

Abstract
Genomes of the plant-pathogenic genus Phytophthora are characterized by small duplicated blocks consisting of two consecutive genes (2HOM blocks) and by an elevated abundance of similarly aged gene duplicates. Both properties, in particular the presence of 2HOM blocks, have been attributed to a whole-genome duplication (WGD) at the last common ancestor of Phytophthora. However, large intraspecies synteny—compelling evidence for a WGD—has not been detected. Here, we revisited the WGD hypothesis by deducing the age of 2HOM blocks. Two independent timing methods reveal that the majority of 2HOM blocks arose after divergence of the Phytophthora lineages. In addition, a large proportion of the 2HOM block copies colocalize on the same scaffold. Therefore, the presence of 2HOM blocks does not support a WGD at the last common ancestor of Phytophthora. Thus, genome evolution of Phytophthora is likely driven by alternative mechanisms, such as bursts of transposon activity.
Keywords: oomycetes, segmental duplication, genome expansion, paleopolyploidy
Phytophthora: a Paleopolyploid Genus?
Whole-genome duplications (WGDs) are powerful facilitators of evolution, because they generate large amounts of raw genetic material. By duplicating the entire set of genes, WGDs pave the way for neo-, subfunctionalization, and reciprocal gene loss (Scannell et al. 2006; Sémon and Wolfe 2007). Therefore, WGDs have been associated with important evolutionary events such as extensive radiation and adaptation towards changing environments (Christoffels et al. 2004; Young et al. 2011). Ancient WGDs have been observed in a variety of eukaryotic lineages, including vertebrates, flowering plants, and the unicellular ciliates (Dehal and Boore 2005; Aury et al. 2006; Amborella Genome Project 2013). Such paleopolyploids are often identified by the presence of intraspecies synteny in addition to many duplicated genes that arose at the same time (Van de Peer 2004). Recently, it has been suggested that a WGD occurred before the speciation of several extant Phytophthora (Martens and Van de Peer 2010), a genus of important plant-infecting eukaryotic microbes (Jiang and Tyler 2012).
The genus Phytophthora encompasses more than a hundred species that infect numerous plant lineages causing devastating diseases, such as potato late blight (Phytophthora infestans) and sudden oak death (P. ramorum) (Govers and Gijzen 2006; Tyler et al. 2006; Haas et al. 2009; Kroon et al. 2012). The extensive radiation within this genus, accompanied by adaptation of its members to novel environments and distinct hosts specificities, makes a WGD in the last common ancestor of Phytophthora a compelling scenario. Moreover, polyploidy has been reported in some extant Phytophthora species, highlighting the potential influence of this process in the evolution of this genus (Sansome 1977; Bertier et al. 2013). Recently, small duplicated blocks consisting of two directly adjacent genes (termed 2HOM blocks) were found to be abundant in Phytophthora (Martens and Van de Peer 2010). This genomic feature has been attributed to an ancient WGD in Phytophthora succeeded by extensive genomic rearrangements. Moreover, a WGD provides a simple explanation for the increased gene duplication frequency at the Phytophthora ancestor (Martens and Van de Peer 2010; Seidl et al. 2012).
However, small-scale duplications cannot be excluded as a possible driving force for the appearance of 2HOM blocks, especially because no larger regions of intraspecies synteny could be detected (Haas et al. 2009; Martens and Van de Peer 2010). Moreover, in-depth re-examination of the 2HOM blocks revealed several features that challenge the WGD hypothesis. For example, many 2HOM blocks have their copies located on the same scaffold or contain transposable elements (TEs) (supplementary table S1, Supplementary Material online), thus most likely reflecting segmental duplications and transposition, respectively.
Because 2HOM blocks nevertheless represent intraspecies synteny in the Phytophthora genomes, widely considered the most important indicator of WGDs, we uncovered their evolutionary origin. We applied two independent methods to estimate the duplication age of 2HOM blocks. First, we determined the timing of 2HOM block duplication by a combination of presence/absence patterns and phylogenetic analysis. Second, we used synonymous substitutions (Ks) as a proxy for duplication time. We established that the majority of 2HOM blocks emerged very recently, following the radiation of Phytophthora. Consequently, the presence of 2HOM blocks is not a remnant of a WGD in the last common ancestor of Phytophthora.
The Origin of 2HOM Blocks Revisited
To infer homology within and across genomes of six divergent Phytophthora lineages and distantly related oomycetes (fig. 1) (Tyler et al. 2006; Haas et al. 2009; Baxter et al. 2010; Lévesque et al. 2010; Lamour et al. 2012; Jiang et al. 2013), we clustered all predicted proteins based on sequence similarity (see supplementary materials and methods, Supplementary Material online). Using the resulting gene families, we identified 2HOM blocks in the Phytophthora species and subsequently screened the other oomycetes for copies of these 2HOM blocks. TEs are common in Phytophthora genomes, and their predicted gene content contains many TE remnants (Haas et al. 2009; Seidl et al. 2011). To exclude blocks in which one or both constituent(s) could have resulted from transposition rather than duplication, we filtered TE-containing 2HOM blocks (supplementary Materials and Methods, Supplementary Material online, and table 1).
Fig. 1.—
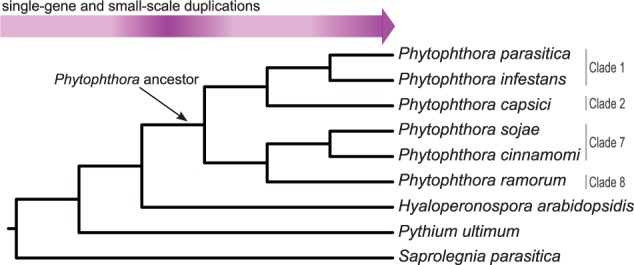
Species tree, as previously established by Blair et al. (2008), with gene (and small-scale) duplication frequency indicated. The gene duplication frequency is elevated at the last common ancestor of Phytophthora and in some extant Phytophthora lineages.
Table 1.
Numbers of 2HOM Blocks in the Examined Genomes
| Species | 2HOM Blocks | 2HOM Blocks Filtered Same Scaffold | Categorization of 2HOM Block Duplications |
|
|---|---|---|---|---|
| Private Duplications | Shared Duplications | |||
| P. capsici | 474 | 98 36 (P<0.0001) | 78 (77%) | 23 |
| P. cinnamomi | 609 | 183 0 | 148 (80%) | 37 |
| P. infestans | 396 | 244 70 (P<0.0001) | 227 (93%) | 16 |
| P. parasitica | 255 | 104 49 (P<0.0001) | 61 (74%) | 21 |
| P. ramorum | 252 | 145 43 (P<0.0001) | 112 (84%) | 22 |
| P. sojae | 282 | 131 62 (P<0.0001) | 99 (80%) | 25 |
Note.—The 2HOM blocks were filtered for large families and for TEs. The number of filtered 2HOM blocks with copies on the same scaffold was assessed for significance by genome randomization. Of each filtered 2HOM block, the timing of underlying duplication(s) was inferred either from phylogenetic trees or from its single-species occurrence and categorized accordingly.
We observed between 98 (P. capsici) and 244 (P. infestans) 2HOM blocks in the Phytophthora genomes (table 1). The numbers of 2HOM blocks in the genomes of P. infestans and P. ramorum are comparable to those that have been found before (Martens and Van de Peer 2010). However, we identified fewer blocks in P. sojae (131 vs. 207). Variations between both analyses are likely caused by our more stringent TE filtering and by differences in determining gene families (see supplementary table S1, Supplementary Material online, for TE content in the previously published 2HOM blocks and supplementary table S2, Supplementary Material online, for a comparison of both 2HOM block sets).
Surprisingly, a high fraction of all 2HOM blocks identified (29%) has at least two copies located on the same scaffold (table 1). For example, both copies of the P. infestans 2HOM block containing a vesicle transporter gene and a riboflavin biosynthesis gene are situated on supercontig 15 (fig. 2A). Such a high fraction of colocalization—up to 47% in P. parasitica and P. sojae—is not expected if blocks are derived from a WGD; those are more likely to be localized on different chromosomes or scaffolds. Although genomic rearrangements could lead to colocalization of some WGD-derived homologous blocks, reshuffling seems unlikely to account for all instances, especially considering the high number of scaffolds in Phytophthora genome assemblies (between 708 and 4,921). Moreover, the number of colocalizing 2HOM blocks in each species (excluding P. cinnamomi) is significantly higher than expected by random localization (P value < 0.0001; supplementary Materials and Methods, Supplementary Material online). Therefore, the colocalized 2HOM blocks are more likely the products of segmental duplication events than of an ancient WGD.
Fig. 2.—

Two examples of phylogenetically analyzed 2HOM blocks. For both 2HOM blocks, phylogenetic trees of the genes and an illustration of the positions on the scaffolds are given. Bootstrap supports based on 100 replicates are displayed on the branches. (A) A Phytophthora infestans 2HOM block, likely the result of a species-specific, possibly segmental duplication, is displayed. (B) A 2HOM block of P. infestans (species-specific duplication), P. ramorum, P. sojae, and P. capsici (derived from a shared, possibly segmental duplication) has a more complex duplication history.
To precisely time the origin of 2HOM blocks in different Phytophthora genomes, we categorized each duplication event that creates a 2HOM block as “private” (species- specific) or “shared” (ancestral). A 2HOM duplication is private if it occurred more recently than any speciation, and it is shared if it precedes a speciation between at least two different Phytophthora species. To categorize the duplications, we inferred the timing of duplications using two complementary methods. The first method is based on presence: If a 2HOM block only occurs in a single Phytophthora species, that is, not a single copy of this block was detected in any of the other genomes, we parsimoniously inferred the number of duplications which created the copies and categorized these as “private.” The second method consists of phylogenetic timing: If a 2HOM block has copies in multiple species, we inferred separate phylogenetic trees for the two genes that form this particular block and interpreted these according to the species tree (fig. 1, see also supplementary Materials and Methods, Supplementary Material online).
Two examples demonstrate the phylogenetic timing of 2HOM duplications (fig. 2). Figure 2A depicts a P. infestans 2HOM block that has copies in five additional species. The duplication that gave rise to this 2HOM block is species-specific (private), and thus this 2HOM block cannot be the result of any ancestral duplication, neither segmental nor whole-genome. In contrast, the genes encoding short chain dehydrogenases and 12-oxophytodienoate reductases constitute a 2HOM block with a more complex evolutionary history (fig. 2B). In P. ramorum, P. sojae, and P. capsici, the 2HOM block is present due to a shared duplication. In P. infestans, this 2HOM block is formed by a private duplication. Even though this block has, at least for three Phytophthora lineages, a shared ancestry, both duplicate copies are located on the same scaffold in their genomes and therefore is likely not a WGD remnant (see discussion above). Evaluating both approaches (single-species occurrence and phylogenetic tree interpretation), the majority (across all species 725 out of 869, at least 74% in individual species) of duplications that gave rise to 2HOM blocks was inferred to be private (table 1).
Interestingly, the relatively young age of 2HOM block duplications revealed by phylogenetics and presence is not in accordance with the age of 2HOM blocks observed by Martens and Van de Peer (2010). Their Ks-based timing revealed a large fraction of 2HOM blocks originating at approximately the same time before speciation, indicated by a peak in the distribution at higher Ks. If duplication rate is relatively constant, one expects to see an exponential decay in the Ks distribution, with a single peak at low Ks constituted by recent duplications that have not been subject to degradation yet. A second peak at higher Ks values, as demonstrated by Martens and Van de Peer (2010), suggests that on top of a relatively constant duplication rate, in the past a high amount of duplications occurred in a very short time interval. Thereby, this peak is potentially indicative of a WGD, as it might be the result of a single duplication event (Lynch and Conery 2000; Blanc and Wolfe 2004). We similarly assessed the age of our 2HOM blocks by calculating Ks of the paralogous gene pairs constituting these blocks. The Ks-inferred age of duplications confirms the duplication categorization, because most paralogs constituting 2HOM blocks have lower Ks values than orthologs, indicating duplications that occurred more recently than speciation (fig. 3A). To further delineate the differences between the two outcomes, we analyzed the original data of Martens and Van de Peer (2010). In-depth reanalyses of these data revealed significant inconsistencies in 2HOM block paralog definition and thus Ks inference. Once correcting for these inconsistencies, their primary data yield Ks distributions pointing to a more recent origin of the 2HOM blocks, similar to the results of this study (supplementary fig. S2, Supplementary Material online). Thus, we conclude that most 2HOM blocks are of recent, species-specific origin. We cannot exclude the possibility that some of the very young 2HOM blocks are in fact alleles and not duplicates (e.g., fig. 2A). However, due to the absence of a substantial number of ancient 2HOM blocks, the 2HOM blocks do not support a WGD in the last common ancestor of Phytophthora.
Fig. 3.—
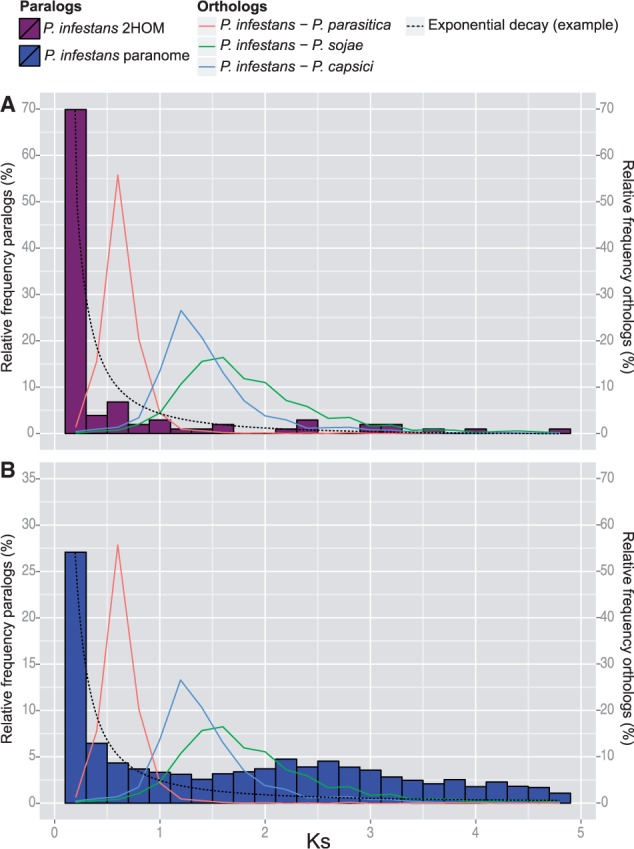
Ks distributions of Phytophthora infestans. (A) 2HOM block paralogs and (B) full paranome. Peaks in the distributions of orthologous sets of gene pairs indicate speciation events. Dashed lines indicate examples of distributions following a pattern of exponential decay. Full paranome distributions of the other Phytophthora spp. are available in supplementary figure S1, Supplementary Material online.
High Frequency of Gene Duplications in Phytophthora Ancestor
In addition to the 2HOM blocks, a second WGD indicator is the observed high frequency of gene duplications at the last common ancestor of Phytophthora (Martens and Van de Peer 2010; Seidl et al. 2012). We investigated whether this proposed increase in gene duplications persists when including additional Phytophthora genomes and applying a more stringent TE filtering.
We calculated Ks for the Phytophthora paranomes, that is, the set of all paralogous gene pairs within a genome (fig. 3B and supplementary fig. S1, Supplementary Material online). We observed a small second peak that deviates from the exponential decay in most Phytophthora genomes, therefore indicating a slightly increased duplication frequency before the radiation of Phytophthora. Martens and Van de Peer (2010) initially showed a more pronounced second peak in the paranome Ks distributions. The observed inconsistency is largely due to differences in the calculation of equilibrium codon frequencies, which one has to estimate to calculate Ks (see supplementary figs. S3–S6, Supplementary Material online, for a comparison of Ks distributions of P. infestans, P. ramorum, and P. sojae under different models of codon frequencies). The method applied by Martens and Van de Peer (2010) assumes that all codons are equally likely to be part of coding sequence. It therefore ignores codon bias, which is a prominent phenomenon in Phytophthora (Jiang and Govers 2006; Tripathy and Tyler 2006). Instead, we applied a method that estimates the codon frequencies based on the average nucleotide frequencies at the three codon positions and thus accommodates codon bias (Yang and Nielsen 1998) yielding more reliable Ks distributions.
To further delineate the precise point of gene duplication and to trace gene duplication in spite of subsequent gene losses, we applied phylogenetic timing to all gene families, like we did for the 2HOM blocks. We constructed phylogenetic trees for the corresponding protein families and automatically reconciled them with the species tree similar to Seidl et al. (2012). This analysis revealed very high numbers of duplications in some of the extant Phytophthora species, and also a burst of gene duplications in the Phytophthora common ancestor (fig. 1 and supplementary fig. S7, Supplementary Material online). This relative abundance of gene duplications persists even if we account for possible wrongly inferred tree topologies by excluding ancestral duplications of which none of the extant species retained both copies (Vilella et al. 2009) (supplementary fig. S7, Supplementary Material online). Given this requirement of gene copy retention, the last common ancestor of P. infestans, P. parasitica, and P. capsici only experienced 281 gene duplications, whereas the Phytophthora ancestor and the today-living lineage P. infestans had 1,651 and 1,353 gene duplications, respectively.
At a first glance, the full paranome Ks distributions and results from the tree reconciliation seem contradictory. However, in contrast to tree reconciliation, Ks-based age estimation can only be accomplished for paralogous gene pairs of which both copies are still present. If for many duplicates one copy was lost over time, the elevated gene frequency of gene duplication would be disguised in the Ks distributions. In the tree reconciliation analysis, the steep decrease in inferred ancient duplications with increasing strictness in duplicate retention likely reflects such gene loss. It suggests that for a high proportion of ancient duplications, the different species lost either one or the other copy over evolutionary time (supplementary fig. S7, Supplementary Material online).
The WGD hypothesis ascribes the high number of gene duplications in the last common ancestor of Phytophthora to a single event. However, this hypothesis demands extensive subsequent reshuffling to explain the loss of syntenic regions longer than one gene. Furthermore, this reshuffling should have occurred in a short time interval before speciation of Phytophthora, because extant divergent Phytophthora species show extensive interspecies colinearity (Tyler et al. 2006; Haas et al. 2009; Lamour et al. 2012). Moreover, even between Pythium ultimum and P. infestans (fig. 1), a considerable level of gene-order conservation has been reported (Adhikari et al. 2013). Therefore, we conclude that a WGD at the last common ancestor with subsequent reshufflings is not very likely and so far not supported by the available data.
Outlook
Even though Phytophthora genomes contain an elevated number of 2HOM blocks, timing by duplication reconstruction and by synonymous substitutions reveals that these are not the result of a genus-shared WGD. Instead, many blocks have a lineage-specific origin and/or are located on the same scaffold. The exponential age distribution of 2HOM block paralogs suggests a constant rate of block duplications followed by decay that continues to date. Nevertheless, many gene duplications occurred in the genome of the last common ancestor of Phytophthora, of which only a small amount of paralogs are present in extant species. The mechanism responsible for this gene duplication burst could in fact be the same as for the occurrence of block duplications. A potential source of such small-scale duplications is the activity of TEs, where so-called transduplication takes non-TE coding sequences along (Jiang et al. 2004). Taking into account proposed roles for TEs in speciation and in generating genetic novelty (Rebollo et al. 2010; Wang et al. 2011) and the high repeat and TE content of many Phytophthora genomes (Tyler et al. 2006; Gijzen 2009; Haas et al. 2009), TEs might have played a significant role in the genome evolution of Phytophthora.
Materials and Methods
Complete information regarding all materials and methods can be found in the supplementary Materials and Methods, Supplementary Material online.
Genome data of nine oomycete species were downloaded. Across these genomes, protein families were defined using BlastP and MCL (Altschul et al. 1997; Enright et al. 2002). Families containing at least one gene with considerable sequence similarity to known TEs were flagged as TE families (Jurka et al. 2005). Each genome was screened for 2HOM blocks with post hoc filtering of blocks comprised of large protein families and/or TE families. Maximum-likelihood phylogenetic trees were inferred for genes comprising 2HOM blocks (Stamatakis 2006). Ks was calculated for the transcripts of all possible pairs in a protein family using codeml (Yang 2007). For each protein family, a neighbor-joining tree was estimated and automatically reconciled with the species tree using NOTUNG (Chen et al. 2000).
Supplementary Material
Supplementary tables S1 and S2, figures S1–S7, and materials and methods are available at Genome Biology and Evolution online (http://ww.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Cindy Martens and Yves van de Peer for kindly providing their raw data. They also thank Lidija Berke, Sjors van der Horst, Alessia Peviani, Eelco Tromer, and Leny van Wijk for their helpful comments on the manuscript. Some of the sequence data and annotation were produced by the U.S. Department of Energy Joint Genome Institute (http://www.jgi.doe.gov) and the Broad Institute of Harvard and the Massachusetts Institute of Technology (http://www.broadinstitute.org). This work was supported by the Centre for BioSystems Genomics (CBSG) which is part of the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research.
Literature Cited
- Adhikari BN, et al. Comparative genomics reveals insight into virulence strategies of plant pathogenic oomycetes. PLoS One. 2013;8:e75072. doi: 10.1371/journal.pone.0075072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amborella Genome Project. The amborella genome and the evolution of flowering plants. Science. 2013;342:1241089. doi: 10.1126/science.1241089. [DOI] [PubMed] [Google Scholar]
- Aury JM, et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444:171–178. doi: 10.1038/nature05230. [DOI] [PubMed] [Google Scholar]
- Baxter L, et al. Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science. 2010;330(6010):1549–1551. doi: 10.1126/science.1195203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertier L, Leus L, D’hondt L, de Cock AW, Höfte M. Host adaptation and speciation through hybridization and polyploidy in Phytophthora. PLoS One. 2013;8:e85385. doi: 10.1371/journal.pone.0085385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair JE, Coffey MD, Park S, Geiser DM, Kang S. A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences. Fungal Genet Biol. 2008;45:266–277. doi: 10.1016/j.fgb.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16:1667–1678. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Durand D, Farach-Colton M. NOTUNG: a program for dating gene duplications and optimizing gene family trees. J Comput Biol. 2000;7:429–447. doi: 10.1089/106652700750050871. [DOI] [PubMed] [Google Scholar]
- Christoffels A, et al. Fugu genome analysis provides evidence for a whole-genome duplication early during the evolution of ray-finned fishes. Mol Biol Evol. 2004;21:1146–1151. doi: 10.1093/molbev/msh114. [DOI] [PubMed] [Google Scholar]
- Dehal P, Boore JL. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005;3:e314. doi: 10.1371/journal.pbio.0030314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijzen M. Runaway repeats force expansion of the Phytophthora infestans genome. Genome Biol. 2009;10:241. doi: 10.1186/gb-2009-10-10-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govers F, Gijzen M. Phytophthora genomics: the plant destroyers’ genome decoded. Mol Plant Microbe Interact. 2006;19:1295–1301. doi: 10.1094/MPMI-19-1295. [DOI] [PubMed] [Google Scholar]
- Haas BJ, et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- Jiang N, Bao Z, Zhang X, Eddy SR, Wessler SR. Pack-MULE transposable elements mediate gene evolution in plants. Nature. 2004;431:569–573. doi: 10.1038/nature02953. [DOI] [PubMed] [Google Scholar]
- Jiang RH, Govers F. Nonneutral GC3 and retroelement codon mimicry in phytophthora. J Mol. Evol. 2006;63:458–472. doi: 10.1007/s00239-005-0211-3. [DOI] [PubMed] [Google Scholar]
- Jiang RH, Tyler BM. Mechanisms and evolution of virulence in oomycetes. Annu Rev Phytopathol. 2012;50:295–318. doi: 10.1146/annurev-phyto-081211-172912. [DOI] [PubMed] [Google Scholar]
- Jiang RHY, et al. Distinctive expansion of potential virulence genes in the genome of the oomycete fish pathogen Saprolegnia parasitica. PLoS Genet. 2013;9(6):e1003272. doi: 10.1371/journal.pgen.1003272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kroon LPNM, Brouwer H, De Cock AWAM, Govers F. The genus Phytophthora anno 2012. Phytopathology. 2012;102:348–364. doi: 10.1094/PHYTO-01-11-0025. [DOI] [PubMed] [Google Scholar]
- Lamour KH, et al. Genome sequencing and mapping reveal loss of heterozygosity as a mechanism for rapid adaptation in the vegetable pathogen Phytophthora capsici. Mol Plant Microbe Interact. 2012;25:1350–1360. doi: 10.1094/MPMI-02-12-0028-R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque CA, et al. Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 2010;11(7):R73. doi: 10.1186/gb-2010-11-7-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Martens C, Van de Peer Y. The hidden duplication past of the plant pathogen Phytophthora and its consequences for infection. BMC Genomics. 2010;11:353. doi: 10.1186/1471-2164-11-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo R, Horard B, Hubert B, Vieira C. Jumping genes and epigenetics: towards new species. Gene. 2010;454:1–7. doi: 10.1016/j.gene.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Sansome E. Polyploidy and induced gametangial formation in British isolates of Phytophthora infestans. J Gen Microbiol. 1977;99:311–316. [Google Scholar]
- Scannell DR, Byrne KP, Gordon JL, Wong S, Wolfe KH. Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature. 2006;440:341–345. doi: 10.1038/nature04562. [DOI] [PubMed] [Google Scholar]
- Seidl MF, Van den Ackerveken G, Govers F, Snel B. A domain-centric analysis of oomycete plant pathogen genomes reveals unique protein organization. Plant Physiol. 2011;155:628–644. doi: 10.1104/pp.110.167841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl MF, Van den Ackerveken G, Govers F, Snel B. Reconstruction of oomycete genome evolution identifies differences in evolutionary trajectories leading to present-day large gene families. Genome Biol Evol. 2012;4:199–211. doi: 10.1093/gbe/evs003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sémon M, Wolfe KH. Consequences of genome duplication. Curr Opin Genet Dev. 2007;17:505–512. doi: 10.1016/j.gde.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Tripathy S, Tyler BM. The repertoire of transfer RNA genes is tuned to codon usage bias in the genomes of phytophthora sojae and phytophthora ramorum. Mol Plant-Microbe Interact. 2006;19:1322–1328. doi: 10.1094/MPMI-19-1322. [DOI] [PubMed] [Google Scholar]
- Tyler BM, et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006;313:1261–1266. doi: 10.1126/science.1128796. [DOI] [PubMed] [Google Scholar]
- Van De Peer Y. Computational approaches to unveiling ancient genome duplications. Nat Rev Genet. 2004;5:752–763. doi: 10.1038/nrg1449. [DOI] [PubMed] [Google Scholar]
- Vilella AJ, et al. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. Modes of gene duplication contribute differently to genetic novelty and redundancy, but show parallels across divergent angiosperms. PLoS One. 2011;6:e28150. doi: 10.1371/journal.pone.0028150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol. 1998;46:409–418. doi: 10.1007/pl00006320. [DOI] [PubMed] [Google Scholar]
- Young ND, et al. The medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011;480:520–524. doi: 10.1038/nature10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.