.25,26 No investigations have yet been performed in pediatric ALL.
CD150 (SLAM) expressed on immune cells are known to be used by MV-Edm as an entry receptor in vivo, while the role of ubiquitously expressed CD46 for in vivo entry of MV-Edm has recently been challenged.27 Nectin-4 (PVRL4) has been shown to be an epithelial cell receptor for both wild-type MV28,29 and MV-Edm.29 After infection and replication, newly formed viruses infect neighboring tumor cells by mechanisms that are not yet completely understood. In some cell types, close cell contact is important for viral spread,30–32 whereas release of MV-Edm by infected cells into the extracellular space appears to be non-essential in those cells.31,32
All human nucleated cells express CD46. Many solid tumor cells over-express CD46.33 This is thought to confer preferential killing of cancer cells versus non-transformed cells following infection by MV-Edm.34 It is unknown whether this mechanism is operative in ALL. Cells infected with paramyxoviruses, such as MV, sense the cytoplasmic presence of virus-specific RNA by the cytoplasmic pattern recognition receptors retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated gene-5 (MDA-5).35–37 Paramyxoviruses attempt to inhibit MDA-5 using their V protein.38 RIG-I binds the adaptor molecule mitochondrial antiviral signaling (MAVS). This activates interferon response factor-3 (IRF-3) and NF-κB, which translocate into the nucleus and activate interferon-α/β (IFN-α/β) genes (reviewed by Randall and Goodbourn39). IFN-α/β thus produced binds to the receptors IFNAR1 and IFNAR2 inducing IFN-stimulated genes, whose products inhibit replication and spread of MV. Deficiency in suppression of the type I IFN response in normal cells is part of the attenuated phenotype of vaccine strains of MV.40,41 Many solid cancer cells are known to have a decreased IFN response (reviewed by Pitha42), which can be exploited for oncolytic virotherapy.43,44 It is unknown whether ALL cells have a deficient type I IFN response impacting on their response to viral infection, whether they harbor defects in the RIG-I/MDA-5 pathway or whether other mechanisms of increased susceptibility to attenuated MV are operative in ALL cells.
In this study, we set out to investigate the hitherto unknown susceptibility of pediatric ALL to attenuated MV. Using our large collection of primary pediatric ALL propagated in immunodeficient mice45 we show that MV-Edm is remarkably effective against acute B-lineage ALL in the pre-clinical setting.
Methods
ALL cell lines, xenografts and patient samples
The ALL cells lines Jurkat, CCRF-CEM, MOLT-4, REH, RS4;11 and NALM-6 were purchased. ALL cells from patients propagated in non-obese diabetic/severe combined immunodeficient mice (NOD/SCID) mice were procured from spleen tissue at a purity of above 90%. Primary patient ALL samples were obtained at diagnosis from pediatric patients with de novo ALL. Most patients were enrolled in the ALL-BFM study protocols. Patients’ characteristics for the xenografts are listed in the Online Supplementary Table S1. The human study protocol was approved by the Ethical Review Board of the University Medical Center Ulm in accordance with the Declaration of Helsinki. Cell lines, xenografts and primary ALL cells were cultured in RPMI 1640 with 10% fetal calf serum, L-glu-tamine, penicillin and streptomycin.
Human ALL NOD/SCID mouse model
NOD/SCID mice at a median age of 8–10 weeks (w) were used. Housing and treatment of animals were in accordance with state guidelines. ALL cells were injected into a lateral tail vein. After grafting, blood samples were evaluated at 1-w or 2-w intervals for human leukemia cells by determining CD45+Ly5− cells using flow cytometry. At necropsy cell suspensions from spleen, bone marrow and meninges were prepared and brains were procured. The presence of leukemic cells in the suspensions was determined by FACS analysis.
Virus infection
Ex vivo cells were infected with MV-Edm at a MOI of 1. ALL cell lines, xenografts, patient samples, peripheral blood mononuclear cells (PBMC), B and T cells were infected in serum-free RPMI 1640 medium at 37°C for 3 hours (h). For hematopoietic stem cells (HSC) serum-free IMDM medium was used. Medium was changed depending on the experiment.
Replication of MV-Edm in Jurkat cells and PBMC
To quantitate viral replication in Jurkat cells, lysates were harvested 3, 24, 48 and 72 h after infection and added to Vero indicator cells. Syncytia were determined 72 h later. To compare replication in Jurkat cells to PBMC, lysates of PBMC were collected 72 h after infection and added to Vero cells. Syncytia formation was documented after 72 h.
Spread and bystander effect of MV-Edm
To assess spread of MV-Edm in the presence of anti-MV-antibodies, infected Jurkat or REH cells were mixed with non-infected Jurkat or REH cells at increasing ratios of 1:20, 1:10, 1:5 and 1:2 in the presence or absence of 10% measles immune serum. Cell viability was determined by forward scatter/side scatter analysis using FACS.
In vivo treatment
NOD/SCID mice were transplanted with B-cell precursor ALL xenograft cells from patients #6, #13, #15 and #19. Once peripheral blast counts reached 5–20%, as determined by FACS, mice received intravenous injections of MV-Edm, heat-inactivated MV-Edm or PBS each day (d) for 5 d. Mice of the treatment group were killed and analyzed when their survival time after therapy was three times the survival time after injection of the animals in the control groups or when they reached the end of their natural lifespan of 250 d.
Materials and Methods are fully described in the Online Supplemental Appendix.
Results
MV-Edm effectively kills cells from ALL cell lines
We first investigated susceptibility of ALL cell lines to MV-Edm. Cells of the T-lineage ALL cell line Jurkat were killed within 72 h by MV-Edm, depending on dose (Figure 1A). Based on these results, a MOI of 1 was chosen for further experiments in vitro. Next, we determined that the T-lineage ALL cell lines Jurkat, CCRF-CEM and MOLT-4, and the B-lineage ALL cell lines REH, NALM-6 and RS4;11 were killed by MV-Edm in a time-dependent manner (Figure 1B and Online Supplementary Figure S1A). B-lineage ALL cells were killed more protractedly compared to T-lineage ALL cells. Within the same experiments we showed good agreement between results of forward scatter/side scatter and Annexin V/PI analyses for all xenografts and all cell lines except for REH and NALM-6 cells, where forward scatter / side scatter analysis underestimated cell kill (Figure 1B and Online Supplementary Figure S1). In addition, we verified that results for inactivated MV-Edm and medium control were comparable (Figure 1B and Online Supplementary Figure S1).
Figure 1.
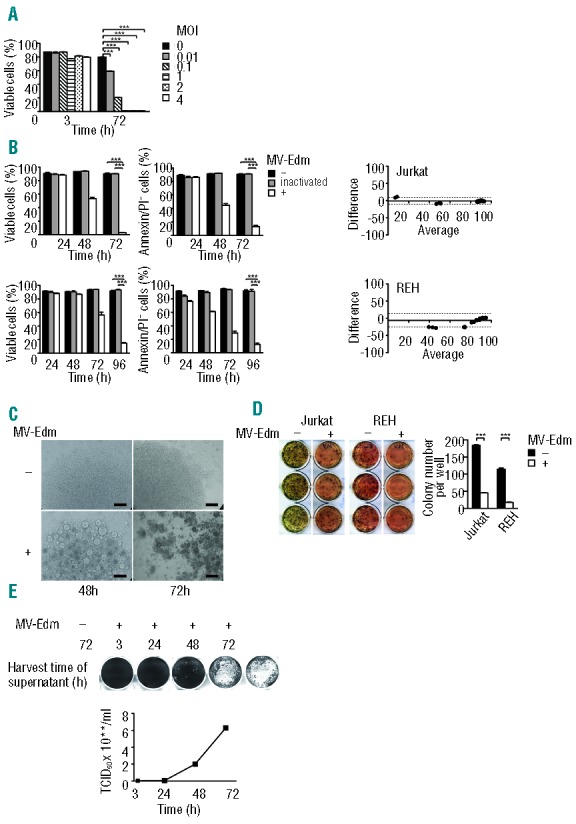
MV-Edm effectively replicates in and kills ALL cell lines (A) Dose-dependent killing of Jurkat cells. Jurkat cells (4 × 104 cells per well in a 96-well plate) were infected with MV-Edm at increasing MOIs. Cell viability was determined at 3 h and 72 h by FACS using FSC/SSC analysis. Results are means +SD of triplicates and are expressed as percentage of total cells. Similar results were obtained in 2 independent experiments. (B) Time-dependent killing of Jurkat and Reh cell lines. 4 × 104 cells per well were incubated with MV-Edm at a MOI of 1 (“+”), with heat-inactivated MV-Edm (“inactivated”) or with medium (“−“). Cell viability was determined by FSC/SSC analysis (left panels) and Annexin V/PI staining (middle panels). Results are means +SD of triplicates and are expressed as percentage of total cells. Agreement between forward/side scatter and Annexin V/PI analyses was determined using Bland-Altman plots (right panels). Mean difference (bias) is indicated by a solid line, ± 1.96 standard deviations of the differences (95% limit of agreement) by dashed lines. Average denotes average of results of both methods. Similar results were obtained in 3 independent experiments. (C) Formation of syncytia in Jurkat cells. Jurkat cells were seeded in 96-well plates (4 × 104 cells per well in a 96-well plate) and infected with MV-Edm at a MOI of 1 or were left uninfected. 48 h and 72 h post infection cells were assessed by light microscopy. Bars equal 200 μm. (D) Reduced clonogenicity of Jurkat and REH cells. REH and Jurkat cells infected with MV-Edm at a MOI of 1 were seeded in 6-well plates (1 × 103 cells per well) in methyl-cellulose-containing medium. 14 d later viable colonies were stained by MTT (left panels) and counted (right panel). Similar results were obtained in 2 independent experiments. (E) MV-Edm replicates in Jurkat cells. Jurkat cells (2.5 × 106 cells per 25 cm2 flask) were infected with MV-Edm at a MOI of 1. Cell supernatants were collected at the time points indicated and added to Vero cells. Syncytia formation was determined (upper panel). To quantitate replication infected Jurkat cells were lysed and the TCID50 was determined using Vero cells (lower panel). ***P<0.001 using the unpaired t-test.
In Jurkat cells growing at high density MV-Edm induced syncytia, the classical cytopathic effect of MV (Figure 1C). Importantly, MV-Edm also markedly decreased clonogenicity of Jurkat and REH cells (Figure 1D). Replication of MV-Edm in Jurkat cells started 24 h post infection and continued at 72 h (Figure 1E).
MV-Edm effectively kills cells of pediatric primary ALL and xenografts
Next, we investigated sensitivity of pediatric primary ALL and xenograft cells to MV-Edm. Of 6 ALLs from the bone marrow of patients 4 could be evaluated, while in 2 leukemias extensive spontaneous apoptosis precluded analysis. Of the evaluable ALL, 3 were sensitive to MV-Edm in a heterogeneous fashion, while one (#4) was resistant (Figure 2A). We extended the analysis to 18 evaluable (out of 21 investigated) primary pediatric ALLs propagated in NOD/SCID mice. Patients’ characteristics of primary ALL and xenografts are listed in the Online Supplementary Table S1. Spontaneous ex vivo apoptosis varied between the ALL xenografts and was marked in some (Figure 2B). Except for xenograft #19, all xenografts were very sensitive to MV-Edm ex vivo.
Figure 2.
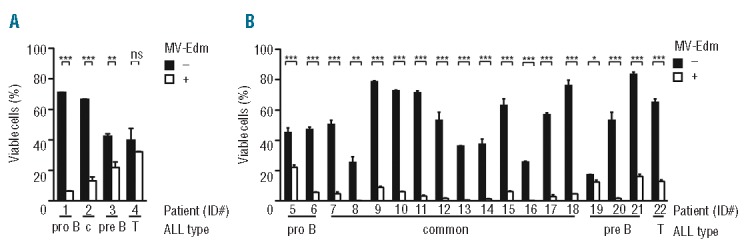
MV-Edm effectively kills cells of pediatric primary ALL and xenografts ex vivo. ALL cells (8 × 104 cells per well in a 96-well plate) were treated at a MOI of 1 or were left untreated. Cell viability was determined 72 h after infection by FACS FSC/SSC. Results are means +SD of triplicates and are expressed as percentage of total cells. ***P<0.001, **P<0.01, *P<0.05 and ns = not significant using the unpaired t-test. (A) Killing of primary ALL cells. Cells were isolated from the bone marrow of 4 patients (ID #1–4) with pro-B, common (c), pre-B and T ALL. (B) Killing of explanted ALL xenografts. Xenograft cells were isolated from the spleen of NOD/SCID mice (#15 refers to the relapse of #9).
As in cell lines, results of FSC/SSC analysis and Annexin V/PI staining in explanted ALL xenografts were comparable, and results for inactivated MV-Edm were similar to medium control (Online Supplementary Figure S1B). Thus, the simpler methods of FSC/SSC analysis and of using medium as control were chosen for further experiments. In summary, MV-Edm was strikingly effective in killing primary ALL cells ex vivo.
MV-Edm spreads effectively in ALL cell lines in the absence of measles antibodies, killing blasts by apoptosis
Effective killing of leukemic cells of systemically administered MV-Edm against ALL hinges on effective spread of MV-Edm between leukemic cells, as initial infection efficiency in vivo is likely to be too low to target every leukemic cell. Viral spread and killing of leukemic bystander cells was modeled in vitro by mixing MV-Edm-treated with untreated leukemic cells and determining cell death in the mixture. A small number of initially infected cells sufficed to kill a large number of initially uninfected cells, e.g. 5% initially infected REH cells killed 50% of the cells in the mixture after 72 h (Online Supplementary Figure S2). This shows that before their demise, infected ALL cells are able to transmit infection to bystander leukemic cells, leading to their death. Spread of MV-Edm in ALL may proceed either by cell-to-cell contact or by release of MV-Edm from infected cells into the extra-cellular space. In either case, MV-Edm could be neutralized if measles antibodies were present. Indeed, in a mixture of infected ALL cells with uninfected bystander leukemic cells, measles immune serum inhibited MV-Edm-induced death in the bystander cells (Online Supplementary Figure S2). Cell death of ALL cell lines and ALL xenograft cells by MV-Edm occurred by apoptosis, while no non-apoptotic death was evident (Online Supplementary Figure S3). Taken together, these results support the notion that, in the absence of measles antibodies, MV-Edm may control ALL even if only a small quantity of blasts is infected initially.
Specific sensitivity of ALL cells to MV-Edm compared to hematopoetic cells does not depend on enhanced expression levels of CD46 or CD150
We have shown that MV-Edm readily kills primary ALL and ALL xenografts, implying efficient viral replication in these cells. We also showed pronounced MV-Edm-induced cell death of ALL cell lines and proved strong replication and intraleukemic spread of MV-Edm in Jurkat cells. Next, we investigated whether normal hematopoietic cells are differentially sensitive to MV-Edm. Indeed, susceptibility of human PBMC from healthy donors was marginal (Figure 3A) compared with primary ALL and ALL xenografts (Figure 2) and ALL cell lines (Figure 1 and Online Supplementary Figure S1). Limited sensitivity of PBMC could be in part explained by limited replication of MV-Edm in PBMC. Indeed, we could not detect infectious viral particles from PBMC lysates 72 h after infection with MV-Edm (Figure 3B). Taken together, these data suggest that preferential replication of MV-Edm in ALL contributed to the enhanced sensitivity of ALL cells.
Figure 3.
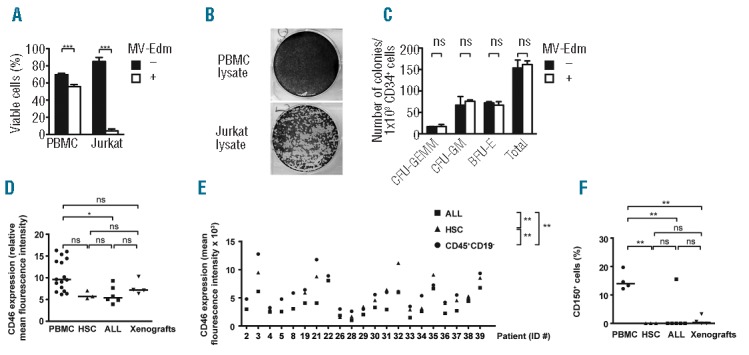
Hematopoetic cells are not sensitive to MV-Edm despite sufficient expression of CD46 and CD150. (A) Limited susceptibility of PBMC. 8 × 104 PBMC per well from buffy coats of 16 donors or 4 × 104 Jurkat cells were infected in 96-well plates with MV-Edm at a MOI of 1 or were left uninfected. Cell viability was determined 72 h after infection by FACS FSC/SSC. ***P<0.001 using the unpaired t-test. (B) No infectious viruses from PBMC treated with MV-Edm after 72 h. 8 × 104 PBMC per well from 3 donors or Jurkat cells were treated with MV-Edm at a MOI of 1. Cell lysates were obtained after 72 h and 15 μL was added to 2.25 × 105 Vero cells. Syncytia formation was determined 72 h later after staining with crystal violet by light microscopy. (C) HSC function is not affected. 2 × 103 CD34+CD38− HSC from human cord blood were infected with MV-Edm at a MOI of 1 or were left uninfected. Thereafter, 1 × 103 cells per dish were seeded in methylcellulose-based medium. 14 d later, CFU-GEMM, CFU-GM and BFU-E colonies were determined. ns = not significant using the unpaired t-test. Similar results were obtained in two independent experiments. (D) ALL cells do not express more CD46 than PBMC and HSC. PBMC from 16 donors, CD34+CD38-HSC from 3 cord blood donors, 6 ALL cell lines (CCRF-CEM, Jurkat, MOLT-4, NALM-6, REH and RS4;11) and xenografts #6, #13, #15 and #19 were analyzed for CD46 expression relative to isotype control using FACS. Horizontal bars depict medians. *P< 0.05, ns = not significant using one-way ANOVA with Tukey’s post hoc test. (E) Less CD46 is expressed on ALL blasts than on hematopoetic cells in patients. CD19+CD10+ ALL blasts, CD34+CD19− HSC and CD45+CD19− hematopoetic cells from the bone marrow of patients at diagnosis were analyzed for expression levels of CD46 by determining mean fluorescence intensity using FACS. 5–10 × 104 cells per sample were analyzed. **P<0.01 comparing medians using the Kruskall-Wallis test. (F) ALL cells do not express CD150 more frequently than PBMC and HSC. CD150 expression levels were determined in the cells described in (D) by mean fluorescence intensity normalized to isotype control using FACS. **P< 0.01, ns = not significant using one-way ANOVA with Tukey’s post hoc test.
During treatment of ALL with MV-Edm hematopoietic stem cells (HSC) will become exposed to MV-Edm, either in the wake of systemic injection or after release of virus from infected leukemic cells. To determine any possible impact on the function of HSC, we asked whether the capacity of HSC to form progenitor colonies would be affected upon infection with MV-Edm. This was not the case, showing that ex vivo human HSC are resistant to MV-Edm (Figure 3C).
Next, we explored potential mechanisms of the differential sensitivity of ALL and hematopoietic cells we had observed. As enhanced expression of CD46, an entry receptor for attenuated MV in vitro, on solid tumor cells contributes to their specific susceptibility to MV compared to normal cells that express less CD46,34 we reasoned that this may also apply to ALL. However, enhanced sensitivity of ALL cells was not due to enhanced expression of CD46, as measured by mean fluorescence intensity. In fact, less CD46 was found on xenografts and ALL cell lines than on PBMC, while HSC from cord blood expressed amounts comparable to leukemic cells (Figure 3D). Along this line, leukemic blasts in the bone marrow of patients always expressed less CD46 than HSC and mature hematopoietic cells (Figure 3E). We then asked whether CD150, a known entry receptor for MV-Edm in vivo, could account for the differential sensitivity of ALL. However, CD150 was expressed on only few of the xenografts and ALL cell lines, on none of the HSC specimens, but on all PBMC samples (Figure 3F). Taken together, the different sensitivity of ALL cells and normal hematopoietic cells to MV-Edm cannot be explained by different expression of the entry receptors CD150 or CD46. Rather, the data suggest that either other entry receptors or post-entry mechanisms modulate efficacy of MV-Edm in ALL.
Intact RIG-I/MDA-5-mediated transcriptional interferon response in B-cell precursor ALL xenograft cells after infection by MV-Edm
As the previous results suggested post-entry mechanisms as possible determinants of differential sensitivity to MV-Edm, we turned our attention to the interferon response. Many cancer cells are defective in mounting a type I IFN response to viral infection. As it was unknown whether ALL cells harbor such a defect, we investigated the pivotal RIG-I/MDA-5 sensing and signaling pathway and subsequent interferon response upon infection with MV-Edm. Investigating this pathway also appeared to be important because paramyxovirus V protein is known to inactivate MDA-5.38 Upon infection by MV-Edm, ALL xenograft cells increased transcription of IFN-β, the interferon target genes RIG-I and MDA-5, and the interferon-responsive genes IRF-1, ISG-15 and IFIT1 (Figure 4). In contrast, infected ALL cell lines did not respond with increased transcription of these genes (Figure 4).
Figure 4.
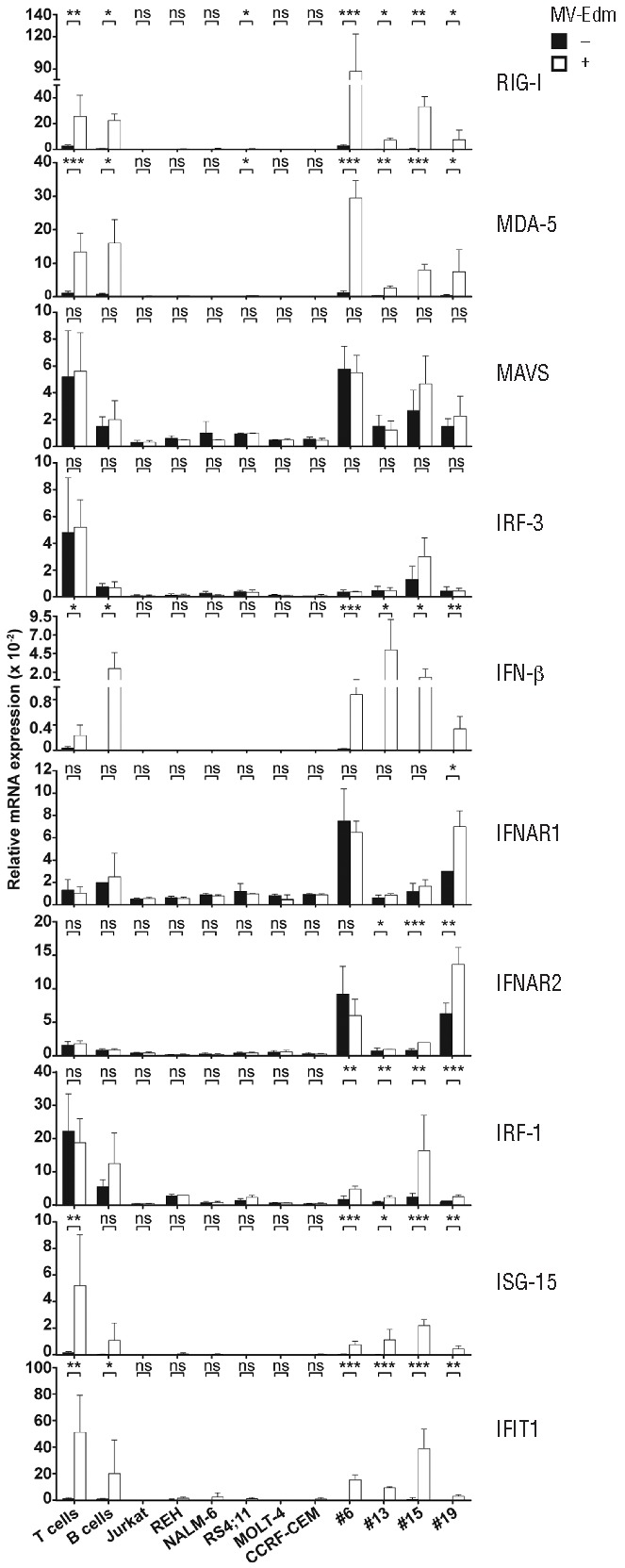
The RIG-I/MDA-5/MAVS/IFN-β axis is defective in ALL cell lines but intact in B-cell precursor ALL xenograft cells. T and B cells isolated from PBMC of 5 and 6 donors, respectively, ALL cell lines and B-cell precursor ALL xenografts #6, #13, #15 and #19 were infected with MV-Edm at a MOI of 1 or were not infected. mRNA was isolated 12 h later. Expression of RIG-I, MDA-5, MAVS, IRF-3, IFN-β, IFNAR1 IFNAR2, IRF-1, ISG-15 and IFIT1 was measured by qRT-PCR and is shown relative to expression of β-actin. ***P<0.001; **P<0.01; *P<0.05 and ns: not sigificant using the unpaired t-test.
These data show that the MV-Edm-induced RIG-I/MDA-5-mediated interferon response was intact on the transcriptional level in B-cell precursor ALL xenograft cells but defective in ALL cell lines.
In vivo, MV-Edm eradicates or markedly decreases peripheral blasts of B-lineage ALL leading to survival of most mice
Given the marked susceptibility of primary ALL to MV-Edm in vitro we set out to determine if MV-Edm can control disseminated patient ALL in the human leukemia NOD/SCID mouse model. We and others have shown that this model faithfully simulates the situation in the pediatric patient, the original characteristics of the patients’ ALL being faithfully preserved even after repeated retransplantations.45,46 We assessed a pro B, a pre B and two common ALL xenografts to cover the spectrum of B-lineage ALL. Upon reaching a high leukemic burden, as determined by the percentage of blasts in the peripheral blood, mice were systemically treated with MV-Edm or were mock-treated. In the mock-treated mice, percentages of peripheral leukemic blasts quickly rose and the general appearance of the mice deteriorated, necessitating their euthanasia (Figure 5A). Thirty-three of the 44 untreated mice across all xenografts could be necropsied, all of these revealed extensive leukemic infiltration of bone marrow and spleen (data not shown). In stark contrast, leukemic blasts in the blood rapidly decreased in all treated mice, regardless of whether they harbored pro B, pre B or common ALL xenografts (Figure 5A). Leukemic blasts remained undetectable in the peripheral blood of all surviving mice. All treated mice with the common ALL xenograft #15 survived until euthanized at the age of 265 d, beyond the life span of 250 d typical for NOD/SCID mice. Necropsy showed no blasts in peripheral blood (Figure 5A), bone marrow, spleen and liver (Online Supplementary Figure S4), indicating complete eradication of leukemic blasts in the hematopoietic system and its restitutio ad integrum. All treated mice with the pro-B xenograft #6 survived until they were 238 d old, when mice died. Necropsy revealed no leukemic blasts in blood, spleen or bone marrow and no obvious cause of death in these 2 visibly aged mice. The remaining #6 mice were euthanized because of advanced age. Upon necropsy, no ALL cells were found in blood, spleen and bone marrow. All but one mouse treated with the pro-B xenograft #19 survived. The mouse that died could not be analyzed. No leukemic blasts were evident in blood, spleen and bone marrow of the surviving mice. Surprisingly, all but one of the treated mice with xenograft #13 died as quickly as the untreated mice harboring this extremely rapidly progressing leukemia (Figure 5A), despite diminished leukemic blasts in the blood and the spleen (Figure 5B). The two bone marrows that could be examined in the treatment group prior to autolysis showed only moderate infiltration by leukemic blasts, extensive areas of necrosis and severe depression of hematopoiesis (Online Supplementary Figure S5). Taken together, MV-Edm eradicated or decreased peripheral leukemia in all of the four different xenografts leading to long-term survival of mice in 3 of the 4 xenografts.
Figure 5.
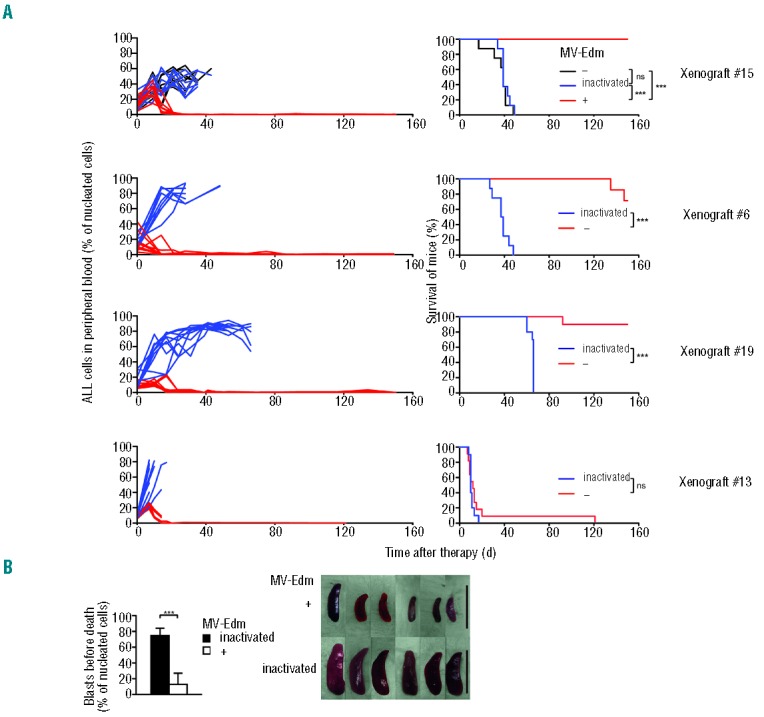
In NOD/SCID mice with B-lineage ALL xenografts MV-Edm eradicates or profoundly decreases peripheral blasts leading to survival of most mice NOD/SCID mice with the common ALL xenografts #15 and #13, the pro B ALL xenograft #6 and the pre B ALL xenograft #19 received MV-Edm (“+”, n = 8), inactivated MV-Edm (“inactivated”, n = 8) or no MV-Edm (“−”, n = 8, #15 only) each d for 5 d once peripheral CD45+Ly5− leukemic blasts reached mice. 5–20 % as determined by FACS. (A) Peripheral leukemic blasts and survival. Peripheral CD45+Ly5− leukemic blasts were monitored by FACS analysis (left panels). Kaplan-Meier survival plots were generated (right panels; *** P<0.001, ns = not significant using the log rank test). (B) Leukemic burden in blood and spleen of mice with xenograft #13 markedly decreases following treatment. The percentages of CD45+Ly5− leukemic blasts in the last blood sample procured before death are compared (left panel; means +SD for each group are depicted, ***P<0.001 using the unpaired t-test). Spleens were procured after death and photographed (right panel, scale bar equals 3 cm).
Efficient intraleukemic replication, spread and impact of MV-Edm in vivo
The kinetics of leukemic cell death induced by MV-Edm was organ-dependent. As shown by FACS analysis, in mice with xenograft #15 peripheral blood was cleared from blasts 18 d after start of therapy while spleens were cleared later at 25 d, a time when bone marrow and meninges were still infiltrated by blasts (Online Supplementary Figure S6). To investigate the kinetics of viral replication and of the blasts’ response in cellular detail histology was performed. Following systemic administration of MV-Edm viral replication was evident within many blasts at 15 d and within most blasts at 18 d (Figure 6A). Cytological, proliferative and apoptotic responses of blasts paralleled viral replication. While 15 d after therapy few morphological changes of the blasts were discernible, at 18 d the boundaries of the blasts disappeared and their nuclei were karyolytic, consistent with formation of syncytia. Apoptosis significantly increased while proliferating cells markedly decreased. Seven days later, at d 25, far fewer blasts were visible and normal mononuclear cells as well as megakaryocytes had reappeared. The apoptosis rate had returned to baseline and far fewer proliferating cells were discernible. Blast clearance from the liver followed a similar kinetic (Figure 6B).
Figure 6.
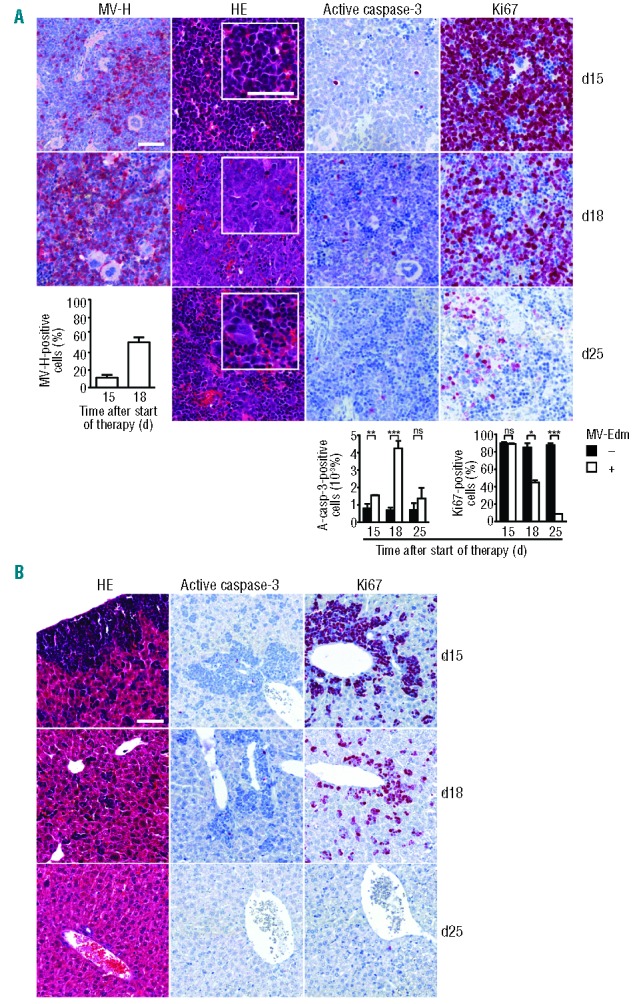
Systemic MV-Edm efficiently replicates, spreads and kills within an ALL xenograft in vivo. NOD/SCID mice with xenograft #15 received MV-Edm (n = 5) or PBS without MV-Edm (n = 5) for 5 d once blasts reached 5–20% of peripheral nucleated blood cells. 15, 18 and 25 d after application of MV-Edm one pair of mice (treated and untreated) was killed. Organs were analyzed by HE staining and immunohistochemistry for MV-H protein, active (cleaved) caspase-3 and Ki67. (A) Replication, spread and kill in spleen. Representative micrographs of treated mice are shown. Inserts depict higher magnification. Scale bars equal 100 μm. Cells positive for active caspase-3, Ki67 and MV-H were counted in representative spleen sections of treated (“+”) and untreated (“−”) mice. Results shown in the graphs are means +SD of three visual fields (magnification 200x) per animal and time point. ***P<0.001 **P<0.01, *P<0.05, ns = not significant using the unpaired t-test. (B) Blast clearance in liver. Representative micrographs of treated mice are shown. Scale bar equals 100 μm.
Thus, MV-Edm readily replicates and spreads in ALL in vivo, eliminating blasts.
Systemic MV-Edm controls CNS leukemia
In 2 mice with xenograft #21 we determined that central nervous system (CNS) disease was already present when therapy was started (Figure 7A). Meningeal leukemic load was over 60% (data not shown) when peripheral blasts had reached 5–20%. Assuming a similar correlation in the other xenografts, efficacy of systemically injected MV-Edm against CNS disease in xenografts #15, #6, #19 and #13 was evaluated. When untreated mice had to be killed, CNS disease in all 4 xenografts had massively progressed to an extent comparable with peripheral disease, i.e. 60–80% (Figure 7B). In stark contrast, when surviving treated mice (none of which exhibited neurological signs) were analyzed 135–150 d after therapy, i.e. near the end of their lifespan, meningeal blasts were markedly decreased in all of the xenografts (Figure 7B). Quantitative histological analysis performed on xenograft #15 proved that MV-Edm had markedly decreased both the number and the extent of infiltration of blasts in treated mice (Figure 7C and Online Supplementary Figure S7).
Figure 7.
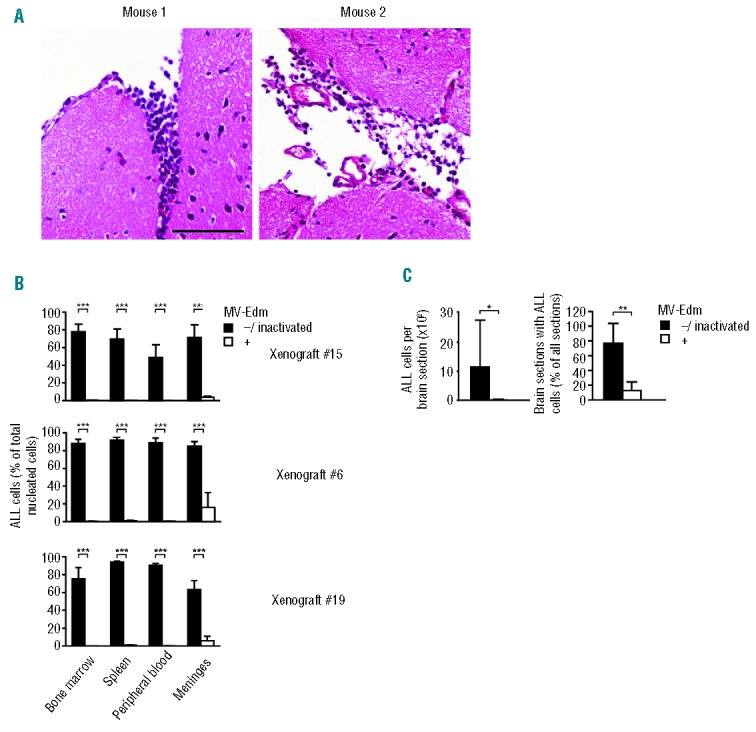
Systemic MV-Edm curbs CNS disease In NOD/SCID mice with ALL xenografts (A) CNS disease is established early. 2 mice with xenograft #21 were killed once peripheral leukemic blasts reached 5–20 % (90 d), as determined by FACS, and CNS disease was assessed by FACS analysis of the meninges and histology of the brain. Scale bars equal 100 μm. (B) Meningeal blasts are controlled while not eradicated by MV-Edm. Mice with xenografts without MV-Edm (“−” and “inactivated”) that thus died from disease, and which could be assessed before onset of autolysis (8 out of 16 mice for xenograft #15, 7 out of 8 for xenograft #6, 10 out of 10 for xenograft #19 and 9 out of 10 for xenograft #13) were compared to mice that were treated with MV-Edm (“+”) and thus died or were sacrificed because of advanced age (8 out of 8 mice for xenograft #15, 7 out of 9 for xenograft #6, 8 out of 9 for xenograft #19 and 2 out of 10 for xenograft #13). Organs were investigated by FACS for the presence of CD45+Ly5− leukemic cells. ***P<0.001 using the unpaired t-test. (C) Marked decrease of blasts infiltrating the brain. Brains of mice with xenograft #15 were serially cut as depicted in Online Supplementary Figure S7 and stained by HE. Blasts were detected by morphology (left panel, scale bar equals 100 μm). The number of leukemic cells per section and the percentage of sections containing leukemic cells were determined (right panel; means +SD for each group are depicted; **P<0.01, *P<0.05 using the unpaired t-test).
We wanted to know whether CNS disease had caused the few spontaneous deaths of treated mice. In the 2 treated mice with xenograft #6 that spontaneously died (Figure 5A), only 16% and 17% leukemic blasts were found in the meninges (data not shown). In the 2 (of 10) treated mice with xenograft #13 that spontaneously died and whose brains could be examined prior to autolysis, leukemic infiltration of the meninges was 3% and 4%. Thus, residual CNS disease is unlikely to have caused the death of the treated mice that died.
Taken together, these data show that MV-Edm controls CNS disease in mice with ALL xenografts. While incomplete, this control most likely contributed to survival.
Discussion
This paper shows that attenuated MV eradicates and controls peripheral and central nervous ALL, respectively, in mice with disseminated primary B-lineage ALL xenografts. Using a large cohort of xenografted or native primary ALL samples investigated ex vivo, we proved general susceptibility of pediatric B-lineage ALL to attenuated MV. Previous knowledge about MV or other viruses as leukemolytic agents addressed adult disease. In mice bearing the Met-1 cell line model of HTLV-1-induced adult T-cell leukemia/lymphoma oncolytic MV decreased tumor burden.47 In vitro, oncolytic MV killed several chronic lymphocytic leukemias (an indolent leukemia of old age) and adult acute lymphocytic leukemias, with differential cytopathology and kinetics.25 MV has been shown to eradicate subcutaneous solid tumors of NALM-6 cells upon intratumoral injection, and to decrease grafting of intravenously injected NALM-6 cells by prompt preemptive injection of naked virus or virus protected within meschenchymal stromal cells.25,26 It was unknown whether MV can eradicate established disseminated patient ALL, in particular pediatric ALL, the most common malignancy in childhood.
A few intravenous injections of MV-Edm at a dose per body weight comparable to the maximal dose administered intravenously to patients in clinical trials for multiple myeloma22 were administered to mice with four established and disseminated patient-derived pediatric B-lineage ALL xenografts that all afflicted the CNS. These leukemias covered the spectrum of B-lineage ALL. The leukemic burden was high at start of treatment, as indicated by a large number of leukemic blasts in bone marrow, blood, spleen, liver and CNS. Our approach thus represented an attempt to cure mice with florid, widely disseminated patient ALL rather than a preemptive approach. Administration of MV-Edm rapidly decreased peripheral leukemic blasts in all four xenografts and eradicated peripheral blasts in three xenografts resulting in survival of the large majority of mice. Of note, the few treated mice with xenografts #15, #6 and #19 that died did so at the end of their natural lifespan. Their death was most likely caused by advanced age, as the number of CNS blasts had markedly decreased to moderate or low levels compatible with life, and since peripheral leukemia was eradicated. The fourth, very rapidly progressing ALL (#13) also responded quickly to MV-Edm, both in the peripheral blood, the bone marrow and the CNS. Nevertheless, all but one of the mice with this xenograft died early. In the mice that could be examined, post mortem death was associated with extensive necrosis in the bone marrow and with hematopoietic depression. As murine cells are not susceptible to MV-Edm, this might represent a leukemia-induced depressive effect that persisted despite regression of this aggressive leukemia, as has recently been described for acute myeloid leukemia.48
MV-Edm was effective ex vivo in a large number of primary and xenografted B-lineage ALL. Analysis of most ALL investigated was not precluded by their intrinsic apoptosis rate evident ex vivo. Marked intrinsic apoptosis is a known characteristic of ALL cells that can be exacerbated ex vivo by culture conditions.49 Extension of ex vivo survival requires the use of bone marrow feeder cells and cytokines. This, however, would introduce additional confounding variables into the assay, including a selection bias for cell death-resistant cells. While T-lineage ALL cells from a xenograft and from cell lines were found to be sensitive to MV-Edm ex vivo and in vitro, the general susceptibility of T-lineage ALL has to be proven in larger studies. An important factor in the potency of MV-Edm against pediatric ALL was its ability to repress CNS disease. Control of CNS disease was, albeit incomplete, sufficient in all treated mice for survival until advanced age without clinical signs of CNS leukemia and with only moderate to low numbers of residual blasts in the CNS. This is remarkable, given that no attempts were made to specifically target CNS leukemia. Of note, extensive CNS disease was already established when MV-Edm was applied, arguing against mere prevention of CNS disease by MV-Edm via eradication of peripheral blasts. Control of leukemia required more time in the CNS than in blood, spleen and liver. This suggests that entry of MV into the CNS, while possibly facilitated by disruption of the blood-brain barrier by meningeal blasts, was nevertheless limited. In addition, spread of MV within the CNS may be more restricted than in the periphery. Given these impediments, future studies will attempt to eradicate CNS disease by modulating the schedule of treatment.
Several additional factors may account for the pronounced efficacy of MV-Edm against ALL in vivo. In contrast to many solid tumor cells, most leukemic cells reside in well-perfused organs like bone marrow, blood, lymphatic system and liver, facilitating viral delivery. No barrier exists between MV and leukemic blasts in the blood, and the sinusoidal capillaries typical for bone marrow, spleen, lymph nodes and liver provide easy access of MV to leukemic cells in these organs. As in mouse solid tumor models, no infection of host cells occurs since they do not express entry receptors for MV-Edm, thus preventing sequestration of the virus and secretion of interferon by these cells. On the other hand, the mouse model may underestimate the efficacy of MV-Edm. Control of leukemia in the patient may be enhanced by several mechanisms, provided that immunosuppression of the patient by the leukemia or prior therapy is not pronounced. Thus, patients may be able to mount a cytotoxic T-cell response to ALL cells infected with attenuated MV, as it has been shown that attenuated MV-infected tumor cells induce tumor antigen cross-presentation by human plasmacytoid dendritic cells.50 In addition, destruction of ALL cells may be enhanced by neutrophils attracted to oncolytic MV-infected ALL cells, as has been described for experimental solid tumors.12 Furthermore, ALL-infiltrating neutrophils may become activated to secrete antitumor cytokines and TRAIL, a notion supported by a recent proof of principle study.51
A further factor-enhancing efficacy of MV-Edm against ALL in the mouse model was efficient intraleukemic viral spread. The absence of immunity against measles virus in mice favored intraleukemic spread and thus leukemolytic efficacy of MV-Edm in vivo. In this regard, it is important to note that 29–60% of children with ALL during chemotherapy do not have protective antibody levels against measles.52 On the one hand, this would facilitate delivery and spread of MV-Edm in this population, on the other hand, it would pose the challenge of safely applying this live, albeit attenuated virus to immunocompromised hosts. As intraleukemic spread of MV-Edm was markedly inhibited in the presence of neutralizing antibodies against measles, clinical application of MV-Edm in measles-immune children with ALL would require transient suppression of antibody production by administering cyclophosphamide, as is currently being done in patients with solid tumors treated with MV-Edm.22,24,53 Another possibility to protect measles virus particles from neutralization is to introduce MV-Edm into cellular vehicles ex vivo before treatment of measles immune hosts, as we and others have shown.26,32,54,55 Yet another determinant of efficacy for oncolytic MV is efficient replication in target cells. Several lines of evidence indicate that MV-Edm strongly replicates in ALL cells. First, the number of leukemic blasts rapidly decreased in all the 4 xenografts treated in vivo. Second, replication-associated MV-H protein was readily detected in a xenograft in vivo. Third, most of the patient xenografts and primary patient ALL assessed ex vivo were susceptible to MV-Edm. Finally, infected Jurkat cells generated a substantial number of MV-Edm. In stark contrast, there was little evidence for replication of MV-Edm in normal hematopoietic cells. Thus, PBMC were just marginally susceptible to MV-Edm and did not generate discernible infectious particles, and HSC were resistant to MV-Edm.
In B-cell precursor ALL xenograft cells replication occurred despite an intact appearing, albeit heterogeneously active RIG-I/MDA-5 sensing and signaling pathway that induced transcription of interferon-β leading to induction of interferon-responsive genes. This suggests that MV-Edm can evade the interferon defense in ALL by mechanisms yet to be elucidated. It remains possible that lack of expression or mutations of genes not assessed or alterations on the protein or posttranslational levels decrease the functional interferon response in ALL. In contrast to xenograft cells, ALL cell lines did not induce response to MV-Edm by mounting an interferon response. This may have been caused by epigenetic silencing of genes within the RIG-1/MDA-5/INF pathway, as has been described in lung cancer cell lines,44 possibly secondary to the conditions of cell culture.
Differential sensitivity of leukemic blasts and normal cells is crucial for safety of leukemolytic MV. While the large majority of ALL was very sensitive to MV-Edm-induced cell death, human hematopoietic progenitors and PBMC were resistant or less sensitive, respectively. With one exception, the entry receptor CD150 was not expressed on ALL cells, thus ruling out that CD150 determined their susceptibility. As leukemic blasts expressed equal or lower levels of the entry receptor CD46 compared to HSC and PBMC, specific sensitivity of ALL can also not be explained by their expression levels of CD46. It remains possible that differential expression of yet unknown entry receptors contribute to the differential sensitivity of ALL. As the transcriptional induction of the interferon response in B-cell precursor ALL xenograft cells was similar to lymphocytes, it is unlikely that their difference in sensitivity can be attributed solely to differential interferon induction. Thus, alternative mechanisms of differential susceptibility remain to be determined. While our results show no hematopoietic toxicity of MV-Edm ex vivo, it is difficult to extrapolate these findings to patients. Infected ALL cells in the bone marrow may generate high local titers of MV causing death of bystander HSC and progenitors. Non-hematopoietic toxicity of attenuated MV-Edm was not investigated in this study. In this regard, MV-Edm given to patients with solid tumors has an excellent safety record, even when administered systemically to immunocompromised hosts. While attenuated MV may potentially cause pneumonitis or encephalitis in children, to date, no molecularly proven case has been reported. However, 2 children with ALL that developed pneumonitis and encephalopathy respectively after MU vaccination have been described.56,57 On the other hand, 10 children with ALL during therapy were vaccinated against MV, without severe side-effects.58
In summary, this work shows that attenuated MV efficiently controls pediatric B-lineage ALL xenografts from patients, supporting the notion of developing attenuated MV as a complementary or alternative therapeutic modality against pediatric ALL of B-lineage. Necessary next steps to this end include molecular dissection of differential sensitivity of leukemic cells versus hematopoietic cells and determination of hematopoietic and neural toxicity of attenuated MV in mouse models with human hematopoiesis harboring ALL and in immunocompromised measles-susceptible mice with human ALL.
Acknowledgments
The authors would like to thank Nicole Heymann and Helgard Knauβ for excellent technical assistance, the Russell lab (Mayo Clinic, Rochester) for MV-Edm, Michaela Feuring-Buske (Internal Medicine III, University Medical Center Ulm) for introducing us to hematopoietic progenitor assays and Klaus Schwarz (Institute for Transfusion Medicine, University Medical Center Ulm) for HSC from cord blood. We appreciate the very helpful discussions with Karsten Stahnke. The authors declare no conflict of interest.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by a grant of the Deutsche Forschungsgemeinschaft within the Sonderforschungsbereich 1074 (to CB).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006;354(2):166–78 [DOI] [PubMed] [Google Scholar]
- 2.Schrappe M, Reiter A, Ludwig WD, Harbott J, Zimmermann M, Hiddemann W, et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 2000;95(11):3310–22 [PubMed] [Google Scholar]
- 3.Schrappe M, Reiter A, Zimmermann M, Harbott J, Ludwig WD, Henze G, et al. Long-term results of four consecutive trials in childhood ALL performed by the ALL-BFM study group from 1981 to 1995. Berlin-Frankfurt-Munster. Leukemia 2000;14(12): 2205–22 [DOI] [PubMed] [Google Scholar]
- 4.Grote D, Russell SJ, Cornu TI, Cattaneo R, Vile R, Poland GA, et al. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood 2001;97(12):3746–54 [DOI] [PubMed] [Google Scholar]
- 5.Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res 2002;62(16):4656–62 [PubMed] [Google Scholar]
- 6.Phuong LK, Allen C, Peng KW, Giannini C, Greiner S, TenEyck CJ, et al. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res 2003; 63(10):2462–9 [PubMed] [Google Scholar]
- 7.Hallak LK, Merchan JR, Storgard CM, Loftus JC, Russell SJ. Targeted measles virus vector displaying echistatin infects endothelial cells via alpha(v)beta3 and leads to tumor regression. Cancer Res 2005;65(12):5292–300 [DOI] [PubMed] [Google Scholar]
- 8.Fielding AK. Measles as a potential oncolytic virus. Rev Med Virol 2005;15(2):135–42 [DOI] [PubMed] [Google Scholar]
- 9.Myers RM, Greiner SM, Harvey ME, Griesmann G, Kuffel MJ, Buhrow SA, et al. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther 2007;82(6):700–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu C, Sarkaria JN, Petell CA, Paraskevakou G, Zollman PJ, Schroeder M, et al. Combination of measles virus virotherapy and radiation therapy has synergistic activity in the treatment of glioblastoma multiforme. Clin Cancer Res 2007;13(23):7155–65 [DOI] [PubMed] [Google Scholar]
- 11.Dingli D, Peng KW, Harvey ME, Greipp PR, O’Connor MK, Cattaneo R, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood 2004;103(5):1641–6 [DOI] [PubMed] [Google Scholar]
- 12.Grote D, Cattaneo R, Fielding AK. Neutrophils contribute to the measles virus-induced antitumor effect: enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res 2003;63(19):6463–8 [PubMed] [Google Scholar]
- 13.Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol 2005;23(2): 209–14 [DOI] [PubMed] [Google Scholar]
- 14.Springfeld C, von Messling V, Frenzke M, Ungerechts G, Buchholz CJ, Cattaneo R. Oncolytic efficacy and enhanced safety of measles virus activated by tumor-secreted matrix metalloproteinases. Cancer Res 2006;66(15):7694–700 [DOI] [PubMed] [Google Scholar]
- 15.Ungerechts G, Springfeld C, Frenzke ME, Lampe J, Johnston PB, Parker WB, et al. Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res 2007;67(22):10939–47 [DOI] [PubMed] [Google Scholar]
- 16.Jing Y, Tong C, Zhang J, Nakamura T, Iankov I, Russell SJ, et al. Tumor and vascular targeting of a novel oncolytic measles virus retargeted against the urokinase receptor. Cancer Res 2009;69(4):1459–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugiyama T, Yoneda M, Kuraishi T, Hattori S, Inoue Y, Sato H, et al. Measles virus selectively blind to signaling lymphocyte activation molecule as a novel oncolytic virus for breast cancer treatment. Gene Ther 2012;20(3):338–47 [DOI] [PubMed] [Google Scholar]
- 18.Myers R, Harvey M, Kaufmann TJ, Greiner SM, Krempski JW, Raffel C, et al. Toxicology study of repeat intracerebral administration of a measles virus derivative producing carcinoembryonic antigen in rhesus macaques in support of a phase I/II clinical trial for patients with recurrent gliomas. Hum Gene Ther 2008;19(7):690–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinzerling L, Kunzi V, Oberholzer PA, Kundig T, Naim H, Dummer R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood 2005;106(7): 2287–94 [DOI] [PubMed] [Google Scholar]
- 20.Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res 2010;70(3):875–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ClinicalTrials.gov. Recombinant Measles Virus Vaccine Therapy and Oncolytic Virus Therapy in Treating Patients With Progressive, Recurrent, or Refractory Ovarian Epithelial Cancer or Primary Peritoneal Cancer. http://clinicaltrials.gov/ct2/show/NCT00408590
- 22.Msaouel P, Dispenzieri A, Galanis E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: an overview. Curr Opin Mol Ther 2009;11(1):43–53 [PMC free article] [PubMed] [Google Scholar]
- 23.ClinicalTrials.gov. Viral Therapy in Treating Patients With Recurrent Glioblastoma Multiforme. http://clinicaltrials.gov/ct/show/NCT00390299
- 24.ClinicalTrials.gov. Vaccine Therapy With or Without Cyclophosphamide in Treating Patients With Recurrent or Refractory Multiple Myeloma. http://clinicaltrials.gov/ct2/show/NCT00450814
- 25.Patel B, Dey A, Ghorani E, Kumar S, Malam Y, Rai L, et al. Differential Cytopathology and Kinetics of Measles Oncolysis in Two Primary B-cell Malignancies Provides Mechanistic Insights. Mol Ther 2011;19(6): 1034–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castleton A, Dey A, Beaton B, Patel B, Aucher A, Davis DM, et al. Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood 2014; 123(9):1327–35 [DOI] [PubMed] [Google Scholar]
- 27.Takeuchi K, Nagata N, Kato SI, Ami Y, Suzaki Y, Suzuki T, et al. Wild-type measles virus with the hemagglutinin protein of the edmonston vaccine strain retains wild-type tropism in macaques. J Virol 2012;86(6):3027–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 2011;480(7378):530–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, et al. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 2011;7(8):e1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Firsching R, Buchholz CJ, Schneider U, Cattaneo R, ter Meulen V, Schneider-Schaulies J. Measles virus spread by cell-cell contacts: uncoupling of contact-mediated receptor (CD46) downregulation from virus uptake. J Virol 1999;73(7):5265–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawrence DM, Patterson CE, Gales TL, D’Orazio JL, Vaughn MM, Rall GF. Measles virus spread between neurons requires cell contact but not CD46 expression, syncytium formation, or extracellular virus production. J Virol 2000;74(4):1908–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei J, Wahl J, Nakamura T, Stiller D, Mertens T, Debatin KM, et al. Targeted release of oncolytic measles virus by blood outgrowth endothelial cells in situ inhibits orthotopic gliomas. Gene Ther 2007; 14(22):1573–86 [DOI] [PubMed] [Google Scholar]
- 33.Jurianz K, Ziegler S, Garcia-Schuler H, Kraus S, Bohana-Kashtan O, Fishelson Z, et al. Complement resistance of tumor cells: basal and induced mechanisms. Mol Immunol 1999;36(13–14):929–39 [DOI] [PubMed] [Google Scholar]
- 34.Anderson BD, Nakamura T, Russell SJ, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res 2004;64(14):4919–26 [DOI] [PubMed] [Google Scholar]
- 35.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006;314(5801):994–7 [DOI] [PubMed] [Google Scholar]
- 36.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006;314(5801):997–1001 [DOI] [PubMed] [Google Scholar]
- 37.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006;441(7089):101–5 [DOI] [PubMed] [Google Scholar]
- 38.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA 2004; 101(49):17264–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 2008;89(Pt 1):1–47 [DOI] [PubMed] [Google Scholar]
- 40.Shingai M, Ebihara T, Begum NA, Kato A, Honma T, Matsumoto K, et al. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J Immunol 2007;179(9):6123–33 [DOI] [PubMed] [Google Scholar]
- 41.Bankamp B, Takeda M, Zhang Y, Xu W, Rota PA. Genetic characterization of measles vaccine strains. J Infect Dis 2011;204(Suppl 1):S533–48 [DOI] [PubMed] [Google Scholar]
- 42.Pitha PM. Introduction: interferon’s connection to cancer. Semin Cancer Biol 2000;10(2):69–72 [DOI] [PubMed] [Google Scholar]
- 43.Krishnamurthy S, Takimoto T, Scroggs RA, Portner A. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J Virol 2006;80(11): 5145–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Q, Tainsky MA. Epigenetic silencing of IRF7 and/or IRF5 in lung cancer cells leads to increased sensitivity to oncolytic viruses. PLoS One 2011;6(12):e28683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer LH, Eckhoff SM, Queudeville M, Kraus JM, Giordan M, Stursberg J, et al. Early relapse in ALL is identified by time to leukemia in NOD/SCID mice and is characterized by a gene signature involving survival pathways. Cancer Cell 2011;19(2):206–17 [DOI] [PubMed] [Google Scholar]
- 46.Meyer LH, Debatin KM. Diversity of human leukemia xenograft mouse models: implications for disease biology. Cancer Res 2011;71(23):7141–4 [DOI] [PubMed] [Google Scholar]
- 47.Parrula C, Fernandez SA, Zimmerman B, Lairmore M, Niewiesk S. Measles virotherapy in a mouse model of adult T-cell leukaemia/lymphoma. J Gen Virol 2011; 92(Pt 6):1458–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miraki-Moud F, Anjos-Afonso F, Hodby KA, Griessinger E, Rosignoli G, Lillington D, et al. Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proc Natl Acad Sci USA 2013;110(33): 13576–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manabe A, Coustan-Smith E, Behm FG, Raimondi SC, Campana D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood 1992;79(9):2370–7 [PubMed] [Google Scholar]
- 50.Guillerme JB, Boisgerault N, Roulois D, Menager J, Combredet C, Tangy F, et al. Measles virus vaccine-infected tumor cells induce tumor antigen cross-presentation by human plasmacytoid dendritic cells. Clin Cancer Res 2013;19(5):1147–58 [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Patel B, Dey A, Ghorani E, Rai L, Elham M, et al. Attenuated, oncolytic, but not wild-type measles virus infection has pleiotropic effects on human neutrophil function. J Immunol 2012;188(3):1002–10 [DOI] [PubMed] [Google Scholar]
- 52.van Tilburg CM, Sanders EA, Rovers MM, Wolfs TF, Bierings MB. Loss of antibodies and response to (re-)vaccination in children after treatment for acute lymphocytic leukemia: a systematic review. Leukemia 2006;20(10):1717–22 [DOI] [PubMed] [Google Scholar]
- 53.Peng KW, Myers R, Greenslade A, Mader E, Greiner S, Federspiel MJ, et al. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther 2013;20(3): 255–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jarmy G, Wei J, Debatin KM, Beltinger C. Apoptosis-inducing cellular vehicles for cancer gene therapy: endothelial and neural progenitors. In: Srivastava R. (ed.) Apoptosis, cell signaling and human diseases: Molecular Mechanisms. Totowa: Humana Press, Inc., 2006:279–320 [Google Scholar]
- 55.Wei J, Jarmy G, Genuneit J, Debatin KM, Beltinger C. Human blood late outgrowth endothelial cells for gene therapy of cancer: determinants of efficacy. Gene Ther 2007; 14(4):344–56 [DOI] [PubMed] [Google Scholar]
- 56.Mitus A, Holloway A, Evans AE, Enders JF. Attenuated measles vaccine in children with acute leukemia. Am J Dis Child 1962; 103:413–8 [DOI] [PubMed] [Google Scholar]
- 57.Valmari P, Lanning M, Tuokko H, Kouvalainen K. Measles virus in the cerebrospinal fluid in postvaccination immunosuppressive measles encephalopathy. Pediatr Infect Dis J 1987; 6(1):59–63 [DOI] [PubMed] [Google Scholar]
- 58.Torigoe S, Hirai S, Oitani K, Ito M, Ihara T, Iwasa T, et al. Application of live attenuated measles and mumps vaccines in children with acute leukemia. Biken J 1981; 24(4):147–51 [PubMed] [Google Scholar]