

Abstract
Meta-analysis of multiple genome-wide association (GWA) studies has become common practice over the last few years. The main advantage of this technique is the maximization of power to detect the subtle genetic effects for common traits. Moreover, one can use meta-analysis to probe and identify heterogeneity in the effect sizes across the combined studies. In this review we systematically appraised and evaluated the characteristics of GWA meta-analyses with 10,000 or more subjects published until June 2012. We overview the current landscape of variants discovered by GWA meta-analyses and we discuss and assess with extrapolations from empirical data the value of larger meta-analyses for the discovery of additional genetic associations and new biology in the future. Finally, we discuss some emerging logistical and practical issues related to the conduct of meta-analysis of GWA studies.
Keywords: variance explained, gene discovery, sample size, common variants, rare variants, missing heritability, consortium
INTRODUCTION
The advent of genome-wide association (GWA) studies has led to the discovery of hundreds of thousands of genotype-phenotype associations with robust statistical significance (42, 66). For example, as of October 2012 the Catalog of Published Genome-Wide Association Studies, hosted by the National Human Genome Research Institute (http://www.genome.gov/gwastudies/) (29), listed more than 7,500 associations between single-nucleotide polymorphisms (SNPs) and complex traits with P<10−5 coming from more than 1,400 GWA publications. These studies survey most of the genome by examining available samples for phenotype associations against up to 3,000,000 polymorphisms. Performance of GWA studies was largely facilitated by technological advances in high-throughput genotyping technologies providing accurate and reproducible genotyping (81) in combination with the progressive drop in genotyping costs. Additionally, the results of the International HapMap Consortium and the 1000 Genomes Project provided further useful insights about human genetic variation by systematically cataloguing common and low-frequency variation, and by characterizing linkage disequilibrium patterns in the human genome (1, 33-35); this information is now used routinely in GWA studies.
Over the last years, GWA studies have made major contributions to the efforts of gene mapping (2, 42) yielding numerous novel genetic associations, many of which have been successfully replicated in subsequent studies (77). However, early studies utilized small sample sizes and were, thus, underpowered to detect the small effect sizes expected for common traits under the common disease-common variant model (83); these variants would require large sample sizes, especially when their frequency is low (76). Consequently, some of the early GWA findings were later disputed in larger studies (18, 36, 68). Although not very common with GWA studies in general, this lack of replication was very common for the results of candidate-gene studies. Results of many such studies had shown surprisingly low reproducibility rates in subsequent larger studies and meta-analyses thereof (38, 39, 63, 96), revealing a large amount of false-positive discoveries (13, 41). Hence, meta-analysis of available GWA data from different studies was soon recognized as the appropriate method in order to achieve adequate sample sizes and optimal power for the discovery of genetic associations with modest effect sizes (19, 87).
As a statistical method which allows the quantitative synthesis of results from different studies in order to estimate a common summary effect (55), the major advantage of meta-analysis is that it maximizes power, i.e. reduces the probability of false negative results (19). Additional potential advantages include the increased precision of the effect estimates for the identified associations, reduction of the extent of the winner’s curse phenomenon (when adequate power is achieved), and opportunities to evaluate and measure the degree of consistency or heterogeneity of the genetic effect across the combined studies (28, 40). Meta-analysis of GWA studies has emerged as an efficient strategy for prioritizing GWA results for further follow-up through functional analyses, or further replication efforts, cross-phenotype checks, or mendelian randomization studies (7). Meta-analysis of multiple GWA datasets is now a well-established and validated strategy for both discovery and replication of genetic association studies (7, 21, 52).
GWA meta-analysis has been made feasible due to technical advances in imputation of missing genotypes between different studies (14, 61), but also due to the creation of consortia that facilitate sharing of data between the different studies. Nevertheless, despite the numerous successes of GWA studies and meta-analyses thereof, the loci discovered so far explain only a small fraction of the estimated total heritability for most common traits (67).
In this review, we demonstrate the power of meta-analysis in GWA studies by summarizing the characteristics of GWA meta-analyses performed so far and estimating the contribution of their discoveries to the genetic variance of the respective common trait. We overview the features of GWA discovered variants and we discuss the potential applications and challenges of even larger GWA meta-analyses in the future.
PART 1: EMPIRICAL APPRAISAL OF PUBLISHED GWA META-ANALYSES
1.1. METHODS
We searched the publicly available database “A Catalog of Published Genome-Wide Association Studies” hosted by the National Human Genome Research Institute. This is an online regularly updated database of SNP-trait associations with P<10−5 extracted from published GWA reports, which attempt to assay at least 100,000 SNPs. The last search was performed on June 15, 2012.
We identified GWA studies with a sample size ≥10,000 in the discovery stage. We used this threshold so as to focus on GWA studies where discovery efforts have had decent power to identify common variants with modest effects (per allele odds ratio >1.20 and minor allele frequency >5%), since with smaller sample sizes many variants with sizeable effects would have been missed and thus there is still substantial uncertainty about the architecture of genetic risks and about what meta-analysis of sufficient data can achieve for the probed phenotypes of interest. Subsequently, we accessed the full-text publications and selected those papers that had performed meta-analyses of 2 or more GWA datasets in the discovery stage regardless of any replication efforts. We included meta-analyses in populations of different ancestries as separate entries. When 2 or more reports were available for the same trait in the same population ancestry, we included the one having the largest sample size.
From each eligible GWA report and the corresponding meta-analyses, we extracted the following information: first author, year and journal of publication, disease/trait, sample size in the discovery and replication stages, population ancestries in the discovery and replication stages, platform used for genotyping and SNPs passing quality control, number of datasets combined through meta-analysis in the discovery stage, method of synthesis (i.e. fixed-effects, random-effects, other), heterogeneity testing, threshold used for determining genome-wide significance, imputation methods and number of imputed SNPs, quality control, criteria for replication, generalizability to diverse ancestry groups, extension to testing for diverse phenotypes and which, phenotypic cross-checks performed, and finally functional analyses performed.
Additionally, from each eligible GWA meta-analysis we extracted the independent SNPs discovered and the corresponding gene loci, chromosomal regions, minor and reference alleles with their corresponding frequencies, effect sizes and measures of uncertainty (i.e. standard error or 95% CI). Subsequently we expressed these effect sizes as the contribution of the respective susceptibility locus to the total genetic variance of the trait using the approach by Park et al. (78).
1.2. RESULTS
As of June 15, 2012, the catalog included a total of 1,271 published GWA reports. Of those, 88 reports (references S1-S88; listed in Supplement) included a total of 139 eligible meta-analyses of GWA datasets published between 2009 and 2012 in 20 different journals (Table 1). The majority (44%) of the reports appeared in Nature Genetics, followed by PLoS Genetics (14%), Circulation Cardiovascular Genetics (7%), Human Molecular Genetics (6%), JAMA (5%), and other journals (24%). 106 (76%) meta-analyses pertained to disease phenotypes and 33 (24%) pertained to quantitative traits. The median sample size of these discovery meta-analyses was 20,611 (IQR, 14,392-38,238). The majority (n=109) had been performed in populations of European ancestry, while the remaining had been performed in Asian (n=20), African (n=2) or mixed populations (n=8). Additional replication efforts had been performed for the findings of 101 meta-analyses. The median sample size used in these replication efforts was 16,378 (IQR, 8,112-48,607). The majority (n=81) had been performed in European populations and only 20 had been in populations of non-European ancestries.
Table 1.
Description of 139 eligible meta-analyses of GWA studies
| Discovery Stage | Replication Stage | |||
|---|---|---|---|---|
| Disease/Trait | Sample Size a | Ancestry | Sample Size a | Ancestry |
| Bone mineral density- Femoral neck (S19) | 32,961 | EUR, AS | 50,933 | EUR, AS |
| Bone mineral density- Lumbar spine (S19) | 31,800 | EUR, AS | 50,933 | EUR, AS |
| Head circumference (infant) (S77) | 10,768 | EUR | 8,321 | EUR |
| Obesity (childhood) (S9) | 13,848 (5,530/8,318) | EUR | 6,901 (2,818/4,083) | EUR |
| Brachial circumference (S7) | 18,753 | EUR | 3,623 | EUR |
| Adiponectin levels (S12) | 35,355 | EUR, AS, AFR | NA | NA |
| Creatinine-based estimated GFR (S55) | 74,354 | EUR | 64,356 | EUR, AFR |
| Cystatine-based estimated GFR (S55) | 22,937 | EUR | 3,709 | EUR |
| Chronic kidney disease (S55) | 68,678 (6,271/62,407) | EUR | 64,356 | EUR, AFR |
| Rheumatoid arthritis (S52) | 20,965 (4,074/16,891) | AS | 52,669 (10,816/41,853) | EUR, AS |
| Platelet count (S59) | 16,388 | AFR | 18,371 | EUR, HIS |
| Metabolic syndrome (S40) | 11,616 (2,637/7,927) | EUR | NA | NA |
| Body mass index (S85) | 27,715 | AS | 55,333 | AS |
| Menopause (age at onset) (S75) | 38,968 | EUR | 14,435 | EUR |
| Atopic dermatitis (S53) | 26,171 (5,606/20,565) | EUR | 25,252 (5,419/19,833) | EUR |
| Ankle-brachial index (S48) | 41,692 | EUR | 16,717 | EUR |
| Multiple sclerosis (S54) | 17,698 (5,545/12,153) | EUR | NA | NA |
| Mean platelet volume (S25) | 18,600 | EUR | 18,838 | EUR |
| Platelet count (S25) | 48,666 | EUR | 18,838 | EUR |
| Aortic stiffness (carotid-femoral pulse wave velocity) (S46) | 20,634 | EUR | 5,306 | EUR |
| Allergic rhinitis (S60) | 12,898 (3,933/8,965) | EUR | NA | NA |
| IgE sensitization to grass (S60) | 12,898 (3,933/8,965) | EUR | NA | NA |
| Alanine aminotransferase (ALT) (S10) | 45,596 | EUR, AS | 8112 | EUR |
| Alkaline phosphatase (ALP) (S10) | 56,415 | EUR, AS | 8112 | EUR |
| Gamma glutamyl transpeptidase (γ-GT) (S10) | 61,089 | EUR, AS | 8112 | EUR |
| Lipoprotein-associated phospholipase A2 mass (S26) | 12,126 | EUR | NA | NA |
| Lipoprotein-associated phospholipase A2 activity (S26) | 11,664 | EUR | NA | NA |
| Height (S49) | 20,427 | AFR | 16,436 | AFR |
| Type 1 diabetes (S8) | 26,890 (9,934/16,956) | EUR | 3,360 (1,120/2,240) | EUR |
| Pulmonary function (S70) | 48,201 | EUR | 46,411 | EUR |
| Bipolar disorder (S64) | 16,731 (7,481/9,250) | EUR | 46,918 (4,496/42,422) | EUR |
| Common carotid intima media thickness (S5) | 31,211 | EUR | 10,403 | EUR |
| Internal carotid intima media thickness (S5) | 10,962 | EUR | NA | NA |
| Plaque (S5) | 25,179 | EUR | 6,013 | EUR |
| Diastolic blood pressure (S31) | 69,395 | EUR | 133,361 | EUR |
| Systolic blood pressure (S31) | 69,395 | EUR | 133,361 | EUR |
| Pulse pressure (S82) | 74,064 | EUR | 48,607 | EUR |
| Mean arterial pressure (S82) | 74,064 | EUR | 48,607 | EUR |
| Coffee consumption (S1) | 18,176 | EUR | 7,929 | EUR |
| Type 2 diabetes (S39) | 20,019 (5,561/14,458) | AS | 85,685 (21,300/64,385) | EUR, AS |
| Proinsulin levels (S76) | 10,701 | EUR | 16,378 | EUR |
| Aging (time to death) (S83) | 25,007 | EUR | 10,411 | EUR |
| Aging (time to event) (S83) | 16,995 | EUR | NA | NA |
| Ankylosing spondylitis (S21) | 11,802 (3,023/8,779) | EUR | 6,594 (2,111/4,483) | EUR |
| Sudden cardiac arrest (S3) | 22,925 (1,283/21,642) | EUR | 14,265 (3,119/11,146) | EUR |
| White blood cell count (S61) | 16,388 | AFR | 37,827 | EUR, AS, HIS |
| Adiposity (% body fat) (S38) | 36,626 | EUR, AS | 39,576 | EUR |
| Diastolic blood pressure (S35) | 19,608 | AS | 30,765 | AS |
| Systolic blood pressure (S35) | 19,608 | AS | 30,765 | AS |
| Glioma (S62) | 11,582 (4,147/7,435) | EUR | NA | NA |
| D-dimer levels (S69) | 21,052 | EUR | NA | NA |
| Dehydroepiandrosterone sulphate (S88) | 14,846 | EUR | NA | NA |
| Caffeine consumption (S11) | 47,431 | EUR | NA | NA |
| Alcohol consumption (S65) | 26,316 | EUR | 21,185 | EUR |
| Alzheimer’s disease (S28) | 20,373 (6,688/13,685) | EUR | 39,343 (13,182/26,161) | EUR |
| Migraine (S42) | 10,980 (2,446/8,534) | EUR | 8,731 (2,957/5,774) | EUR |
| Coronary heart disease (S66) | 86,995 (22,233/64,762) | EUR | 56,508 (28,834/27,674) | EUR |
| Urinary albumin excretion (S6) | 31,580 | EUR | 38,258 | EUR, AFR |
| C-reactive protein (S14) | 66,185 | EUR | 16,540 | EUR |
| Ulcerative colitis (S2) | 26,405 (6,687/19,718) | EUR | 22,545 (9,628/12,917) | EUR |
| Parkinson’s disease (S32) | 17,352 (5,333/12,019) | EUR | 16,060 (7,053/9,007) | EUR |
| IGF-1 (S34) | 10,280 | EUR | NA | NA |
| IGFBP-3 (S34) | 10,018 | EUR | NA | NA |
| Personality dimensions (S13) | 17,375 | EUR | 3,294 | EUR |
| Renal cell carcinoma (S58) | 12,277 (3,772/8,505) | EUR | 7,116 (2,198/4,918) | EUR |
| Menarche (age at onset) (S18) | 87,802 | EUR | 14,731 | EUR |
| Crohn’s disease (S22) | 21,389 (6,333/15,056) | EUR | 29,720 (15,694/14,026) | EUR |
| QRS duration (S72) | 40,407 | EUR | 7,170 | EUR |
| Osteoarthritis (knee) (S20) | 38,280 (2,371/35,909) | EUR | 15,296 (5,521/9,775) | EUR, AS |
| Retinal venular caliber (S30) | 15,358 | EUR | 6,673 | EUR |
| Retinal arteriolar caliber (S30) | 15,358 | EUR | 6,673 | EUR |
| Waist-hip ratio (S27) | 77,167 | EUR | 113,636 | EUR |
| Body mass index (S73) | 123,865 | EUR | 125,931 | EUR |
| Urate levels (S86) | 28,283 | EUR | NA | NA |
| Gout (S86) | 28,283 | EUR | NA | NA |
| Height (S41) | 133,653 | EUR | 50,074 | EUR |
| Asthma (S47) | 26,475 (10,365/16,110) | EUR | NA | NA |
| Glycated hemoglobin (S71) | 46,368 | EUR | 10448 | EUR |
| Calcium levels (S50) | 20,611 | EUR | NA | NA |
| Magnesium levels (S45) | 15,366 | EUR | 8,463 | EUR |
| Sodium (S45) | 11,552 | EUR | 8,463 | EUR |
| Potassium (S45) | 13,683 | EUR | 8,463 | EUR |
| Triglycerides (S78) | 96,598 | EUR | 51,875 | EUR, AS, AFR |
| HDL cholesterol (S78) | 99,900 | EUR | 51,875 | EUR, AS, AFR |
| LDL cholesterol (S78) | 95,454 | EUR | 51,875 | EUR, AS, AFR |
| Total cholesterol (S78) | 100,184 | EUR | 51,875 | EUR, AS, AFR |
| Resting heart rate (RR interval) (S17) | 38,991 | EUR | NA | NA |
| Type 2 diabetes (S81) | 47,117 (8,130/38,987) | EUR | 94,337 (34,412/59,925) | EUR |
| Phosphorus levels (S36) | 16,264 | EUR | 5,444 | EUR |
| Vitamin D insufficiency (S84) | 16,125 | EUR | 17,871 | EUR |
| Rheumatoid arthritis (S74) | 25,708 (5,539/20,169) | EUR | 15,574 (6,768/8,806) | EUR |
| Heart failure (S68) | 20,926 | EUR | NA | NA |
| Smoking behavior - Cigarettes per day (S79) | 38,181 | EUR | 35,672 | EUR |
| Smoking behavior - Ever vs never smoker (S79) | 69,409 (39,022/30,387) | EUR | 68,988 (NR/NR) | EUR |
| Smoking behavior - Former vs current smokers (S79) | 35,845 (20,619/15,226) | EUR | 23,646 | EUR |
| Smoking behavior - Age of smoking initiation (S79) | 22,438 | EUR | NA | NA |
| Birth weight (S23) | 10,623 | EUR | 27,591 | EUR |
| Intracranial aneurysm (S87) | 15,295 (2,780/12,515) | EUR | 4,777 (3,111/1,666) | AS |
| Urinary bladder cancer (S37) | 41,199 (1,889/39,310) | EUR | 8,650 (2,691/5,959) | EUR |
| Coagulation factor VII (S67) | 15422 | EUR | 3678 | EUR |
| Von Wilenbrand factor (S67) | 17596 | EUR | 6307 | EUR |
| Coagulation factor VIII (S67) | 15279 | EUR | 2381 | EUR |
| Height (S51) | 19,633 | AS | NA | NA |
| White blood cell count (S33) | 14,677 | AS | NA | NA |
| Red blood cell count (S33) | 14,392 | AS | NA | NA |
| Hemoglobin (S33) | 14,402 | AS | NA | NA |
| Hematocrit (S33) | 14,395 | AS | NA | NA |
| Mean corpuscular volume (S33) | 14,364 | AS | NA | NA |
| Mean corpuscular hemoglobin (S33) | 14,362 | AS | NA | NA |
| Mean corpuscular hemoglobin concentration (S33) | 14,377 | AS | NA | NA |
| Platelet count (S33) | 14,806 | AS | NA | NA |
| Gamma glutamyl transpeptidase (γ-GT) (S33) | 10,090 | AS | NA | NA |
| Aspartate aminotransferase (AST) (S33) | 13,898 | AS | NA | NA |
| Alanine aminotransferase (ALT) (S33) | 13,696 | AS | NA | NA |
| Total protein (S33) | 10,047 | AS | NA | NA |
| Blood urea nitrogen (S33) | 14,347 | AS | NA | NA |
| Serum creatinine (S33) | 14345 | AS | NA | NA |
| Fasting glucose (S16) | 46,186 | EUR | 76,558 | EUR |
| Fasting insulin (S16) | 38,238 | EUR | 76,558 | EUR |
| HOMA-B (S16) | 36,466 | EUR | 76,558 | EUR |
| HOMA-IR (S16) | 37,037 | EUR | 76,558 | EUR |
| Major mood disorders (S44) | 15,754 (6,686/9,068) | EUR | 4,677 (1,930/2,747) | EUR |
| 2h glucose challenge (S63) | 15,234 | EUR | 30,620 | EUR |
| PR interval (S57) | 28,517 | EUR | NA | NA |
| Hemoglobin (S24) | 24,731 | EUR | 9,456 | EUR |
| Hematocrit (S24) | 24,731 | EUR | 9,456 | EUR |
| Mean corpuscular volume (S24) | 21,456 | EUR | 9,456 | EUR |
| Mean corpuscular hemoglobin (S24) | 13,329 | EUR | 9,456 | EUR |
| Mean corpuscular hemoglobin concentration (S24) | 13,329 | EUR | 9,456 | EUR |
| Red blood cell count (S24) | 13,329 | EUR | 9,456 | EUR |
| Atrial fibrillation (S4) | 40,518 (3,413/37,105) | EUR | 6,218 (2,145/4,073) | EUR |
| Left ventricular mass (S80) | 12,612 | EUR | 4,094 | EUR |
| Left ventricle internal diastolic dimensions (S80) | 12,612 | EUR | 4,094 | EUR |
| Left ventricle wall thickness (S80) | 12,612 | EUR | 4,094 | EUR |
| Left ventricle systolic dysfunction (S80) | 12,612 | EUR | 4,094 | EUR |
| Adiposity- waist circumference (S43) | 38,580 | EUR | 102,064 | EUR |
| Stroke (S29) | 19,602 (3,810/15,792) | EUR | 7,269 (935/6,334) | EUR, AFR |
| Fibrinogen (S15) | 22,096 | EUR | 17,686 | EUR |
| QT interval (S56) | 15,842 | EUR | 13,602 | EUR |
AFR, African; AS; Asian; EUR, European; GFR, glomerular filtration rate; GWA, genome-wide association; HIS, Hispanic; HOMA-B, homeostatic model assessment beta cell function; HOMA-IR, homeostatic model assessment insulin resistance
NA, not applicable because no replication was performed
Sample size in discovery and replication stages is presented as total number of participants for continuous traits and as total number of participants (number of cases/number of controls) for binary traits.
As shown in Supplementary Table 1, for all but 2 meta-analyses the respective individual studies included in the meta-analysis had performed imputation of non-genotyped SNPs. The imputation method used different types of hidden Markov models depending on the specific imputation software. Additionally, in all meta-analyses the respective studies used HapMap data (33-35) to impute missing genotypes, except for 2 meta-analyses (1 %), which used exclusively data from the 1000 Genome Project (1), and another one that used data from both HapMap and the 1000 Genome Project. Commercial Affymetrix and Illumina platforms were the most common genotyping platforms used, with Perlegen platforms being used in 23 meta-analyses. The median number of datasets combined through meta-analysis was 9 (IQR, 5-15; range, 2-46). Fixed-effects model had been used in all 139 meta-analyses as the primary method of data synthesis across the different datasets, while additional models had been used in a total of 16 meta-analyses pertaining to random-effects (n=13), Bayesian approaches (n=1), both random-effects and Bayesian approaches (n=1), and P-value based methods (n=1). Fixed-effects models assume that there is a common true genetic effect across all studies and any variation in it is attributed to random error; on the other hand, random-effects models assume that there are different effect sizes in the included studies and any variation, i.e. heterogeneity, is due to real population differences (15, 40). In this context, the observed domination of fixed-effects synthesis is expected as this approach is more powerful than random-effects for discovery purposes, while the latter is preferable when the aim is to determine and generalize the magnitude of the genetic effect size (80). The method of weighting was reported in a total of 137 meta-analyses. Of those, 127 meta-analyses used inverse-variance weighting; 7 used sample-size weighting; 1 used both inverse-variance and sample-size; and 2 meta-analyses used the Mantel-Haenszel effect-size based method. The median number of SNPs discovered, i.e. passing the respective significance or other thresholds, in these 139 meta-analyses was 4 (IQR, 2-9). Of note, 18 meta-analyses had made no discoveries. The maximum number of discovered SNPs was 115 (S41).
Supplementary Table 2 shows the SNP quality control criteria used in the eligible 88 GWA reports. In 69 of those, the individual studies had used non-identical quality control criteria and in only 18 there was apparent uniformity among the individual studies (report did not provide details but referred to previous publications). Minor allele frequency (MAF), SNP call rate and deviation from Hardy-Weinberg equilibrium (HWE) were among the most common parameters applied. A MAF cut-off of 1% was used for inclusion of SNPs in the analyses in 41 reports, while 4 reports included SNP with MAF of 5% or more, 3 included SNPs with MAF of 2% or more and 1 report included SNPs with MAF of 0.1% or more. When information was provided, 11 reports used a SNP call rate cut-off of 95%, 4 used a cut-off of 90%, 1 used 97%, 4 used 98%, and 2 used 99%. The P-value used to denote deviation from HWE varied from 10−4 to 10−7, with only 1 study using P=5×10−20. Twenty-one reports used some additional stated criterion other than MAF, HWE and SNP call rate. Finally, in most reports the individual studies included in the respective meta-analyses retained data with high imputation quality, with variable quality thresholds and different software used for imputation.
Supplementary Table 3 shows the selected genome-wide significance thresholds, the methods of investigating heterogeneity across studies, and the replication processes applied in the eligible met-analyses. In all meta-analyses, investigators used primarily P-value thresholds to claim genome-wide significance. In two of them, they used additionally Bayesian methods (Bayes factor and posterior probability of association). The P-values thresholds used were P=5×10−8 for 123 meta-analyses; P=5×10−7 for 8 meta-analyses; P=1×10−8 for 3 meta-analyses; P=7.2×10−8 for 2 meta-analyses; and P=2.5×10−8, P=4×10−7 and P=1.6×10−7 for each one of the remaining 3 meta-analyses. Methods of investigating heterogeneity were reported in 103 meta-analyses. These pertained to Cochran’s Q test alone (n=37), I2 alone (n=27), or both Q and I2 (n=37), while one meta-analysis reported Q, I2 and τ2 and another one had used the Breslow-Day test. Cochran’s Q statistic follows a χ2 distribution and tests whether the observed differences in results are compatible with chance. I2 measures the percentage of variability in effect estimates that is attributed to heterogeneity rather than chance (28). Heterogeneity in GWA studies can be attributed to differences between the included studies such as different populations, different linkage disequilibrium patterns, different environmental exposures, different genotyping platforms and different imputation accuracies, or it may represent unexplained statistical heterogeneity (27, 40, 80, 103).
Replication efforts had been performed for the results of 104 meta-analyses, showing that the practice is widely accepted in the field (9, 38). The rationale beyond SNP selection for replication testing is described in detail in Supplementary Table 3. It included mostly statistical significance criteria and to a lesser extent biological and/or functional plausibility.
As shown in Supplementary Table 4, the results of only 27 (19%) meta-analyses used populations of mixed ancestry in the discovery stage or followed up the meta-analyses findings in populations of different ancestries than the discovery population. Loci and even more effect sizes may be difficult to generalize across different ancestries (75). For a total of 79 meta-analyses, investigators had performed further association analyses between SNPs discovered in the discovery GWA meta-analysis and traits or diseases other than those examined in the meta-analyses. Additionally, for 19 (14%) meta-analyses investigators had extended their association testing to diverse phenotypes. These diverse phenotypes resulted from using cut-offs in order to transform a continuous trait to binary disease outcome (ankle-brachial index and peripheral artery disease; systolic blood pressure and hypertension; diastolic blood pressure and hypertension; BMI and obese/overweight; urinary albumin excretion and microalbuminuria; common and internal carotid intima media thickness and plaque); from using different definitions of the indexed trait (waist circumference and waist to hip ratio to measure adiposity; forced expiratory volume in 1 second (FEV1) and FEV1/FVC (forced vital capacity) to measure pulminary function; time to event and time to death to measure aging); from using other products of the same metabolic pathway (proinsulin and 32,33-split proinsulin/insulinogenic index/C-peptide; urinary albumin excretion and urinary albumin to creatinine ratio); and finally from using sub-phenotypes included and measured the indexed traits (different types of white blood cell count; different types of stroke; components of the metabolic syndrome; types of personality dimensions; Alzheimer’s disease and age of onset of the same disease).
Table 2 describes the 45 GWA reports where at least one type of functional work accompanying the respective meta-analyses had been performed in the same paper where the GWA meta-analysis was published. Expression quantitative trait loci (eQTL) analyses were the most common type of functional testing (mentioned in 26 reports). Other expression analyses included gene expression (n=19 reports) and RNA expression (n=2). Animal models (50) had been used in 9 reports and pathway analyses in another 13. Three reports had investigated protein-protein interactions for the products of discovered gene loci. Examination of the relationships between genes in different diseases using GRAIL (Gene Relationships Across Implicated Loci) analysis (82) was included in 8 reports, while the role of coding variation in the respective loci was listed in another 8 reports. Finally, other reported functional tests included analyses using the Online Mendelian Inheritance in Man (OMIM) database (n=3 reports); gene-interactions and gene ontology (n=2 reports for each); gene-environment interactions, DNA methylation, mutation analysis, and transcript profiling (n=1 report for each). 28 reports (62%) reported more than 1 functional analysis; 15 (33%) reported more than 2; and 6 reported more than 3. The extent of selective reporting of functional analyses in these papers (e.g. whether only specific methods/analyses with most interesting results are reported) is unknown.
Table 2.
Functional work accompanying meta-analyses
| GWA report |
Gene expression |
eQTL | RNA expression |
Animal models |
Pathway analyses |
Protein- protein interactions |
GRAIL | Coding variation |
Other |
|---|---|---|---|---|---|---|---|---|---|
| (S19) | X | X | X | X | OMIM | ||||
| (S77) | X | X | |||||||
| (S12) | X | ||||||||
| (S55) | X | X | |||||||
| (S52) | X | ||||||||
| (S40) | X | ||||||||
| (S85) | X | ||||||||
| (S75) | X | X | |||||||
| (S53) | X | ||||||||
| (S54) | X | X | X | ||||||
| (S25) (S46) | X | X | X | X | X | Transcript profiling |
|||
| (S10) | X | X | X | ||||||
| (S49) | X | ||||||||
| (S70) | X | X | X | GxE interactions | |||||
| (S64) | X | Gene ontology | |||||||
| (S31) | X | X | X | ||||||
| (S1) | X | ||||||||
| (S39) | X | ||||||||
| (S76) | X | X | |||||||
| (S83) | X | X | GxG interactions | ||||||
| (S21) | GxG interactions | ||||||||
| (S38) | X | X | |||||||
| (S35) | X | ||||||||
| (S88) | X | X | Gene ontology | ||||||
| (S65) | X | X | |||||||
| (S28) | X | ||||||||
| (S66) | X | ||||||||
| (S2) | X | X | X | ||||||
| (S32) | X | X | DNA methylation | ||||||
| (S18) | X | X | |||||||
| (S22) | X | X | X | ||||||
| (S72) | X | X | X | ||||||
| (S20) | X | X | |||||||
| (S27) | X | X | X | X | X | ||||
| (S73) | X | X | |||||||
| (S41) | X | X | X | X | OMIM | ||||
| (S50) | X | ||||||||
| (S78) | X | X | |||||||
| (S81) | X | X | X | X | |||||
| (S74) | X | ||||||||
| (S37) | X | Mutations | |||||||
| (S16) | X | X | |||||||
| (S44) | X | ||||||||
| (S24) | X |
eQTL, expression quantitative trait loci; GxG, gene-gene; GxE, gene-environment; GRAIL software, software for Gene Relationships Across Implicated Loci (http://www.broadinstitute.org/mpg/grail/): OMIM, Online Mendelian Inheritance in Man (www.ncbi.nlm.nih.gov/omim)
Finally, Table 3 lists the contribution of the discovered loci to the total genetic variance of the respective traits across the 62 meta-analyses with suitable data available for estimating the variance. Ankylosing spondylitis was the trait with the largest portion of genetic variance explained, as the 7 loci reaching genome-wide significance explained 79% of the variance. This is largely attributed to the effect of rs4349859 in HLA-B with OR=56 and P<10−200. This locus is a well-established risk factor and diagnostic test for the disease. Several other phenotypes had over 20% of their variance explained: Crohn’s disease (71%), rheumatoid arthritis (52% in Europeans and 33% in Asians), multiple sclerosis (49%), ulcerative colitis (39%), type 1 diabetes (36%), and Parkinson’s disease (20%). There was a weak correlation (r=0.28, P=0.03) between the number of discovered/validated SNPs and the proportion of variance explained, e.g. 115 validated SNPs explained less than 7% of the variance of height. Many phenotypes had very low proportions of variance explained. The lowest explained variance was found for ankle-brachial index (only 1 SNP was genome-wide significant and explained <0.1% of the total genetic variance). For most phenotypes there is apparently a substantial number of additional but yet undiscovered loci, which could increase the portion of the genetic variance explained (78), as we discuss in Part 2.
Table 3.
Genetic variance explained by the total number of independent loci discovered in the eligible GWA meta-analyses
| Disease/Trait | SNPs Discovered | % Genetic Variance Explained |
|---|---|---|
| Stroke (S29) | 2 | 4.97 |
| Atrial fibrillation (S4) | 2 | 4.23 |
| Major mood disorders (S44) | 1 | 0.94 |
| Alanine aminotransferase (ALT) (S33) | 2 | 0.60 |
| Aspartate aminotransferase (AST) (S33) | 2 | 0.84 |
| Blood urea nitrogen (S33) | 3 | 0.89 |
| Gamma glutamyl transpeptidase (y-GT) (S33) | 2 | 1.96 |
| Hemoglobin (S33) | 2 | 0.61 |
| Hematocrit (S33) | 2 | 0.56 |
| Mean corpuscular hemoglobin (S33) | 15 | 6.98 |
| Mean corpuscular hemoglobin concentration (S33) | 7 | 2.07 |
| Mean corpuscular volume (S33) | 15 | 6.56 |
| Platelets count (S33) | 6 | 2.19 |
| Red blood cells count (S33) | 8 | 3.47 |
| Total protein (S33) | 2 | 0.69 |
| White blood cells count (S33) | 5 | 1.28 |
| Serum creatinine (S33) | 1 | 0.22 |
| Height a (S51) | 8 | 1.78 |
| Intracranial aneurysm (S87) | 6 | 12.20 |
| Birth weight (S23) | 2 | 0.51 |
| Heart failure (S68) | 1 | 1.12 |
| Rheumatoid arthritis b (S74) | 6 | 51.65 |
| Vitamin D insufficiency (S84) | 3 | 12.86 |
| Phosphorus levels (S36) | 7 | 0.39 |
| Type 2 diabetes b (S81) | 10 | 13.09 |
| Calcium levels (S50) | 1 | 0.09 |
| Asthma (S47) | 6 | 5.71 |
| Height b (S41) | 115 | 6.56 |
| Gout (S86) | 2 | 14.34 |
| Waist-hip ratio (S27) | 12 | 0.60 |
| Crohn’s disease (S22) | 52 | 71.21 |
| Renal cell carcinoma (S58) | 6 | 5.38 |
| Parkinson’s disease (S32) | 11 | 20.17 |
| Ulcerative colitis (S2) | 33 | 39.17 |
| Coronary heart disease (S66) | 13 | 9.01 |
| Alzheimer’s disease (S28) | 6 | 5.78 |
| Glioma (S62) | 6 | 12.19 |
| Adiposity (% body fat) (S38) | 2 | 0.27 |
| Sudden cardiac arrest (S3) | 2 | 6.757 |
| Ankylosing spondylitis (S21) | 7 | 78.63 |
| Proinsulin levels (S76) | 7 | 0.67 |
| Type 2 diabetes a (S39) | 1 | 2.09 |
| Coffee consumption (S1) | 1 | 0.005 |
| Plaque (S5) | 3 | 2.48 |
| Bipolar disorder (S64) | 4 | 3.70 |
| Pulmonary function (S70) | 8 | 0.69 |
| Type 1 diabetes (S8) | 19 | 36.42 |
| Height c (S49) | 2 | 0.30 |
| Allergic rhinitis (S60) | 1 | 1.22 |
| IgE sensitization to grass (S60) | 3 | 5.54 |
| Aortic stiffness (carotid-femoral pulse wave velocity) (S46) | 1 | 0.26 |
| Multiple sclerosis (S54) | 9 | 49.08 |
| Atopic dermatitis (S53) | 2 | 1.76 |
| Ankle-brachial index (S48) | 1 | 0.002 |
| Metabolic syndrome (S40) | 1 | 2.07 |
| Rheumatoid arthritis a (S52) | 7 | 32.90 |
| Chronic kidney disease (S55) | 2 | 2.74 |
| Adiponectin levels (S12) | 10 | 0.72 |
| Obesity (childhood) (S9) | 7 | 10.15 |
| Head circumference (infant) (S77) | 3 | 0.77 |
| Bone mineral density- Femoral neck (S19) | 22 | 3.89 |
| Bone mineral density- Lumbar spine (S19) | 26 | 5.62 |
GWA, genome-wide association; SNP, single-nucleotide polymorphism
It pertains to populations of Asian ancestry
It pertains to populations of European ancestry
It pertains to populations of African ancestry
PART 2: THE FUTURE OF GWA META-ANALYSIS
2.1. FEATURES OF GWA VARIANTS DISCOVERED AND TO BE DISCOVERED
With such a large number of discovered common variants in GWA studies and meta-analyses thereof, one can empirically characterize their features to understand what we have learned and what more we can expect to discover in the future. There has been substantial criticism of GWA studies, and the most commonly cited justification is the inability of discovered SNPs to account for all of the heritability of a disease or trait. On the other hand, proponents of GWA studies cite the numerous novel loci and novel biological mechanisms, which are arising from the study of these traits and diseases in humans. It’s the typical ‘glass half-full or half-empty’ argument. Whether the cost of the GWA experiments that resulted in these biological findings justified the effort is difficult to quantitate, but what is abundantly clear is that the number of novel loci identified from large-scale unbiased association tests of representative common markers across the genome could not have been identified using other approaches such as candidate gene approaches or linkage studies in Mendelian pedigrees. Meta-analysis of GWA datasets has been essential for accomplishing these discoveries.
Several features of the genome led to the ability to perform GWA studies; i) that most common genetic variation is shared amongst individuals, particularly within the same ethnic group, and also ii) that segments of chromosomes are often inherited intact, resulting in batches of correlated markers. However, these features also bias this approach towards detecting association with common genetic markers (44). To examine this empirically, we examined the frequency distribution of SNPs that reach genome-wide association in studies of > 10,000 individuals, SNPs typically examined in GWAS studies including imputation and HapMap SNPs (Figure 1A) and there is a clear excess of common variants in the category of associated variants.
Figure 1.


SNPs associated via GWA studies in > 10,000 individuals were compared to the entire set of SNPs typically examined in a GWA study including imputation and to the HapMap Phase II SNPs. A) The proportion of SNPs in each minor allele frequency bin are shown in and B) the cumulative proportion of SNPs that show a gene within a certain distance (kb) is plotted.
It has been proposed that distant, rare variants may be responsible for association signals with common variants (17) and there are several examples of rare functional variants that were not assayed in the GWA studies because of an absence from the HapMap SNP dataset, such as the PCSK9 R46L variant (85) and three rare coding variants in NOD2 (31) that are not tagged by GWA arrays (6). However, the prevalent theory currently is that most loci that show association with common variants will ultimately identify a functional variant from amongst the most strongly associated SNPs (3) that impacts regulation of nearby genes, as recently described for the SORT1 functional variant at an LDL locus (71) identified by GWA studies (45, 98).
The hypothesis that most GWA discovered loci will have an underlying common variant that impacts regulation of gene expression was recently tested by Stamatoyannopoulos and colleagues (69). They first examined regions of the genome where the chromatin is unpacked and available for binding transcription factors, as marked by hypersensitivity to cleavage by DNase I enzymes. The next step was to look for an enrichment of disease and trait-associated loci from GWA studies in these DNase hypersensitive regions. The authors identified a 40% enrichment of these disease- and trait-associated SNPs from GWAS (P < 10−55) in these DNase hypersensitive regions indicating regulatory function. We also assessed, as a simple proxy for function, the distance to the nearest gene for three categories of variants; those that show association from GWA studies, those examined in GWA studies, and HapMap SNPs (Figure 1B). For the first category, 90% of variants are within 87 kb of a gene, however, for the third category, 90% of variants are within 367 kb of a gene.
The increased commonality of GWA discovered variants relative to those in the genome (Figure 1A) is likely a result of the chips used to assay the genome and properties of linkage disequilibrium. Which leaves us all wondering whether there are additional rare variants waiting to be identified if we could just examine them in large numbers of individuals. These variants have been much more difficult to test using GWA panels.
The ability of GWA meta-analyses to identify rare associated variants can be improved with imputation (61). The development of methods for imputing markers from fully genotyped reference panels into phenotyped samples with GWA markers has improved the genomic coverage, and therefore power of GWA meta-analyses. However, there are many more rare variants in the genome than common variants, and these variants typically have fewer highly correlated proxies. This makes rare variants much more difficult to impute from GWA marker panels. Methods for estimating the accuracy of imputed markers are also somewhat less informative for rare variants than for common. However, in spite of these limitations, preliminary imputation experiments have shown that rare variants can be imputed and may play at least some role in common diseases (65).
Another interim approach between GWA studies and sequencing is to assay rare (and common) variants in coding regions of the genome using inexpensive chip technology. A chip assaying most variation identified from exome sequencing >12,000 individuals is available from Illumina and soon from Affymetrix (http://genome.sph.umich.edu/wiki/Exome_Chip_Design). This will allow for testing of rare variants (except for private mutations and the ~15% of variants that cannot be designed for chip-based assays) that are the most likely to be functional by changing the amino acid make-up of a protein. There are still hurdles with experiments of this type. In the early stage of an experiment, one challenge is to call rare variants using clustering algorithms designed for common variants, and some progress towards rare-variant-specific approaches has been made (23). A challenge in the later stages of an experiment is to use statistical tests that aggregate the evidence for association across all functional variants in a biologically-equivalent unit (gene or pathway (4)). We expect to find a mix of functional and non-functional variants in most genes, but identifying which variants are likely to be functional has been challenging. Although prediction methods such as Polyphen2 (90) and SIFT (53) can predict that a variant might be functionally deleterious, these approaches are not completely accurate. Sensitivity and specificity may be low for subtle functional effects. A combined score might be more accurate than any single score (24, 64). Others have used frequency as a predictor of deleteriousness and either use thresholds so that only rare variants are considered (60) or use weights such that common variants are downweighted (56).
There have been some early success stories from sequencing of candidate genes for common diseases and traits, e.g. IFIH1 rare alleles provide some protection from type 1 diabetes (73) and sequencing candidate genes LDLR and PCSK9 (12) has identified rare variants associated with high and low LDL levels, respectively. Despite much debate and statistical modeling, it is currently unknown how substantial a role rare variants will play in common diseases. Large-scale sequencing studies in tens of thousands of individuals may be needed to resolve this debate and meta-analysis or mega-analysis of data from many investigators may be essential, as discussed below.
2.2. WHY DO LARGER META-ANALYSES?
It is important to consider whether further increases in sample size will continue to yield new, worthwhile insights. The new discoveries achieved by larger GWA studies take the form of new associated loci, and of new variants at known loci. Each of these has different potential benefits. New loci can aid in the identification of new causal genes or pathways, new variants can help define mechanism or causal genes at associated loci, and both new loci and new variants increase the phenotypic variance explained, which could aid in prediction.
In theory, GWA studies could at some point reach saturation for discovering new loci. It is becoming clear that many associated loci have multiple causal common variants, so at some point new variants could fall largely or exclusively in already-identified loci. This outcome is similar to a saturation mutagenesis scan in model organisms, where discovery of multiple alleles at each locus suggests that few new loci will be discovered by analyzing more mutants. If the main goal of GWA studies is to identify new loci and hence new biology (30) , then a diminishing return in new loci would strongly suggest that little would be gained by continuing. However, if the goal is to account for as yet unexplained heritability (67) then larger studies may yield more information, even if few new loci emerge. In either case, a key consideration is whether the new information – either new loci or increased variance explained – will be helpful in understanding the underlying biology and in treating or preventing disease. Often, these considerations will depend in an idiosyncratic fashion on the particular clinical features of the disease and on available treatments and preventive measures.
2.3. CAN ONE PREDICT THE OUTCOME OF LARGER META-ANALYSES?
The ability to discover a new association depends on the effect size and allele frequency for the associated variant and on the sample size encompassed by the meta-analysis. Because the effect sizes and frequencies for as yet undiscovered associations are by definition unknown, it is not easy to predict precisely what a given sample size will yield in new associations. Different groups have made different assumptions about the distributions of effect sizes (22, 100). Some of these attempts to fit mathematical distributions to the effect sizes based on results from early association studies produced inaccurate predictions of the effect sizes seen in subsequent, larger meta-analyses (54). However, consideration of empirical results across a number of different polygenic diseases and traits can provide some sense of the general pattern of outcomes as meta-analyses increase in size, even if the actual distribution of effect sizes does not fit a simple mathematical formula.
One approach to predicting the outcome of future studies rests on the observation that most GWA meta-analyses are actually quite underpowered to discover the loci that reached genome-wide significance in those studies. In other words, given the variance explained of these loci when estimated in independent samples, the power to reach genome-wide significance was low, and the investigators were “lucky” to find the association. Besides the discovered loci, many more loci with similar effect sizes remained undiscovered. For example, if there are 20 variants that explain equal small amounts of phenotypic variance, then a study with 10% power for any one variant will identify 2 new associations on average, and miss the remaining 18. From these considerations of power, it is possible to extrapolate from current results to future studies, at least for variants that have an effect size (variance explained) that is at least as large as some of the variants discovered in the earlier study (54, 78, 99). These methods have been reasonably accurate in predicting future discovery of loci with similar effect sizes to those identified in existing studies, although they cannot estimate the number of loci that have even smaller effect sizes than any of those already discovered.
It is also possible to use genome-wide association data to estimate the total amount of phenotypic variation that can be explained by common genetic variation (101). However, it is not possible to extrapolate from the total amount of heritability accounted for in toto to the variance explained by any individual undiscovered locus, and therefore difficult to use these estimates to predict the likely number of new loci or variants that would be identified by future GWA studies. Bayesian approaches have been used to infer the effect size (and frequency) distribution of undiscovered variants (89), although predictions from this method have not yet been compared to results from subsequent studies. Of course, if the total variance explained already approaches the estimated total heritability, further studies are unlikely to be useful. The variance explained in the usual additive models may also underestimate the total heritability accounted for, both because of multiple variants within each locus (20, 26, 54, 84, 102), and also in theory because of complex interactions that could lead to underestimation of the variance explained (104).
Although current theoretical and empirical estimates of future success are fraught with uncertainty, it is possible to use traits where large meta-analyses have already been performed to try to predict outcomes for GWA meta-analyses of traits and diseases where the sample size has not yet increased as rapidly. Such an analysis may be a useful guide for deciding whether to put new resources into increasing sample size. In two empirical attempts to catalogue the relationship between increasing sample size and number of loci discovered across multiple traits (48, 93), it appears that, once some loci have been discovered, the number of loci tends to increase more or less linearly with sample size.
We repeated this analysis for GWA studies where at least one locus had reached genome-wide significance for any of three quantitative traits: height (8, 11, 25, 43, 54, 57, 72, 84, 88, 92, 94, 95), lipid levels (triglycerides, HDL-cholesterol and LDL-cholesterol; (5, 10, 16, 45, 46, 49, 51, 58, 91, 98)) and blood pressure (systolic and diastolic blood pressure (32, 47, 59, 74)). For multistage studies (where top results from an initial GWAS are taken forward into additional samples), we extracted the sample sizes at each stage and the statistical threshold for moving SNPs forward into the next stage. We then calculated the sample size for which a one-stage design would have equivalent power as the actual study, and plotted this “effective” sample size against the number of loci that reached genome-wide significance (P<5×10−8 (70, 79)). As had been seen previously, the number of loci reaching genome-wide significance increases with sample size (Figure 2), and, where there is a broad range of sample sizes, the increase appears to be approximately linear. Furthermore, if we omit the largest study and use the remaining studies to estimate the linear relationship between effective sample size and number of genome-wide significant loci, we can predict the number of loci that should reach genome-wide significance in the largest study. As seen in Figure 2, the observed number of loci in the largest study matches (for lipids) or exceeds (for height and blood pressure) the predicted number of loci.
Figure 2.

The effective sample size (see text for details) is plotted against the number of loci reaching genome wide significance for genome-wide association studies for height (top), the combination of three lipid traits (middle; HDL-cholesterol, LDL-cholesterol, and triglycerides), and the combination of two blood pressure traits (bottom; systolic and diastolic blood pressure). In each case, the largest study is removed, and a line through the origin is fitted to the remaining studies (circles). The number of loci in the largest study (filled triangle) is greater than or equal to that predicted by extrapolating the line to the effective sample size of the largest study (open triangle).
Based on these observations, it is reasonable to assume that, once GWA studies of a disease or trait have begun to yield associated loci, then larger sample sizes will yield additional associated loci, and that the number of loci will scale at least linearly with sample size. Of course, at some point increasing sample size will yield diminishing returns. This point might be recognized by a fall-off of associated loci from a linear pattern (this presumably would happen because new variants are being largely found in already-discovered loci). Alternatively, if new loci consistently contain genes in already recognized pathways, then the gain in biological insight may be limited even if large numbers of new loci remain to be discovered. Moreover, the effect sizes (variance explained) of new discovered loci will tend to be on average smaller than those of early discovered loci.
2.4. META-ANALYSES VERSUS MEGA-ANALYSES
Currently, most consortia in human genome epidemiology perform meta-analysis: individual cohorts are analyzed separately, and then summary statistics are combined (typically with a fixed-effects model, such as that implemented in METAL (97)). The alternative, in which individual-level genotypes are combined into a single dataset before analysis, is sometimes called “mega-analysis.” In theory, mega-analysis may avoid whatever information loss (and hence a small power loss) is inherent in sharing summary statistics rather than pooling individual level data. Although the power loss from this loss of information is minimal for studies of common variants (62), it may be more sizeable for rare/uncommon variants. There are also other potential advantages of mega-analysis. For example, in meta-analyses, any analyses, whether simple associations or more complex ones (such as interactions or conditional analyses) need to be coordinated across each of the analysts of the participating teams. In addition, meta-analysis would not be able to detect cryptic relatedness between individuals from different studies, while this is feasible in mega-analysis. Mega-analysis may be particularly valuable for studies of rare variants, where pooling individuals may both increase the number of minor allele carriers in a given analysis (which lends robustness to statistics) and where more complex analytical methods (4) will require more complex methods of meta-analysis.
However, there are also several practical advantages to meta-analysis. For example, in meta-analysis the association testing of a particular study is more likely to be done by an analyst who is quite familiar with the genotype and phenotype data from that study. It may be computationally more feasible to analyze studies separately rather than as single cohorts of >100,000 samples, and the distribution of effort across multiple investigative groups may also ease the computational burden. Finally, it is more appropriate to analyze studies separately if there is heterogeneity in their ascertainment, ancestry, phenotyping, etc. Such heterogeneity is almost always present when the constituent studies have been designed independently in the past. Thus, meta-analysis would still be preferred even if all the individual data became centrally available.
2.5. META-ANALYSIS AND CONSORTIUM CULTURE
The participation of multiple investigative groups requires careful attention to the creation of a community of collaborating investigators within a consortial setting. Because the consortial approach (37, 86) is a new way of doing science for most biologists, investigators sometimes need to adapt to a new scientific culture characterized by data sharing and openness. Many well-functioning consortia share several guiding principles, including “no surprises” (declaring to other consortium members the intent to initiate projects, or submit abstracts, publications, grants, etc.) and “don’t use other people’s data to gain a competitive advantage over them.” In addition, a general sense of trust, common purpose and willingness to share credit is essential, and care must be taken to manage or avoid participation in separate but overlapping efforts. Typically, consortia work best when investigators recognize that the advantages of increased power to make discoveries and access to a large shared data set outweighs the perceived cost of sharing data and credit. Academic institutions need to recognize properly the contributions made by investigators who may not be (co-)first or (co-)last authors. It is important that tenure committees, dissertation committees, and other groups that evaluate academic achievement be willing and able to recognize that many authors make key, invaluable contributions to consortium-style papers.
It is important to stress that the presence of a consortium need not preclude projects performed by smaller groups or based on ideas from individual investigators. Indeed, such “side projects” are often enabled by providing many groups access to a large, shared data set, so that new ideas can be tested in a more powerful way. If multiple investigators have similar interests, then open communication of ideas and plans can permit collaboration or at least coordination of efforts that take advantage of the large, shared resource. Finally, broad access to summary results and individual level data maximizes the utility of both meta-analytic efforts and of the underlying data, permitting use of the data and results not only by members of the consortium but also by the broader scientific community.
2.6. CONCLUDING COMMENTS
In summary, almost all of what we presently know about the genetics of human polygenic disorders and quantitative traits has derived from meta-analysis of genome-wide association data. Continued meta-analyses on larger samples are likely to continue to yield useful information. Meta-analysis is also likely to remain a key approach as studies extend into rarer variation, although some methodological changes may be necessary. Consortia or other forms of collaboration are likely to continue to be vital to generate, assemble and analyze large genetic and genomic data sets. The academic community should understand the nature and value of individual contributions to these shared efforts and provide the appropriate academic recognition for collaborative work. Fostering collaboration will continue to enable the shared efforts like meta-analysis of genome-wide association studies that have helped propel human genetics forward over the last several years.
Supplementary Material
Mini-Glossary.
Cochran’s Q: a χ2 statistic for detecting between-study heterogeneity by testing whether the observed differences in results are compatible with chance
common variants: polymorphisms with frequency of >5% in a given population
exome sequencing: the process of sequencing the exons (i.e. coding sequence) of all genes across the genome
fixed-effects model: a method of combining individual studies under the assumption of common genetic effect; variation is attributed to chance
heterogeneity: the variation in the genetic effects observed among the combined studies
HapMap Project: an extensive catalogue of the common variation in the human genome
HWE (Hardy-Weinberg equilibrium): a theoretical description according to which the genotype frequencies are equal to the product of the allele frequencies
I2: a measure of quantifying between-study heterogeneity; it describes the percentage of variability in effect estimates attributed to heterogeneity rather than chance
imputation: a method used to infer the genotypes of SNPs that have not been directly genotyped
linkage disequilibrium: a measure of the extent that two SNPs are found together in a population more often than expected by chance.
MAF (minor allele frequency): the frequency of the less common of two alleles in a given population
mega-analysis: an approach where individual data at the genome-wide level are pooled before the analysis
meta-analysis: a statistical method that allows the combination of multiple different datasets estimating a common summary effect
power: the probability of rejecting the null hypothesis of no association when the null hypothesis is false
random-effects model: a method of combining individual studies under the assumption of different genetic effects; variation is attributed to genuine differences
rare variants: polymorphisms with frequency of 5% or less in a given population
variance explained: the proportion of the variability in a complex trait that can be explained by genetic factors
Summary Points.
Meta-analysis of GWA datasets has become increasingly common for the detection of subtle genetic effects on complex traits with most of them being performed within international consortia achieving very large sample sizes.
Among 139 GWA meta-analyses published to-date, each GWA meta-analysis has discovered on average 4 SNPs (range 0-115) with many more SNPs being followed-up in replication and functional studies.
For most traits, the GWA-discovered SNPs to-date explain <20% of the genetic variance and there is a weak correlation between the number of SNPs and the proportion of variance explained.
Larger GWA meta-analyses could increase our knowledge by identifying new loci thus increasing the proportion of variance explained and potentially providing also new insights into the biology of human disease.
The number of loci reaching genome-wide significance seems to increase at least linearly with increasing sample size.
Meta-analysis methods can be extended for application in sequencing studies and those focusing on rare variants.
Meta-analysis is preferable to mega-analyses of GWA data (pooling individual data before analysis), when the combined studies are potentially heterogeneous, but the latter might also have some uses under select circumstances.
Future Issues.
Meta-analyses of GWA data are expected to become even more common in the future as individual teams worldwide create more consortia and share data.
Given the wide-spread role of consortia, mega-analyses of individual GWA data could increase power for detecting subtle effects, especially for rare variants, and could also detect cryptic relatedness, overcoming some of the limitations of GWA meta-analyses.
Detection of rare variation will be greatly enhanced by advancements in imputation of these variants and by sequencing thousands of individuals within the consortial setting.
The potential for clinical translation of this information will largely depend in an idiosyncratic fashion on the particular clinical features of the disease and on available treatments and preventive measures.
Continued meta-analyses on larger samples are likely to continue to yield useful information, being a key approach besides imputation and sequencing for extending into rarer variation, although some methodological changes may be necessary.
Contributions from individual teams to shared efforts and recognition for collaborative work within the consortial setting are vital for maximizing the potential of future discoveries.
LITERATURE CITED
- 1.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–8. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson CA, Soranzo N, Zeggini E, Barrett JC. Synthetic associations are unlikely to account for many common disease genome-wide association signals. PLoS Biol. 2011;9:e1000580. doi: 10.1371/journal.pbio.1000580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asimit J, Zeggini E. Rare variant association analysis methods for complex traits. Annu Rev Genet. 2010;44:293–308. doi: 10.1146/annurev-genet-102209-163421. [DOI] [PubMed] [Google Scholar]
- 5.Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009;41:47–55. doi: 10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: A review of statistical methods and recommendations for their application. Am J Hum Genet. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carty CL, Johnson NA, Hutter CM, Reiner AP, Peters U, et al. Genome-wide association study of body height in African Americans: the Women’s Health Initiative SNP Health Association Resource (SHARe) Hum Mol Genet. 2012;21:711–20. doi: 10.1093/hmg/ddr489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, et al. Replicating genotype-phenotype associations. Nature. 2007;447:655–60. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 10.Chasman DI, Pare G, Mora S, Hopewell JC, Peloso G, et al. Forty-three loci associated with plasma lipoprotein size, concentration, and cholesterol content in genome-wide analysis. PLoS Genet. 2009;5:e1000730. doi: 10.1371/journal.pgen.1000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet. 2009;41:527–34. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 12.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–5. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 13.Colhoun HM, McKeigue PM, Davey Smith G. Problems of reporting genetic associations with complex outcomes. Lancet. 2003;361:865–72. doi: 10.1016/s0140-6736(03)12715-8. [DOI] [PubMed] [Google Scholar]
- 14.de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17:R122–8. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7:177–88. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 16.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT. Lund University, Novartis Institutes of BioMedical Research. Saxena R, Voight BF, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–6. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 17.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elbaz A, Nelson LM, Payami H, Ioannidis JP, Fiske BK, et al. Lack of replication of thirteen single-nucleotide polymorphisms implicated in Parkinson’s disease: a large-scale international study. Lancet Neurol. 2006;5:917–23. doi: 10.1016/S1474-4422(06)70579-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evangelou E, Maraganore DM, Ioannidis JP. Meta-analysis in genome-wide association datasets: strategies and application in Parkinson disease. PLoS One. 2007;2:e196. doi: 10.1371/journal.pone.0000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42:1049–51. doi: 10.1038/ng.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gogele M, Minelli C, Thakkinstian A, Yurkiewich A, Pattaro C, et al. Methods for meta-analyses of genome-wide association studies: critical assessment of empirical evidence. Am J Epidemiol. 2012;175:739–49. doi: 10.1093/aje/kwr385. [DOI] [PubMed] [Google Scholar]
- 22.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–8. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein JI, Crenshaw A, Carey J, Grant GB, Maguire J, et al. zCall: a rare variant caller for array-based genotyping: Genetics and population analysis. Bioinformatics. 2012;28:2543–45. doi: 10.1093/bioinformatics/bts479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–9. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gudbjartsson DF, Walters GB, Thorleifsson G, Stefansson H, Halldorsson BV, et al. Many sequence variants affecting diversity of adult human height. Nat Genet. 2008;40:609–15. doi: 10.1038/ng.122. [DOI] [PubMed] [Google Scholar]
- 26.Haiman CA, Patterson N, Freedman ML, Myers SR, Pike MC, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet. 2007;39:638–44. doi: 10.1038/ng2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet. 2011;88:586–98. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–60. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirschhorn JN. Genomewide association studies--illuminating biologic pathways. N Engl J Med. 2009;360:1699–701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 31.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 32.International Consortium for Blood Pressure Genome-Wide Association Studies. Ehret GB, Munroe PB, Rice KM, Bochud M, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–9. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.International HapMap Consortium. Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–8. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.International HapMap Consortium. Frazer KA, Ballinger DG, Cox DR, Hinds DA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ioannidis JP. Non-replication and inconsistency in the genome-wide association setting. Hum Hered. 2007;64:203–13. doi: 10.1159/000103512. [DOI] [PubMed] [Google Scholar]
- 37.Ioannidis JP, Bernstein J, Boffetta P, Danesh J, Dolan S, et al. A network of investigator networks in human genome epidemiology. Am J Epidemiol. 2005;162:302–4. doi: 10.1093/aje/kwi201. [DOI] [PubMed] [Google Scholar]
- 38.Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29:306–9. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- 39.Ioannidis JP, Panagiotou OA. Comparison of effect sizes associated with biomarkers reported in highly cited individual articles and in subsequent meta-analyses. JAMA. 2011;305:2200–10. doi: 10.1001/jama.2011.713. [DOI] [PubMed] [Google Scholar]
- 40.Ioannidis JP, Patsopoulos NA, Evangelou E. Heterogeneity in meta-analyses of genome-wide association investigations. PLoS One. 2007;2:e841. doi: 10.1371/journal.pone.0000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ioannidis JP, Tarone R, McLaughlin JK. The false-positive to false-negative ratio in epidemiologic studies. Epidemiology. 2011;22:450–6. doi: 10.1097/EDE.0b013e31821b506e. [DOI] [PubMed] [Google Scholar]
- 42.Ioannidis JP, Thomas G, Daly MJ. Validating, augmenting and refining genome-wide association signals. Nat Rev Genet. 2009;10:318–29. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansson A, Marroni F, Hayward C, Franklin CS, Kirichenko AV, et al. Common variants in the JAZF1 gene associated with height identified by linkage and genome-wide association analysis. Hum Mol Genet. 2009;18:373–80. doi: 10.1093/hmg/ddn350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jorgenson E, Witte JS. A gene-centric approach to genome-wide association studies. Nat Rev Genet. 2006;7:885–91. doi: 10.1038/nrg1962. [DOI] [PubMed] [Google Scholar]
- 45.Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40:189–97. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet. 2011;43:531–8. doi: 10.1038/ng.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim Y, Zerwas S, Trace SE, Sullivan PF. Schizophrenia genetics: where next? Schizophr Bull. 2011;37:456–63. doi: 10.1093/schbul/sbr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim YJ, Go MJ, Hu C, Hong CB, Kim YK, et al. Large-scale genome-wide association studies in East Asians identify new genetic loci influencing metabolic traits. Nat Genet. 2011;43:990–5. doi: 10.1038/ng.939. [DOI] [PubMed] [Google Scholar]
- 50.Kitsios GD, Tangri N, Castaldi PJ, Ioannidis JP. Laboratory mouse models for the human genome-wide associations. PLoS One. 2010;5:e13782. doi: 10.1371/journal.pone.0013782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kooner JS, Chambers JC, Aguilar-Salinas CA, Hinds DA, Hyde CL, et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat Genet. 2008;40:149–51. doi: 10.1038/ng.2007.61. [DOI] [PubMed] [Google Scholar]
- 52.Kraft P, Zeggini E, Ioannidis JP. Replication in genome-wide association studies. Stat Sci. 2009;24:561–73. doi: 10.1214/09-STS290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 54.Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–8. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lau J, Ioannidis JP, Schmid CH. Quantitative synthesis in systematic reviews. Ann Intern Med. 1997;127:820–6. doi: 10.7326/0003-4819-127-9-199711010-00008. [DOI] [PubMed] [Google Scholar]
- 56.Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13:762–75. doi: 10.1093/biostatistics/kxs014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet. 2008;40:584–91. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lettre G, Palmer CD, Young T, Ejebe KG, Allayee H, et al. Genome-wide association study of coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. PLoS Genet. 2011;7:e1001300. doi: 10.1371/journal.pgen.1001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–87. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–21. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin DY, Zeng D. On the relative efficiency of using summary statistics versus individual-level data in meta-analysis. Biometrika. 2010;97:321–32. doi: 10.1093/biomet/asq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33:177–82. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- 64.Lopes MC, Joyce C, Ritchie GR, John SL, Cunningham F, et al. A combined functional annotation score for non-synonymous variants. Hum Hered. 2012;73:47–51. doi: 10.1159/000334984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Magi R, Asimit JL, Day-Williams AG, Zeggini E, Morris AP. Genome-Wide Association Analysis of Imputed Rare Variants: Application to Seven Common Complex Diseases. Genet Epidemiol. 2012 doi: 10.1002/gepi.21675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, et al. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet. 2005;77:685–93. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 71.Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–9. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.N’Diaye A, Chen GK, Palmer CD, Ge B, Tayo B, et al. Identification, replication, and fine-mapping of Loci associated with adult height in individuals of african ancestry. PLoS Genet. 2011;7:e1002298. doi: 10.1371/journal.pgen.1002298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–76. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ntzani EE, Liberopoulos G, Manolio TA, Ioannidis JP. Consistency of genome-wide associations across major ancestral groups. Hum Genet. 2012;131:1057–71. doi: 10.1007/s00439-011-1124-4. [DOI] [PubMed] [Google Scholar]
- 76.Panagiotou OA, Evangelou E, Ioannidis JP. Genome-wide significant associations for variants with minor allele frequency of 5% or less--an overview: A HuGE review. Am J Epidemiol. 2010;172:869–89. doi: 10.1093/aje/kwq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Panagiotou OA, Ioannidis JP, for the Genome-Wide Significance Project What should the genome-wide significance threshold be? Empirical replication of borderline genetic associations. Int J Epidemiol. 2012;41:273–86. doi: 10.1093/ije/dyr178. [DOI] [PubMed] [Google Scholar]
- 78.Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–5. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–5. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 80.Pereira TV, Patsopoulos NA, Salanti G, Ioannidis JP. Discovery properties of genome-wide association signals from cumulatively combined data sets. Am J Epidemiol. 2009;170:1197–206. doi: 10.1093/aje/kwp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ragoussis J. Genotyping technologies for genetic research. Annu Rev Genomics Hum Genet. 2009;10:117–33. doi: 10.1146/annurev-genom-082908-150116. [DOI] [PubMed] [Google Scholar]
- 82.Raychaudhuri S, Plenge RM, Rossin EJ, Ng AC, International Schizophrenia C et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–10. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 84.Sanna S, Jackson AU, Nagaraja R, Willer CJ, Chen WM, et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat Genet. 2008;40:198–203. doi: 10.1038/ng.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sanna S, Li B, Mulas A, Sidore C, Kang HM, et al. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet. 2011;7:e1002198. doi: 10.1371/journal.pgen.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seminara D, Khoury MJ, O’Brien TR, Manolio T, Gwinn ML, et al. The emergence of networks in human genome epidemiology: challenges and opportunities. Epidemiology. 2007;18:1–8. doi: 10.1097/01.ede.0000249540.17855.b7. [DOI] [PubMed] [Google Scholar]
- 87.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Optimal designs for two-stage genome-wide association studies. Genet Epidemiol. 2007;31:776–88. doi: 10.1002/gepi.20240. [DOI] [PubMed] [Google Scholar]
- 88.Soranzo N, Rivadeneira F, Chinappen-Horsley U, Malkina I, Richards JB, et al. Meta-analysis of genome-wide scans for human adult stature identifies novel Loci and associations with measures of skeletal frame size. PLoS Genet. 2009;5:e1000445. doi: 10.1371/journal.pgen.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stahl EA, Wegmann D, Trynka G, Gutierrez-Achury J, Do R, et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet. 2012;44:483–9. doi: 10.1038/ng.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–7. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 91.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tonjes A, Koriath M, Schleinitz D, Dietrich K, Bottcher Y, et al. Genetic variation in GPR133 is associated with height: genome wide association study in the self-contained population of Sorbs. Hum Mol Genet. 2009;18:4662–8. doi: 10.1093/hmg/ddp423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. 2008;40:575–83. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weedon MN, Lettre G, Freathy RM, Lindgren CM, Voight BF, et al. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat Genet. 2007;39:1245–50. doi: 10.1038/ng2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Willer CJ, Bonnycastle LL, Conneely KN, Duren WL, Jackson AU, et al. Screening of 134 single nucleotide polymorphisms (SNPs) previously associated with type 2 diabetes replicates association with 12 SNPs in nine genes. Diabetes. 2007;56:256–64. doi: 10.2337/db06-0461. [DOI] [PubMed] [Google Scholar]
- 97.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–9. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17:1520–8. doi: 10.1101/gr.6665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang J, Ferreira T, Morris AP, Medland SE, Genetic Investigation of ATC et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44:369–75. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zaitlen N, Eskin E. Imputation aware meta-analysis of genome-wide association studies. Genet Epidemiol. 2010;34:537–42. doi: 10.1002/gepi.20507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109:1193–8. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Annotations to References
- Asimit J, Zeggini E. Rare variant association analysis methods for complex traits. Annu Rev Genet. 2010;44:293–308. doi: 10.1146/annurev-genet-102209-163421.
 A comprehensive review on the statistical methods for the analysis of rare variants
A comprehensive review on the statistical methods for the analysis of rare variants - McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–69. doi: 10.1038/nrg2344.
 A review on the value of GWA studies for detecting genetic associations and fine mapping.
A review on the value of GWA studies for detecting genetic associations and fine mapping. - DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7:177–88. doi: 10.1016/0197-2456(86)90046-2.
 The paper introducing the most commonly used random-effects model for meta-analysis
The paper introducing the most commonly used random-effects model for meta-analysis - Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294.
 A paper supporting the contested hypothesis that signals from common variants may arise from synthetic associations of rare variants.
A paper supporting the contested hypothesis that signals from common variants may arise from synthetic associations of rare variants. - Evangelou E, Maraganore DM, Ioannidis JP. Meta-analysis in genome-wide association datasets: strategies and application in Parkinson disease. PLoS One. 2007;2:e196. doi: 10.1371/journal.pone.0000196.
 The first study to combine different GWA datasets with different meta-analysis methods (fixed and random effects) showing the merits and disadvantages of each.
The first study to combine different GWA datasets with different meta-analysis methods (fixed and random effects) showing the merits and disadvantages of each. - Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–60. doi: 10.1136/bmj.327.7414.557.
 A comprehensive analysis of popular heterogeneity metrics in meta-analysis.
A comprehensive analysis of popular heterogeneity metrics in meta-analysis. - Ioannidis JP, Patsopoulos NA, Evangelou E. Heterogeneity in meta-analyses of genome-wide association investigations. PLoS One. 2007;2:e841. doi: 10.1371/journal.pone.0000841.
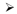 A useful discussion on the importance of considering between-study heterogeneity in GWA meta-analysis.
A useful discussion on the importance of considering between-study heterogeneity in GWA meta-analysis. - Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–5. doi: 10.1038/ng.610.
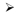 Methods for predicitng the number of susceptibility loci and their effect-sizes distribution for a complex trait
Methods for predicitng the number of susceptibility loci and their effect-sizes distribution for a complex trait - Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–5. doi: 10.1002/gepi.20303.
 Theoretical evaluation of the optimal genome-wide significance threshold in difference ancestral groups.
Theoretical evaluation of the optimal genome-wide significance threshold in difference ancestral groups. - Pereira TV, Patsopoulos NA, Salanti G, Ioannidis JP. Discovery properties of genome-wide association signals from cumulatively combined data sets. Am J Epidemiol. 2009;170:1197–206. doi: 10.1093/aje/kwp262.
 Simulation study proving the power of different synthesis models for discovery and generalizabilty purposes in GWA meta-analysis.
Simulation study proving the power of different synthesis models for discovery and generalizabilty purposes in GWA meta-analysis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.