

Abstract
In diploid organisms, selfing reduces the efficiency of selection in removing deleterious mutations from a population. This need not be the case for all organisms. Some plants, for example, undergo an extreme form of selfing known as intragametophytic selfing, which immediately exposes all recessive deleterious mutations in a parental genome to selective purging. Here, we ask how effectively deleterious mutations are removed from such plants. Specifically, we study the extent to which deleterious mutations accumulate in a predominantly selfing and a predominantly outcrossing pair of moss species, using genome-wide transcriptome data. We find that the selfing species purge significantly more nonsynonymous mutations, as well as a greater proportion of radical amino acid changes which alter physicochemical properties of amino acids. Moreover, their purging of deleterious mutation is especially strong in conserved regions of protein-coding genes. Our observations show that selfing need not impede but can even accelerate the removal of deleterious mutations, and do so on a genome-wide scale.
Keywords: high throughput sequencing, haploid, diploid, haploid-dominant life cycle, intragametophytic selfing, outcrossing, deleterious mutations
Introduction
Heritable genetic variation, the substrate of natural selection (Maynard Smith and Szathmáry 1995), is ultimately caused by mutations. Because few mutations are beneficial and most are deleterious when they first arise, populations of organisms carry a genetic load of deleterious mutations in their genomes. Whether natural selection can remove such deleterious mutations from a population depends on various factors, such as how strongly deleterious the mutations are, how large the population is, whether an organism is diploid or haploid, and whether it reproduces sexually or asexually (Glémin 2007; Otto and Gerstein 2008; Charlesworth and Willis 2009; Glémin and Galtier 2012). Among sexually reproducing organisms, the mating system—whether organisms reproduce primarily through selfing or outcrossing—is a key factor that determines how effectively deleterious mutations can be removed.
On the one hand, selfing reduces a population’s effective size Ne, and thus selection’s ability to remove weakly deleterious mutations (Charlesworth and Wright 2001; Wright et al. 2008; Wright et al. 2013). In particular, in organisms with a diploid-dominant life phase, selfing increases genome-wide homozygosity, which in turn reduces the effective number of independently sampled gametes in a population, an effect that can cut the effective population size in half (Nordborg 2000). In addition, high levels of homozygosity cause low effective recombination rates (Charlesworth and Wright 2001) that further decrease Ne and promote the accumulation of deleterious mutations (Felsenstein 1974). Finally, the very population structure of selfers, which is characterized by small subpopulations in which founder events can be frequent, can also reduce the effective population size (Charlesworth and Wright 2001).
On the other hand, selfing can facilitate the purging of recessive deleterious mutations (Glémin 2007; Glémin and Galtier 2012). Such mutations can be completely masked in diploids if they are paired with a wild-type allele in a heterozygote. Selfing increases the incidence of homozygotes where two recessive deleterious alleles are paired (Charlesworth and Willis 2009). It can thus expose deleterious mutations and facilitate their purging.
Both theoretical and experimental evidence suggest that the Ne-reducing effects of selfing are usually stronger than the enhanced exposure of deleterious mutations caused by selfing (Bustamante et al. 2002; Slotte et al. 2010, 2013; Qiu et al. 2011; Ness et al. 2012; Hazzouri et al. 2013). In other words, selfers with a diploid-dominant life cycle should overall remove deleterious mutations less well than outcrossers (Charlesworth and Wright 2001; Glémin 2007).
However, this may not be the case in plants with a haploid-dominant life cycle or with a free-living haploid phase for two reasons. First, theory suggests that effective population size reduction caused by selfing should be less pronounced in haploid-dominant than in diploid-dominant plants because their life cycle poorly fit the assumptions underlying Wright–Fisher populations (Stenøien and Såstad 2001; Bengtsson and Cronberg 2009). More specifically, in bryophytes, the automatic 2-fold reduction of Ne caused by selfing does not operate. Ne is thus primarily reduced by a decrease in effective recombination rates (Hedrick 1987a, 1987b).
In addition to that, plants with an independent haploid phase or with a haploid-dominant life cycle can undergo “intragametophytic selfing,” which can be more effective in purging deleterious mutations than regular selfing in diploid-dominant plants. Intragametophytic selfing occurs between genetically identical male and female gametes produced by the same gametophyte (bisexual gametophyte) (fig. 1; Shaw 2000). It leads to a complete loss of heterozygosity in merely one generation (Shaw 2000; Barner et al. 2011; Billiard et al. 2012). In the resulting diploids, all deleterious mutations are exposed to selection. In this way, intragametophytic selfing can facilitate the purging of deleterious mutations (Eppley et al. 2007). This extreme form of selfing is not possible in plants with unisexual gametophytes, where female and male gametes are always produced on genetically separate haploid individuals. Such plants must undergo outcrossing or intergametophytic selfing—a less extreme form of selfing among siblings produced by the same diploid (an analogous situation to selfing in a hermaphroditic flowering plant, see fig. 1), which reduces heterozygosity only by a factor one-half per generation (Charlesworth and Wright 2001).
Fig. 1.—
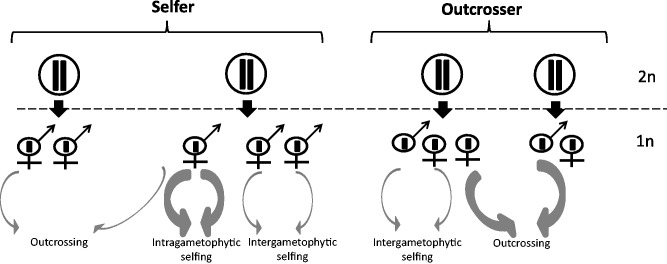
Schematic representation of the mating system of selfer and outcrosser organisms with a haploid-dominant life cycle. Solid black arrows refer to meiotic events and the dashed horizontal line separates the haploid and diploid phases. Thickness of the gray arrows indicates the relative frequency of selfing and outcrossing events in selfers and outcrossers.
Taken together, the available body of theory (Hedrick 1987a, 1987b; Holsinger 1987) suggests that haploid-dominant plants with frequent intragametophytic selfing might purge their genetic load more effectively than outcrossers. This prediction is supported by some indirect experimental evidence, for example, less severe inbreeding depression in plants capable of intragametophytic selfing (Klekowski 1973, 1982; Lloyd 1974; Taylor et al. 2007; Eppley et al. 2007).
However, the proportion of the genome subject to purging via homozygous exposure is not known. Yet, this information is crucial to understand the extent at which intragametophytic selfing can affect the accumulation of deleterious mutations on a genome-wide scale. If we assume that only diploid-specific genes are subject to purging, then the genome-wide effect of intragametophytic selfing on the accumulation of deleterious mutations should be minor (Szövényi et al. 2011). This is because only very few genes are diploid-specific (2–3%; Szövényi et al. 2011, 2013) and because genes also expressed in the haploid phase may not be affected by purging due to effective haploid selection (Klekowski 1973, 1982; Lloyd 1974; McCauley et al. 1985; Taylor et al. 2007; Eppley et al. 2007; Billiard et al. 2012). Alternatively, if genes expressed in both phases can also be affected by purging via homozygous exposure, intragametophytic selfing is expected to have a genome-wide effect because the majority of the genes is shared by the two phases (97%; Szövényi et al. 2011, 2013). Therefore, no experimental evidence specifically addresses the question: Do intragametophytic selfers accumulate a lower number of deleterious mutations on a genome-wide scale?
To answer this question, we here use mosses as model systems for plants with a haploid-dominant life cycle capable of intragametophytic selfing. We compare the genome-wide accumulation of deleterious mutations in protein-coding genes between two pairs of moss species that either predominantly self or outcross. We find that selfing mosses accumulate fewer deleterious mutations, and especially so in conserved positions of protein-coding genes. Thus, intragametophytic selfing in haploid-dominant plants can indeed facilitate effective purging of deleterious mutations.
Materials and Methods
Study Species
We use transcriptomes of two pairs of moss species to compare the number of deleterious mutations accumulated between species that predominantly self or outcross. The first pair comprises Physcomitrella patens and its relative Funaria hygrometrica. Both P. patens and F. hygrometrica have haploid-dominant life cycles, and their haploid individuals with combined sexes primarily undergo intragametophytic selfing (Shaw 1991; Eppley et al. 2007; Perroud et al. 2011). Because they undergo more selfing, and a more extreme form of selfing than the other species pairs, we refer to them as the “selfers.” Both species are genetically and functionally haploid and neither of them has an allo- or autopolyploid origin (Rensing et al. 2007; Shaw et al. 2010; Liu et al. 2012).
Our pair of outcrossing organisms (hereinafter referred to as “outcrossers”) includes two peat moss species Sphagnum subsecundum and Sphagnum cribrosum. Their haploid life stages can be either females or males, that is, individual plants can bear either male or female gametangia, but never both. They can outcross or undergo intergametophytic selfing, that is, fertilization between products of a single meiosis, comparable to selfing in diploid plants (Shaw 2000). Although they may therefore experience some amount of selfing (Eppley et al. 2007; Szövényi et al. 2009), we refer to them as outcrossers to distinguish them from the more extreme selfers P. patens and F. hygrometrica. Importantly, the divergence times between the selfer and outcrosser species pairs are similar (McDaniel et al. 2010; Shaw et al. 2010; Liu et al. 2012; Szövényi et al. 2013).
Available evidence suggests that the selfer and outcrosser species pairs have comparable range size and population structure. Both the selfer and the outcrosser pairs consist of a broadly and a more narrowly distributed species. The selfer F. hygrometrica is world-wide distributed, whereas P. patens occurs primarily in Europe. Similarly, the outcrosser S. subsecundum is widely distributed across the Northern Hemisphere whereas S. cribrosum only occurs in the United States (Anderson et al. 2009). Both the selfer and the outcrosser species pairs have well-interconnected populations (via long-distance spore dispersal) which is evidenced by weak among-population genetic differentiation both at the local and global scales (Shaw 1991; Shaw et al. 2008; Szövényi et al. 2008). Within-population genetic diversity is considerably higher in the outcrossers indicating greater effective population size of local populations (Shaw 1991; Shaw et al. 2008; Szövényi et al. 2008). Finally, both species pairs are effective colonizers with small, temporary populations (Shaw 1991; Shaw et al. 2008; Szövényi et al. 2008; Anderson et al. 2009). Therefore, range size and population structures of the selfer and the outcrosser species pairs are comparable, rendering mating system differences the primary candidate factor to affect mutation purging.
We focus our analysis on among-species divergence data because molecular traces of selfing are predicted to be more pronounced for among-species divergence data than for within-population polymorphism data when mating systems have been stable for a long time (Glémin 2007). Phylogenetic evidence suggests that the breeding systems of our selfer and outcrosser species pairs have indeed been stable, so the mating system had enough time to affect the accumulation of mutations (Shaw et al. 2010; Liu et al. 2012).
Sequence Data and Read Filtering
As a proxy of the incidence of mutation purging, we use the accumulation of likely deleterious mutations between selfer and outcrosser species pairs. To estimate the incidence of such mutations, we used high-throughput sequencing to generate two genome-wide transcriptome data sets for this study (S. subsecundum and S. cribrosum) and employed another such data set from our previous work (F. hygromerica) (Szövényi et al. 2013). Briefly, for F. hygrometrica we had collected samples of three haploid and three diploid developmental stages, extracted RNA, and performed sequencing on an Illumina GAIIx machine that generated 173,542,944 raw reads. We then discarded all reads containing low quality base calls (Q < 20 and one or more undefined nucleotides [N], FASTQ Quality Trimmer, http://hannonlab.cshl.edu/fastx_toolkit/ , last accessed May 20, 2014) and/or showing adaptor contamination (Tagdust, false discovery rate threshold of 0.01; Lassmann et al. 2009). We used 133,851,302 quality-filtered reads in the final assembly. Detailed description of data collection, read filtering and assembly are described in Szövényi et al. (2013). Sequence data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress, last accessed May 16, 2014) under accession number E-MTAB-1664.
For S. subsecundum and S. cribrosum, we used the Spectrum Plant Total RNA kit (Sigma) to extract total RNA from 100 mg of gametophytic tissue (capitulum) obtained from 6-month-old cultivated plants originally collected in Swanville, Waldo County, Maine, USA. We note that at the time of collection no sporophytic tissues were available and thus we extracted RNA from purely gametophytic material. Because only few genes are expected to show sporophyte-specific expression, differential tissue sampling in the selfer and outcrosser species pairs is unlikely to significantly bias our analyses (Szövényi et al. 2011, 2013). We subjected Poly-A selected RNA to paired-end sequencing (insert size: 300 bp) on one full lane and half a lane of an Illumina Hiseq2000 sequencer for S. subsecundum and S. cribrosum, respectively. The sequencing runs provided 163,776,364 and 94,818,996 raw paired end reads for S. subsecundum and S. cribrosum, respectively. Prior to assembly, we filtered out duplicate reads using an in-house Python script, trimmed the 3′-end of each read in a pair based on quality using the FASTX-Toolkit (FASTQ Quality Trimmer, http://hannonlab.cshl.edu/fastx_toolkit/, last accessed May 16, 2014) with a quality score threshold of 25, clipped off Illumina sequencing adapters, when present, with the FASTX-Toolkit (FASTQ/A Clipper, http://hannonlab.cshl.edu/fastx_toolkit/, last accessed May 16, 2014), and finally kept only paired-end reads for which both sequences were longer than 40 bp after trimming and clipping. After these steps, 117,836,947 and 81,917,656 cleaned paired end reads were left for S. subsecundum and S. cribrosum, respectively. Raw sequence data obtained are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress, last accessed May 16, 2014) under accession number E-MTAB-2482.
For the fourth species, P. patens, we retrieved high quality genomic data (Rensing et al. 2008) for transcript sequences and their corresponding protein translations from a public database (http://www.phytozome.net/version8.0, last accessed October 8, 2013).
Transcriptome Assembly
We assembled the S. subsecundum, S. cribrosum, and F. hygrometrica RNA-seq data into virtual transcripts using the assembler Trinity with default options (Grabherr et al. 2011), which has high sensitivity and leads to the most contiguous transcriptome assembly, for example, the highest number of full-length transcripts for nonmodel species. In S. subsecundum and S. cribrosum, we assembled a total of 117,836,947 and 81,917,656 cleaned paired end reads into 541,929 and 508,499 contigs, respectively (371,788 and 261,128 “components” according to Grabherr et al. 2011). In F. hygrometrica, we obtained 133,851,302 cleaned single end reads and assembled them into 192,665 transcripts (102,856 “components” according to Grabherr et al. 2011).
Protein Translation, Orthology, and Pairwise Alignment of Orthologs
In order to predict coding frame and to generate protein translations, we compared all contigs of the assembly against the National Center for Biotechnology Information (NCBI) nr database plant division (ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr.xx.tar.gz , last accessed May 10, 2013) with the aid of BLASTX (Altschul et al. 1997). We filtered the Blast (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.2.25+-ia32-linux.tar.gz, last accessed May 10, 2013; Altschul et al. 1997) output and kept only transcripts with BLAST hits showing at least 30% identity over at least 150 amino acids. Using these filtering criteria, 21,101 (S. subsecundum), 16,991 (S. cribrosum), and 34,119 (F. hygrometrica) transcripts had a valid match against the database. After that we provided each transcript and its best hit protein as input to the program GeneWise2 (http://www.ebi.ac.uk/∼birney/wise2/Wise2.4.0, last accessed March 8, 2013; Birney et al. 2004), and used the best scoring protein-to-cDNA alignment to predict the encoding gene’s ORF (Open Reading Frame) and generate a protein translation. We also discarded all transcripts with internal stop codons. The various filtering steps and the homology-guided translation ensured a high-confidence transcript set (we discarded transcripts without homology to the database) and helped exclude artifactual or contaminant sequences that are expected to occur in de novo transcriptome assemblies (Vijay et al. 2013).
We next identified orthologous gene pairs from the sets of proteins thus identified in each species, and did so separately for the outcrosser (S. subsecundum vs. S. cribrosum) and for the selfer species pair (F. hygrometrica vs. P. patens). In order to establish orthology, we first kept the transcript with the longest protein translation for each putative gene (referred to as a “component” in the software Trinity). We then used the reciprocal best hits approach (Bork et al. 1998) to establish orthologous protein pairs, keeping only pairs showing 30% identity over at least 150 amino acids in a BLASTP search (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.2.25+-ia32-linux.tar.gz, last accessed May 10, 2013; Altschul et al. 1997).
To generate DNA alignments of the coding sequence of orthologous proteins identified, we used Muscle (Edgar 2004) with default parameters. After that we used PAL2NAL (Suyama et al. 2006) to map protein alignments onto nucleotide alignments. We used the “remove gaps” option of PAL2NAL in order to generate codon alignments without gaps.
Estimating the Accumulation of Deleterious Mutations
To determine whether deleterious mutations accumulate at different rates in selfers compared with outcrossers, we used three complementary approaches.
First, we followed well-established precedence (Wright and Andolfatto 2008; Gossmann et al. 2010; Slotte et al. 2011; Yang and Gaut 2011) by assuming that the majority of nonsynonymous mutations is deleterious, and estimated their incidence relative to silent mutations. Specifically, we used our pairwise alignments to obtain estimates for the number of nonsynonymous substitutions per nonsynonymous sites (dN), as well as the number of synonymous substitutions per synonymous (dS) sites, and computed their ratio dN/dS using the KaKs_Calculator (Zhang et al. 2006) with the YN (Yang and Nielsen) model of sequence evolution (Yang and Nielsen 2000). We filtered the pairwise alignments by excluding entries with dS > 2, dS = 0, and dN/dS > 2. We used these thresholds because alignments with greater dS values are likely to become saturated with synonymous substitutions (Axelsson et al. 2008; Mank et al. 2010). Furthermore, visual inspection indicated that alignments with dS > 2 or dN/dS > 2 are frequently unreliable and thus cannot be trusted. Subsequently, we asked whether the dN/dS ratios differed between selfer and outcrosser species pairs. Furthermore, we also tested whether any such difference persists after excluding genes potentially under positive selection. For the latter analysis, we kept only gene pairs with dN/dS < 1, following the method of Subramanian (2013). With this approach we tried to distinguish relaxed from positive selection, because both can accelerate the accumulation of nonsynonymous mutations and thus lead to elevated dN/dS ratios.
Different amino acids differ in their physicochemical properties, such as size, charge, or aromaticity. Conservative amino acid changes, which alter an amino acid into one with similar physicochemical properties, are usually less detrimental to protein function than radical changes, which alter these properties (Zhang 2000; Eyre-Walker et al. 2002; Hughes and Friedman 2009). The ratio of the number of radical to conservative amino acid changes that a protein has experienced can thus be used as a proxy for the incidence of deleterious mutations (Zhang 2000; Eyre-Walker et al. 2002; Smith 2003). We used a complex classification of amino acids established by Miyata et al. (1979), which group amino acids into acidic, basic, neutral small, and neutral large categories. We treated all amino acid replacements that change an amino acid into one of a different category as radical, whereas we classified category-preserving changes as conservative. This classification has performed well in multiple studies and provides a reliable way to describe how strongly an amino acid change affects amino acid properties (Hanada et al. 2007; Wernegreen 2011). We estimated the proportion of radical (Dr) and conservative (Dc) amino acid changes using the hon-new program (Zhang 2000), taking into account the transition/transversion bias for each pair of genes, as estimated by PAML (pairwise option, model: F3×4, kappa). Before calculating these statistics, we again filtered alignments based on dN/dS ratios, as explained above (0 < dS < 2, dN/dS < 2). Furthermore, we discarded all alignments with no amino acid change (Dr = Dc = 0) or with no radical change (Dr = 0). We also repeated the analysis by excluding genes that may be subject to positive selection, that is, by retaining only alignments with dN/dS < 1 (Subramanian 2013). We determined significant differences in dN/dS and Dr/Dc ratios among outcrossers and selfers with a Mann–Whitney U test (Sokal and Rohlf 2012).
Finally, we also used a third approach to estimate the accumulation of deleterious mutations. It relies on the observation that the lower effective population size (Ne) potentially caused by selfing will allow slightly deleterious mutations accumulate more rapidly, and thus lead to an elevated dN/dS ratio between-species. Theory predicts that the effect of Ne is more pronounced on sites that are under more intense purifying selection, where the average selection coefficient of deleterious mutations is greater (Subramanian 2013). This prediction is a consequence of the theoretical relationship between ω (dN/dS), Ne, and the selection coefficient (s), ω = S/(1 − e−S) where S = 4Ne × s (Kimura 1983; Nielsen and Yang 2003; Subramanian 2013).
Because the rate at which mutations accumulate depends on the intensity of selection, the number of accumulated deleterious mutations is expected to be lower in conserved than in nonconserved parts of proteins (sconserved > snonconserved). Furthermore, the relationship among Ne, s, and ω predicts that a unit increase in purifying selection (s) will result in a steeper decrease in ω in the species pair with the larger effective population size (Ne). That is, given that Ne (selfer) considerably differs from Ne (outcrosser) the difference in the number of accumulated deleterious mutations between outcrossers and selfers should be more pronounced in conserved parts of proteins, which are under more intense purifying selection. Furthermore, differences in the dN/dS and Dr/Dc ratios between conserved and nonconserved regions within genes should also differ between outcrossers and selfers. The effect of purging via homozygous exposure should be also more pronounced in conserved than in nonconserved domains of proteins, because deleterious mutations with large effect can be more effectively purged (Wang et al. 1999). For example, if purging is more efficient in selfers, we expect to see not only lower dN/dS and Dr/Dc ratios in selfers but also the differences to outcrossers should be more pronounced in conserved protein regions. Furthermore, dN/dS and Dr/Dc ratios between conserved and nonconserved regions within genes should differ more markedly in selfers if they experience more effective purging.
To perform this analysis, we searched protein translations of assembled transcripts against the conserved domain database (Marchler-Bauer and Bryant 2004; Marchler-Bauer et al. 2013) to identify highly conserved regions of the genes in our data set. We used the default search parameters and kept only the best match for each protein. After that, we merged multiple conserved regions per protein into a contiguous stretch of amino acids, likewise for nonconserved regions. To ensure that our analysis is based on sufficiently many amino acid changes, we only used those pairwise alignments where each protein contained a conserved and nonconserved region of at least 100 amino acids each.
Furthermore, as in the previous analyses, we discarded uninformative or unreliable protein pairs (dS = 0, dS > 2, dN/dS > 2). Finally, we used the Ka/Ks estimator (Zhang et al. 2006) and the hon-new program (Zhang 2000) to obtain the ratios dN/dS and Dr/Dc, and did so separately for the conserved and nonconserved parts of each gene. We determined significant differences in dN/dS and Dr/Dc ratios among outcrossers and selfers for conserved and nonconserved regions separately with a Mann–Whitney U test (Sokal and Rohlf 2012). One limitation of this test is that we treat conserved and nonconserved regions as independent entities. This is because no meaningful pairing exists when analyzing the total data set. However, dependency of conserved and nonconserved regions was fully accounted for (Wilcoxon matched pairs tests, see below) when analyzing the one-to-one ortholog data set. We also used Wilcoxon matched pairs tests (Sokal and Rohlf 2012) to determine whether conserved and nonconserved regions (the matched pairs) within each gene showed significantly different dN/dS and Dr/Dc ratios for each species pair separately. We performed all statistical analyses in R (R Development Core Team 2011).
Comparing functionally different gene sets could obscure the effect of mating system on the evolutionary rate of genes. To account for this bias, we identified proteins that showed one-to-one orthology across all four of our study species because orthologous proteins tend to be more similar in function than paralogs (Altenhoff et al. 2012). We identified orthologs using the algorithm provided in ProteinOrtho (Lechner et al. 2011) and repeated all the above analyses on this data set. To establish orthology across the four species gene set, we executed ProteinOrtho using all predicted proteins of S. subsecundum, S. cribrosum, F. hygrometrica, and P. patens (http://www.phytozome.net/version 8.0, last accessed October 8, 2013) with the following parameters: e value threshold of 10−4, 30% identity, adaptive best alignments similarity of f = 0.95, algebraic connectivity > 0.1. Subsequently, we kept only those co-orthologs containing one single protein per species in order to reduce the risk of including paralogous relationships. Finally, we used the four-way orthology relationship to identify corresponding pairwise orthologous genes (S. subsecundum vs. S. cribrosum; F. hygrometrica vs. P. patens).
Results
Selfers Purge Deleterious Mutations Better than Outcrossers
Our analysis of complete quality-filtered transcriptomes (see Materials and Methods) identified 10,038 orthologous gene pairs in the selfer species (F. hygrometrica and P. patens) and 6,089 orthologous gene pairs in the outcrosser species pair (S. subsecundum and S. cribrosum) (table 1).
Table 1.
Accumulation of Deleterious Mutations among the Selfer and the Outcrosser Species Pairs across the Full Coding Region of Proteins
| Type of Analysis | Mating System | Species Pair | Number of Pairwise Alignments | Length of Alignment in Base Pairs, Mean (median) | Lower–Upper Quartiles | dN or Dr, Mean (median) | Lower–Upper Quartiles | dS or Dc, Mean (median) | Lower–Upper Quartiles | dN/dS or Dr/Dc, Mean (median) | Lower–Upper Quartiles | Significance (selfer vs. outcrosser) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Synonymous–nonsynonymous mutations (dN/dS) | ||||||||||||
| Selfer (S) | Funaria hygrometrica versus Physcomitrella patens | |||||||||||
| 10,038 | 1,065 (879) | 624–1,293 | 0.0539 (0.0359) | 0.0202–0.0601 | 0.2926 (0.2649) | 0.2218–0.3270 | 0.1957 (0.1339) | 0.0742–0.2174 | O (P < 0.00001) | |||
| dN/dS < 1 | 10,038 | 1,065 (879) | 624–1,293 | 0.0539 (0.0359) | 0.0202–0.0601 | 0.2926 (0.2649) | 0.2218–0.3270 | 0.1957 (0.1339) | 0.0742–0.2174 | O (P < 0.00001) | ||
| Outcrosser (O) | Sphagnum subsecundum versus S. macrophyllum | |||||||||||
| 6,089 | 1,195 (963) | 675–1,452 | 0.0727 (0.0226) | 0.0096–0.0571 | 0.2455 (0.0516) | 0.0291–0.1214 | 0.4222 (0.4163) | 0.2376–0.6030 | ||||
| dN/dS < 1 | 5,780 | 1,213 (975) | 681–1,470 | 0.0703 (0.0220) | 0.0095–0.0577 | 0.2540 (0.0537) | 0.0307–0.1234 | 0.3757 (0.3866) | 0.2334–0.5657 | |||
| Radical-conservative amino acid changes (Dr/Dc) | ||||||||||||
| Selfer (S) | F. hygrometrica versus P. patens | |||||||||||
| 9,913 | 1,075 (885) | 624–1,308 | 0.0421 (0.0360) | 0.0200–0.0580 | 0.0740 (0.0680) | 0.0430–0.0940 | 0.5590 (0.5526) | 0.4000–0.7391 | O (P < 0.00001) | |||
| dN/dS < 1 | 9,900 | 1,082 (891) | 630–1,317 | 0.0419 (0.0360) | 0.0200–0.0580 | 0.0737 (0.0680) | 0.0430–0.0940 | 0.5590 (0.5526) | 0.4000–0.7391 | O (P < 0.00001) | ||
| Outcrosser (O) | S. subsecundum versus S. macrophyllum | |||||||||||
| 5,643 | 1,084 (876) | 633–1,305 | 0.0539 (0.0220) | 0.0100–0.0610 | 0.0810 (0.0300) | 0.0150–0.1030 | 0.7576 (0.6364) | 0.4930–0.8889 | ||||
| dN/dS < 1 | 5,338 | 1,233 (987) | 690–1,500 | 0.0530 (0.0220) | 0.0100–0.0580 | 0.0806 (0.0290) | 0.0140–0.1000 | 0.7475 (0.6309) | 0.4880–0.8468 | |||
Note.—dS, proportion of synonymous mutations per synonymous sites; dN, proportion of nonsynonymous mutations per nonsynonymous sites; Dc, proportion of conserved amino acid replacements; Dr, proportion of radical amino acid replacements; Significance, statistical significance according to a Mann–Whitney U test. We conducted two separate tests: One for the unfiltered and one for the filtered (dN/dS < 1) data sets.
Overall, we found that the mean dN/dS ratios among orthologous genes in both species pairs fell into the interval 0.1–0.4, a range that is typical for such genome-wide analyses (Slotte et al. 2011; Yang and Gaut 2011). Importantly, the dN/dS ratio in the outcrosser species pair was twice as high than in the selfer species pair, and this difference was statistically significant, according to a Mann–Whitney U test (fig. 2 and table 1; zdf=1 = 53.48, P < 0.00001). After removing gene pairs that may be subject to positive selection (dN/dS > 1), the dN/dS ratio was still much higher in the outcrosser species (table 1; zdf=1 = 50.01, P < 0.00001). Repeating the above analyses on the data set containing only genes that are one-to-one orthologous across our four study species led to the very same conclusions (supplementary table S1, Supplementary Material online). In sum, deleterious mutations are purged more efficiently from the genomes of the selfer species.
Fig. 2.—
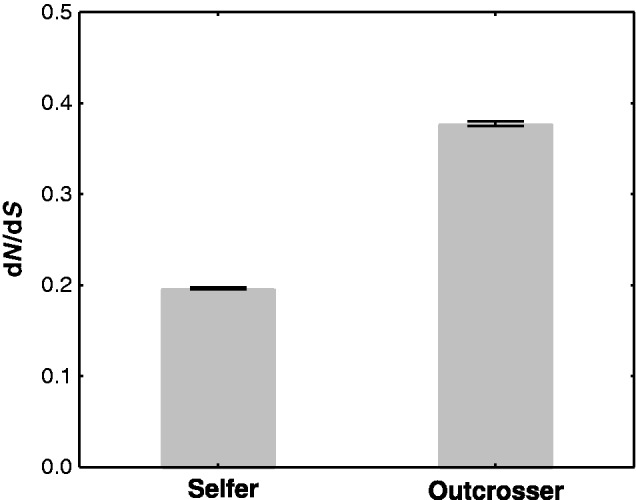
Ratio of nonsynonymous mutations per nonsynonymous sites to synonymous mutations per synonymous sites (dN/dS, mean, and SE) in the selfer and in the outcrosser species pairs. dN/dS ratios are significantly greater in the outcrosser than in the selfer species pair (P < 0.00001).
Selfers Purge Radical Amino Acid Changes More Efficiently than Outcrossers
Many radical changes are deleterious and eliminated by natural selection (Zhang 2000). In a complementary analysis which focused on the ratio of radical to conservative amino acid changes Dr/Dc, we found that the Dr/Dc ratio was about 36% greater in the outcrosser than in the selfer species pair (fig. 3 and table 1; zdf=1 = 25.62, P < 0.00001). We repeated this analysis by excluding those genes that may be subject to positive selection (dN/dS > 1) which led to the same outcome (table 1, selfer vs. outcrosser zdf=1 = 23.59, P < 0.00001). Finally, repeating these analyses on one-to-one orthologs led to the very same conclusions (supplementary table S1, Supplementary Material online). Analysis of radical and conserved amino acid changes between selfer and outcrosser species pairs thus shows that selfers are more efficient in purging deleterious mutations than outcrossers, which is consistent with our observations based on dN/dS ratios.
Fig. 3.—
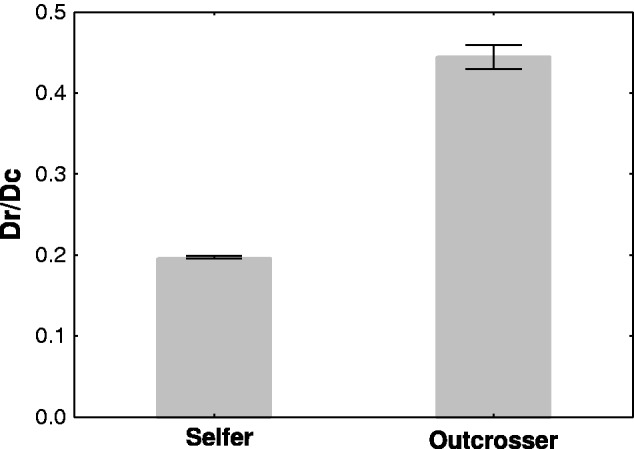
Ratio of radical to conserved amino acid changes (Dr/Dc, mean, and SE) in the selfer and in the outcrosser species pairs. Dr/Dc ratios are highly significantly greater in the outcrosser than in the selfer species pair (P < 0.00001).
Comparison of Conserved and Nonconserved Stretches of Genes
In our next analysis, we contrasted dN/dS or Dr/Dc ratios in conserved and nonconserved regions of genes (see Materials and Methods). We first investigated whether the difference in dN/dS values between conserved and nonconserved regions within each gene differs more considerably in selfers than in outcrossers, which would imply more effective purging in selfers. We found that dN/dS ratios were smaller in conserved than in nonconserved regions of genes in both the selfer and the outcrosser species pairs. Importantly, in selfing species, this difference was more pronounced (table 2 and fig. 4). Specifically, mean dN/dS ratios were on average 116% greater in nonconserved regions of selfers (zS[conserved vs. nonconserved] = 40.03, P < 0.00001), and only 73% greater in outcrossers (zO[conserved vs. nonconserved] = 21.97, P < 0.00001), which is also evident from the z scores of the Wilcoxon matched pairs test (zS[conserved vs. nonconserved] = 40.03, zO[conserved vs. nonconserved] = 22.33; table 2 and fig. 4). We repeated this analysis after exclusion of genes that may have been subject to positive selection (dN/dS > 1), which leads to the same conclusion (table 2; zS[conserved vs. nonconserved] = 40.03, P < 0.00001; zO[conserved vs. nonconserved] = 22.17, P < 0.00001), as did the same analysis for one-to-one orthologous genes (supplementary table S2, Supplementary Material online).
Table 2.
Accumulation of Deleterious Mutations (dN/dS) between the Selfer and the Outcrosser Species Pairs in Conserved Domain and Nonconserved Domain Stretches of Proteins
| Sequence Stretch | Mating System | Species Pair | Number of Pairwise Alignments | Length of Alignment in Base Pairs, Mean (median) | Lower– Upper Quartiles | dN, Mean (median) | Lower–Upper Quartiles | dS, Mean (median) | Lower–Upper Quartiles | dN/dS, Mean (median) | Lower–Upper Quartiles | Significance (selfer vs. outcrosser; conserved and nonconserved domains separately tested) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conserved domains | ||||||||||||
| Selfer (S) | Funaria hygrometrica versus Physcomitrella patens | |||||||||||
| 2,995 | 768 (639) | 459–930 | 0.0321 (0.0219) | 0.0124–0.0362 | 0.2835 (0.2504) | 0.2123–0.3120 | 0.1260 (0.0814) | 0.0454–0.1496 | O (P < 0.00001) | |||
| dN/dS < 1 | 2,995 | 768 (639) | 459–930 | 0.0321 (0.0219) | 0.0124–0.0362 | 0.2835 (0.2504) | 0.2123–0.3120 | 0.1260 (0.0814) | 0.0454–0.1496 | O (P < 0.00001) | ||
| Outcrosser (O) | Sphagnum subsecundum versus S. macrophyllum | |||||||||||
| 1,699 | 786 (666) | 465–969 | 0.0420 (0.0079) | 0.0041–0.0199 | 0.1679 (0.0361) | 0.0246–0.0702 | 0.2651 (0.1904) | 0.1036–0.3575 | ||||
| dN/dS < 1 | 1,665 | 804 (678) | 468–987 | 0.0309 (0.0078) | 0.0040–0.0184 | 0.0922 (0.0361) | 0.0246–0.0702 | 0.2448 (0.1889) | 0.1020–0.3527 | |||
| Nonconserved domains | ||||||||||||
| Selfer (S) | F. hygrometrica versus P. patens | |||||||||||
| 2,995 | 1,376 (765) | 456–1500 | 0.0661 (0.0513) | 0.02883–0.0843 | 0.2761 (0.1559) | 0.2109–0.2991 | 0.2629 (0.1975) | 0.1092–0.3296 | O (P < 0.001) | |||
| dN/dS < 1 | 2,995 | 1,376 (765) | 456–1500 | 0.0661 (0.0513) | 0.02883–0.0843 | 0.2761 (0.2560) | 0.2109–0.2991 | 0.2629 (0.1975) | 0.1092–0.3296 | O (P < 0.001) | ||
| Outcrosser (O) | S. subsecundum versus S. macrophyllum | |||||||||||
| 1,699 | 970 (720) | 456–1152 | 0.0760 (0.0204) | 0.0104–0.0591 | 0.1860 (0.0467) | 0.0283–0.1273 | 0.4523 (0.4170) | 0.2515–0.6220 | ||||
| dN/dS < 1 | 1,665 | 1,016 (736) | 465–1215 | 0.0638 (0.0202) | 0.0099–0.0590 | 0.1153 (0.0461) | 0.0282–0.1274 | 0.4034 (0.4083) | 0.2460–0.6183 | |||
Note.—dS, proportion of synonymous mutations per synonymous sites; dN, proportion of nonsynonymous mutations per nonsynonymous sites; Significance, statistical significance according to a Kruskal–Wallis ANOVA. We conducted two separate tests: One for the unfiltered and one for the filtered (dN/dS < 1) data sets. We show results of the two tests for conserved domain and nonconserved domain stretches of proteins separately.
Fig. 4.—
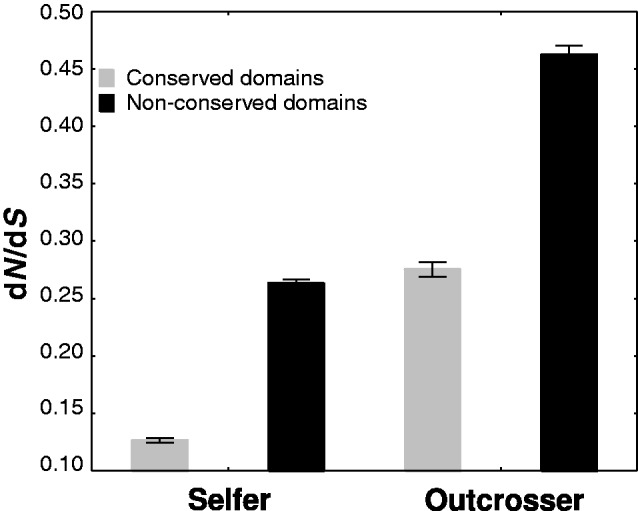
Ratio of nonsynonymous mutations per nonsynonymous sites to synonymous mutations per synonymous sites (dN/dS, mean, and SE) for conserved and nonconserved domains of genes in the selfer and in the outcrosser species pairs. dN/dS ratios in conserved and nonconserved domains of genes are highly significantly different both within the selfer and the outcrosser species pairs studied (P < 0.00001). However, the difference between conserved and nonconserved domains is less pronounced in the outcrosser comparison. Finally, dN/dS ratios are significantly (P < 0.00001) greater in the outcrosser than in the selfer species pair both in conserved and in nonconserved domains with a greater effect in conserved domains.
Because the ratio of radical to conservative amino acid changes (Dr/Dc) also reflects mutational purging, we reasoned that it should show a pattern similar to dN/dS in conserved and nonconserved gene regions (table 3 and fig. 5). Indeed, the proportion of radical amino acid changes was significantly smaller (15%) in conserved regions for selfers (zS[conserved vs. nonconserved] = 18.38, P < 0.00001). This was also true for outcrossers but to a significantly lesser extent (10%, zO[conserved vs. nonconserved] = 5.51, P < 0.00001). This conclusion did not change when we excluded genes that may have been subject to positive selection (dN/dS > 1; table 3; zS[conserved vs. nonconserved] = 18.38, P < 0.00001; zO[conserved vs. nonconserved] = 5.36, P < 0.00001) and when we restricted our analysis for one-to-one orthologous genes (supplementary table S3, Supplementary Material online). Altogether, this means that conserved regions accumulate fewer radical amino acid changes in the selfers than in the outcrossers. This supports the hypothesis that purging of deleterious mutations through homozygous exposure is more efficient in selfers than in outcrossers.
Table 3.
Accumulation of Deleterious Mutations (Dr/Dc) in the Selfer and the Outcrosser Species Pair in Conserved Domain and Nonconserved Domain Stretches of Proteins
| Sequence Stretch | Mating System | Species Pair | Number of Pairwise Alignments | Length of Alignment in Base Pairs, Mean (median) | Lower– Upper Quartiles | Dr, Mean (median) | Lower– Upper Quartiles | Dc, Mean (median) | Lower–Upper Quartiles | Dr/Dc, Mean (median) | Lower– Upper Quartiles | Significance (selfer vs. outcrosser; conserved and nonconserved domains separately tested) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conserved domains | ||||||||||||
| Selfer (S) | Funaria hygrometrica versus Physcomitrella patens | |||||||||||
| 2,746 | 768 (639) | 459–930 | 0.0254 (0.0160) | 0.0070–0.0280 | 0.0534 (0.0390) | 0.0240–0.0570 | 0.5304 (0.4130) | 0.2389–0.6027 | O (P < 0.001) | |||
| dN/dS < 1 | 2,746 | 768 (639) | 459–930 | 0.0254 (0.0160) | 0.0070–0.0280 | 0.0534 (0.0390) | 0.0240–0.0570 | 0.5304 (0.4130) | 0.2389–0.6027 | O (P < 0.001) | ||
| Outcrosser (O) | Sphagnum subsecundum versus S. macrophyllum | |||||||||||
| 1,203 | 858 (750) | 504-1,050 | 0.0389 (0.0090) | 0.0050–0.0310 | 0.0637 (0.0170) | 0.0090–0.0540 | 0.6677 (0.5455) | 0.3514–0.8462 | ||||
| dN/dS < 1 | 1,168 | 866 (759) | 504–1,065 | 0.0388 (0.0090) | 0.0050–0.0290 | 0.0638 (0.0160) | 0.0090–0.0530 | 0.6605 (0.5455) | 0.3571–0.8358 | |||
| Nonconserved domains | ||||||||||||
| Selfer (S) | F. hygrometrica versus P. patens | |||||||||||
| 2,746 | 1,374 (765) | 456–1,500 | 0.0551 (0.0400) | 0.0190–0.0680 | 0.0893 (0.0730) | 0.0490–0.1100 | 0.6160 (0.5294) | 0.3871–0.7086 | O (P < 0.00001) | |||
| dN/dS < 1 | 2,746 | 1,374 (765) | 456–1,500 | 0.0551 (0.0400) | 0.0190–0.0680 | 0.0894 (0.0730) | 0.0490–0.1100 | 0.6160 (0.5294) | 0.3871–0.7086 | O (P < 0.00001) | ||
| Outcrosser (O) | S. subsecundum versus S. macrophyllum | |||||||||||
| 1,203 | 1,047 (783) | 486–1,263 | 0.0680 (0.0220) | 0.0100–0.0690 | 0.1004 (0.0300) | 0.0160–0.1150 | 0.7383 (0.6667) | 0.4750–0.8726 | ||||
| dN/dS < 1 | 1,168 | 1,061 (792) | 492–1,281 | 0.0681 (0.0210) | 0.0100–0.0680 | 0.1013 (0.0320) | 0.0160–0.1150 | 0.7285 (0.6667) | 0.4737–0.8587 | |||
Note.—Dc, proportion of conservative amino acid replacements; Dr, proportion of radical amino acid replacements; Significance, statistical significance according to a Kruskal–Wallis ANOVA. We conducted two separate tests: One for the unfiltered and one for the filtered (dN/dS < 1) data sets. We show results of the two tests for conserved domain and nonconserved domain stretches of proteins separately.
Fig. 5.—
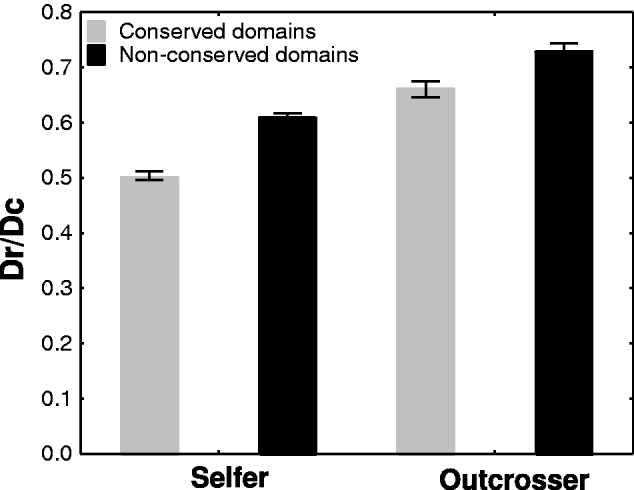
Ratio of radical to conserved amino acid changes (Dr/Dc, mean, and SE) for conserved and nonconserved domains of genes in the selfer and in the outcrosser species pairs. Dr/Dc ratios in conserved and nonconserved domains of genes are significantly different both within the selfer (P < 0.00001) and in the outcrosser species pairs (P < 0.00001). In both conserved and nonconserved domains, Dr/Dc ratios are significantly greater in the outcrosser (P < 0.00001) than in the selfer species pair. Nevertheless, this difference is more pronounced in the conserved than in the nonconserved domain stretches.
The preceding analysis focused on differences in dN/dS and Dr/Dc ratios among conserved and nonconserved regions “within” genes for the selfer and outcrosser species pairs. The following analysis focuses on analogous predictions “between” species pairs. We found that the dN/dS ratio was smaller in the selfer than in the outcrosser species pair and this difference was more pronounced in conserved gene regions (table 2 and fig. 4). Specifically, in the conserved domains of genes, the dN/dS ratio was approximately 116% (PS vs. O < 0.00001) higher in the outcrosser than in the selfer species pair. In contrast, in nonconserved regions of genes the difference in dN/dS ratios between selfer species and outcrosser species, while significant (PS vs. O < 0.00001), was less pronounced (73% greater in outcrosser species than in selfing species). Repeating this analysis by excluding genes that may have been subject to positive selection (dN/dS > 1) did not affect this observation (table 2; dN/dS in conserved stretches: PS vs. O < 0.00001; dN/dS in nonconserved stretches: PS vs. O < 0.001), and neither did the analysis of one-to-one orthologous gene sets (supplementary table S2, Supplementary Material online).
Finally, we performed an analogous analysis for the ratio of radical to conservative amino acid changes (Dr/Dc). Again we found that the difference in Dr/Dc values between selfer and outcrosser species pairs was more pronounced in conserved than in nonconserved regions of genes (table 3 and fig. 5). Specifically, Dr/Dc ratios in conserved regions of genes were approximately 26% (PS vs. O < 0.00001) greater in the outcrosser species pair and this difference was statistically highly significant. In contrast, in nonconserved regions of genes Dr/Dc values were only approximately 18% (PS vs. O < 0.0001) greater between outcrossers and selfers. This observation was not affected by excluding genes that may have been subject to positive selection (table 3; Dr/Dc in conserved stretches: PS vs. O < 0.00001; Dr/Dc in nonconserved stretches: PS vs. O < 0.00001), nor by considering only one-to-one orthologs (supplementary table S3, Supplementary Material online). All these observations imply that deleterious mutations are more effectively purged in the selfers than in the outcrossers.
Discussion
Selfing has two opposing effects on the efficacy of purifying selection. On the one hand, selfing reduces effective population size, because it increases genome-wide homozygosity (Nordborg 2000) and lowers effective recombination rates (Wright et al. 2008, 2013). These effects reduce the efficacy of purifying selection. On the other hand, selfing exposes recessive deleterious mutations in a homozygous state and can thus also enhance purging (Glémin 2007). In selfers with a diploid-dominant life cycle the first effect dominates, thus leading to a net reduction in purifying selection (Slotte et al. 2013; Wright et al. 2013). Here we show that in plants with a haploid-dominant life cycle undergoing intragametophytic selfing, an extreme form of selfing, homozygous exposure of deleterious mutations can lead to enhanced purging on a genome-wide scale.
This assertion is supported by three lines of evidence. First, we found that selfers accumulate fewer nonsynonymous mutations (dN/dS) and radical amino acid replacements (Dr/Dc) than outcrossers. Second, within genes, significantly fewer deleterious mutations (dN/dS and Dr/Dc) accumulate in conserved than in nonconserved regions, and this effect is greater in selfers than in outcrossers. Finally, by contrasting the accumulation of deleterious mutations (estimated again through dN/dS and Dr/Dc) among the two species pairs, we found that the difference between selfers and outcrossers is more pronounced in conserved than in nonconserved gene regions.
Previous studies provided indirect evidence that intragametophytic selfing can facilitate purging, but did not directly assess the effect of intragametophytic selfing on the accumulation of deleterious mutations at the level of genes and genomes. For instance, they found a lower incidence of lethality in homozygous embryos of ferns and a lack of inbreeding depression in homozygous sporophytes of mosses capable of intragametophytic selfing (Klekowski 1973, 1982; Lloyd 1974; Hedrick 1987a, 1987b; Taylor et al. 2007). By studying constrained sequence evolution on a genome-wide scale, our study goes beyond previous phenotypic observations and provides direct evidence that purifying selection under intragametophytic selfing can balance and even outweigh the effect of effective population size reduction caused by selfing (Hedrick 1987a, 1987b; Holsinger 1987). Furthermore, our observations also suggest that the correlation between intragametophytic selfing and a lack of both homozygous lethality and inbreeding depression may be explained by a decreased accumulation of deleterious mutations in the genome. Any direct connection between deleterious mutations and observed phenotypic effects, however, has to be experimentally demonstrated in future work.
It is generally assumed that the potential masking of deleterious mutations is restricted to diploid-specific genes of a plant’s lifecycle. The reason is that deleterious mutations in haploid-specific genes (as well as in genes needed in both stages) are always exposed to selection. Previous work showed that only approximately 2% of genes expressed in F. hygrometrica and P. patens show diploid-specific expression (Szövényi et al. 2011; O’Donoghue et al. 2013). Such a minute fraction of the genome subject to effective purging by intragametophytic selfing might go undetected. However, the significant effect of intragametophytic selfing on purging we observed shows otherwise. Together with our previous finding that diploid- and haploid-specific genes experience similar efficacy of purging (Szövényi et al. 2013), it contradicts the notion that only diploid-specific genes are affected by intragametophytic selfing.
To explain this apparent contradiction, we note that a gene’s exposure to selection in the haploid phase does not necessarily lead to more effective selection (Gerstein et al. 2011; Gerstein and Otto 2011; Billiard et al. 2012; Arunkumar et al. 2013; Szövényi et al. 2013; Zörgö et al. 2013). Therefore, contrary to previous assumptions, genes expressed in both phases can experience the effect of intragametophytic selfing on purging. One possible mechanism is that homozygous exposure of deleterious mutations via intragametophytic selfing may extend the time span over which deleterious mutations are directly exposed to selection (once as haploids and once as diploid homozygotes). Deleterious mutations, which may escape effective purifying selection in the haploid stage, may no longer escape natural selection when exposed in both stages. Moreover, the same mutation may have different deleterious effects in haploid and diploid stages, and their compound effect may be more severe, which can further contribute to its efficient purging. Because existing theory does not fully account for complications like this, more work is needed to fully understand the evolutionary dynamics of deleterious mutations in organisms with complex life cycles.
Because our findings are in stark contrast to the observation that selfing leads to reduced selection efficacy (Glémin and Galtier 2012), it is important to address the effect of confounding factors that may bias our analyses. The first of two major factors is that selfer and outcrosser species pairs may express different gene sets that could bias our analysis (Yang and Gaut 2011). This gene set bias may be partially caused by differential tissue sampling in the selfer and outcrosser species pairs: RNA was obtained only from gametophytic tissues for the outcrosser whereas both gametophytic and sporophytic tissues were used for the selfer species pair. For this reason we repeated our analyses with genes that were one-to-one orthologous across all four study species, which yielded the same conclusions (see supplementary material, Supplementary Material online). Therefore, gene set bias cannot explain the differences we see between the selfer and the outcrosser species pairs.
Second, it is well-known that estimates of synonymous divergence dS (a measure of divergence time) and dN/dS are negatively correlated. That is, dN/dS values are higher between recently diverged species, because intraspecific polymorphisms contribute prominently to dN/dS (Rocha et al. 2006; Kryazhimskiy and Plotkin 2008; Wolf et al. 2009; Mugal et al. 2014). Being aware of this issue, we selected species pairs (selfer and outcrosser) that exhibit similar divergence times. More specifically, both the outcrosser and the selfer species pairs are estimated to have diverged about 14–20 Ma (Shaw et al. 2010; Szövényi et al. 2013) which corresponds to an approximate divergence time of 23–33 Ne generations (assuming an Ne of 600,000 [McDaniel, van Baren et al. 2013] and a mutation rate of 1.8 × 10−8 [Rensing et al. 2007]). Previous work suggests that the contribution of polymorphisms to dN/dS estimates within this time range is small and the overall influence of divergence time on total dN/dS is thus weak (Mugal et al. 2014). Because this argument depends on the parameter estimates used, we also conducted a second, complementary analysis. Specifically, we divided gene alignments into five categories according to their dS values in both species pairs and compared dN/dS values for each category separately. We found that dN/dS ratios were significantly greater for the outcrosser species pair in all dS categories investigated, both for the full and for the one-to-one ortholog data set (supplementary table S4, Supplementary Material online). All this suggests that the difference in dN/dS ratios between selfers and outcrossers is not caused by the difference in synonymous divergence.
Conclusions, Limitations, and Open Questions
Although our study provides evidence that purging under intragametophytic selfing can be more effective than under intergametophytic selfing, multiple questions remain to be resolved. First, our current analysis is based on the comparison of one selfer and one outcrosser species pair. Therefore, with the current data set in hand it is difficult to assess the generality of our findings. Including multiple selfer and outcrosser lineages in future studies will be necessary to test the generality of our findings. Second, the extent to which diploid-specific genes or genes expressed in both ploidal phases are affected by purging has to be determined. This is critical to understand the evolutionary mechanism of purging. Third, our data suggest that intragametophytic selfing is advantageous, because it can efficiently prevent the accumulation of deleterious mutations in organism with extreme selfing. Theory predicts, however, that extreme linkage disequilibrium established by repeated events of intragametophytic selfing may hinder adaptive evolution (Felsenstein 1974; Birky and Walsh 1988; Charlesworth 2012). Nevertheless, McDaniel, Atwood et al. (2013) showed that hermaphroditic lineages of haploid-dominant plants have an elevated diversification rate compared with dioecious lineages. Therefore, the better purging in haploid selfers may also have long-term macroevolutionary consequences that need to be explored further. Thus, the extent to which intragametophytic selfing constrains the adaptive potential of species remains to be determined.
Supplementary Material
Supplementary tables S1–S4 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
Three anonymous reviewers provided helpful comments on an earlier version of this manuscript. This work was supported by the Swiss National Science Foundation, Ambizione grant number PZ00P3_131726 to P.S.Z. and by a URPP pilot grant of the Systems Biology/Functional Genomics priority program of the University of Zurich to P.S.Z. This work also received support through Swiss National Science Foundation grants 315230-129708 to A.W., and grant 31003A_140917 to K.K.S., as well as by the University Priority Research Program in Evolutionary Biology at the University of Zurich to A.W. and K.K.S. This work was also supported by an NSF grant no. DEB-0918998 to A.J.S. and Blanka Shaw.
Literature Cited
- Altenhoff AM, Studer RA, Robinson-Rechavi M, Dessimoz C. Resolving the ortholog conjecture: orthologs tend to be weakly, but significantly, more similar in function than paralogs. PLoS Comput Biol. 2012;8:e1002514. doi: 10.1371/journal.pcbi.1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LE, Shaw AJ, Shaw B. Peatmosses (Sphagnum) of the southeastern United States. New York: New York Botanical Garden Press; 2009. [Google Scholar]
- Arunkumar R, Josephs EB, Williamson RJ, Wright SI. Pollen-specific, but not sperm-specific, genes show stronger purifying selection and higher rates of positive selection than sporophytic genes in Capsella grandiflora. Mol Biol Evol. 2013;30:2475–2486. doi: 10.1093/molbev/mst149. [DOI] [PubMed] [Google Scholar]
- Axelsson E, et al. Natural selection for avianprotein-coding genes expressed in brain. Mol Ecol. 2008;17:3008–3017. doi: 10.1111/j.1365-294X.2008.03795.x. [DOI] [PubMed] [Google Scholar]
- Barner AK, Pfister CA, Wootton JT. The mixed mating system of the sea palm kelp Postelsia palmaeformis: few costs to selfing. Proc Biol Sci. 2011;278:1347–1355. doi: 10.1098/rspb.2010.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson BO, Cronberg N. The effective size of bryophyte populations. J Theor Biol. 2009;258:121–126. doi: 10.1016/j.jtbi.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Billiard S, López-Villavicencio M, Hood ME, Giraud T. Sex, outcrossing and mating types: unsolved questions in fungi and beyond. J Evol Biol. 2012;25:1020–1038. doi: 10.1111/j.1420-9101.2012.02495.x. [DOI] [PubMed] [Google Scholar]
- Birky CW, Walsh JB. Effects of linkage on rates of molecular evolution. Proc Natl Acad Sci U S A. 1988;85:6414–6418. doi: 10.1073/pnas.85.17.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, et al. Predicting function: from genes to genomes and back. J Mol Biol. 1998;283:707–725. doi: 10.1006/jmbi.1998.2144. [DOI] [PubMed] [Google Scholar]
- Bustamante CD, et al. The cost of inbreeding in Arabidopsis. Nature. 2002;416:531–534. doi: 10.1038/416531a. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. The effects of deleterious mutations on the evolution of linked sites. Genetics. 2012;190:5–22. doi: 10.1534/genetics.111.134288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Willis JH. The genetics of inbreeding depression. Nat Rev Genet. 2009;10:783–796. doi: 10.1038/nrg2664. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, Wright SI. Breeding system and genome evolution. Curr Opin Genet Dev. 2001;11:685–690. doi: 10.1016/s0959-437x(00)00254-9. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppley SM, Taylor PJ, Jesson LK. Self-fertilization in mosses: a comparison of heterozygote deficiency between species with combined versus separate sexes. Heredity. 2007;98:38–44. doi: 10.1038/sj.hdy.6800900. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD, Smith NG, Gaffney D. Quantifying the slightly deleterious mutation model of molecular evolution. Mol Biol Evol. 2002;19:2142–2149. doi: 10.1093/oxfordjournals.molbev.a004039. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78:737–756. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Cleathero LA, Mandegar MA, Otto SP. Haploids adapt faster than diploids across a range of environments. J Evol Biol. 2011;24:531–540. doi: 10.1111/j.1420-9101.2010.02188.x. [DOI] [PubMed] [Google Scholar]
- Gerstein AC, Otto SP. Cryptic fitness advantage: diploids invade haploid populations despite lacking any apparent advantage as measured by standard fitness assays. PLoS One. 2011;6:e26599. doi: 10.1371/journal.pone.0026599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glémin S. Mating systems and the efficacy of selection at the molecular level. Genetics. 2007;177:905–916. doi: 10.1534/genetics.107.073601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glémin S, Galtier N. Genome evolution in outcrossing versus selfing versus asexual species. Methods Mol Biol. 2012;855:311–335. doi: 10.1007/978-1-61779-582-4_11. [DOI] [PubMed] [Google Scholar]
- Gossmann TI, et al. Genome wide analyses reveal little evidence for adaptive evolution in many plant species. Mol Biol Evol. 2010;27:1822–1832. doi: 10.1093/molbev/msq079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Shiu SH, Li WH. The nonsynonymous/synonymous substitution rate ratio versus the radical/conservative replacement rate ratio in the evolution of mammalian genes. Mol Biol Evol. 2007;24:2235–2241. doi: 10.1093/molbev/msm152. [DOI] [PubMed] [Google Scholar]
- Hazzouri KM, et al. Comparative population genomics in Collinsia sister species reveals evidence for reduced effective population size, relaxed selection, and evolution of biased gene conversion with an ongoing mating system shift. Evolution. 2013;67:1263–1278. doi: 10.1111/evo.12027. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. Genetic load and the mating system in homosporous ferns. Evolution. 1987a;41:1282–1289. doi: 10.1111/j.1558-5646.1987.tb02466.x. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. Population genetics of intragametophytic selfing. Evolution. 1987b;41:137–144. doi: 10.1111/j.1558-5646.1987.tb05776.x. [DOI] [PubMed] [Google Scholar]
- Holsinger KE. Gametophytic self-fertilization in homosporous plants: development, evaluation, and application of a statistical method for evaluating its importance. Am J Bot. 1987;74:1173–1183. [Google Scholar]
- Hughes AL, Friedman R. More radical amino acid replacements in primates than in rodents: support for the evolutionary role of effective population size. Gene. 2009;440:50–56. doi: 10.1016/j.gene.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge: Cambridge University Press; 1983. [Google Scholar]
- Klekowski EJ. Genetic load in Osmunda regalis populations. Am J Bot. 1973;60:146–154. [Google Scholar]
- Klekowski EJ. Genetic load and soft selection in ferns. Heredity. 1982;49:191–197. [Google Scholar]
- Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann T, Hayashizaki Y, Daub CO. TagDust-a program to eliminate artifacts from next generation sequencing data. Bioinformatics. 2009;25:2839–2840. doi: 10.1093/bioinformatics/btp527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner M, et al. Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Budke JM, Goffinet B. Phylogenetic inference rejects sporophyte based classification of the Funariaceae (Bryophyta): rapid radiation suggests rampant homoplasy in sporophyte evolution. Mol Phylogenet Evol. 2012;62:130–145. doi: 10.1016/j.ympev.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Lloyd RM. Mating systems and genetic load in pioneer and non-pioneer Hawaiian Pteridophyta. Bot J Linn Soc. 1974;69:23–35. [Google Scholar]
- Mank JE, Nam K, Brunström B, Ellegren H. Ontogenetic complexity of sexual dimorphism and sex-specific selection. Mol Biol Evol. 2010;27:1570–1578. doi: 10.1093/molbev/msq042. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32(Web Server issue):W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, et al. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41:348–352. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J, Szathmáry E. The major transitions in evolution. Oxford: Oxford University Press; 1995. [Google Scholar]
- McCauley DE, Whittier DP, Reilly LM. Inbreeding and the rate of self-fertilization in a grape-fern, Botrychium dissectum. Am J Bot. 1985;72:1978–1981. [Google Scholar]
- McDaniel SF, et al. The speciation history of the Physcomitrella–Physcomitrium complex. Evolution. 2010;64:217–231. doi: 10.1111/j.1558-5646.2009.00797.x. [DOI] [PubMed] [Google Scholar]
- McDaniel SF, Atwood J, Burleigh JG. Recurrent evolution of dioecy in bryophytes. Evolution. 2013;67:567–572. doi: 10.1111/j.1558-5646.2012.01808.x. [DOI] [PubMed] [Google Scholar]
- McDaniel SF, van Baren MJ, Jones KS, Payton AC, Quatrano RS. Estimating the nucleotide diversity in Ceratodon purpureus (Hedw.) Brid. from 208 conserved intron-spanning nuclear loci. Appl Plant Sci. 2013;1:1200387. doi: 10.3732/apps.1200387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Miyazawa S, Yasunaga T. Two types of amino acid substitutions in protein evolution. J Mol Evol. 1979;12:219–236. doi: 10.1007/BF01732340. [DOI] [PubMed] [Google Scholar]
- Mugal CF, Wolf JBW, Kaj I. Why time matters: codon evolution and the temporal dynamics of dN/dS. Mol Biol Evol. 2014;31:212–231. doi: 10.1093/molbev/mst192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Siol M, Barrett SC. Genomic consequences of transitions from cross- to self-fertilization on the efficacy of selection in three independently derived selfing plants. BMC Genomics. 2012;13:611. doi: 10.1186/1471-2164-13-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. Estimating the distribution of selection coefficients from phylogenetic data with applications to mitochondrial and viral DNA. Mol Biol Evol. 2003;20:1231–1239. doi: 10.1093/molbev/msg147. [DOI] [PubMed] [Google Scholar]
- Nordborg M. Linkage disequilibrium, gene tress and selfing: an ancestral recombination graph with partial self-fertilization. Genetics. 2000;154:923–929. doi: 10.1093/genetics/154.2.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donoghue MT, et al. Genome-wide transcriptomic analysis of the sporophyte of the moss Physcomitrella patens. J Exp Bot. 2013;64:3567–3581. doi: 10.1093/jxb/ert190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP, Gerrsein AC. The evolution of haploidy and diploidy. Curr Biol. 2008;18:1112–1124. doi: 10.1016/j.cub.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Perroud PF, Cove DJ, Quatrano RS, McDaniel SF. An experimental method to facilitate the identification of hybrid sporophytes in the moss Physcomitrella patens using fluorescent tagged lines. New Phytol. 2011;191:301–306. doi: 10.1111/j.1469-8137.2011.03668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Zeng K, Slotte T, Wright SI, Charlesworth D. Reduced efficacy of natural selection on codon usage bias in selfing Arabidopsis and Capsella species. Gen Biol Evol. 2011;3:868–880. doi: 10.1093/gbe/evr085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2011. [cited 2014 May 16]. Available from: http://www.R-project.org/ [Google Scholar]
- Rensing SA, et al. An ancient genome duplication contributed to the abundance of metabolic genes in the moss Physcomitrella patens. BMC Evol Biol. 2007;7:130. doi: 10.1186/1471-2148-7-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensing SA, et al. The genome of the moss Physcomitrella patens reveals evolutionary insights into the conquest of land by plants. Science. 2008;319:64–69. doi: 10.1126/science.1150646. [DOI] [PubMed] [Google Scholar]
- Rocha EPC, et al. Comparisons of dN/dS are time dependentforclosely related bacterial genomes. J Theor Biol. 2006;239:226–235. doi: 10.1016/j.jtbi.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Shaw AJ. The genetic structure of sporophytic and gametophytic populations of the moss, Funaria hygrometrica Hedw. Evolution. 1991;45:1260–1274. doi: 10.1111/j.1558-5646.1991.tb04391.x. [DOI] [PubMed] [Google Scholar]
- Shaw AJ. Population ecology, population genetics, and microevolution. In: Goffinet B, Shaw AJ, editors. Bryophyte biology. 1st ed. New York: Cambridge University Press; 2000. pp. 403–448. [Google Scholar]
- Shaw AJ, et al. Genetic structure and genealogy in the Sphagnum subsecundum complex (Sphagnaceae: Bryophyta) Mol Phylogenet Evol. 2008;49:304–317. doi: 10.1016/j.ympev.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Shaw AJ, et al. Peatmoss (Sphagnum) diversification associated with Miocene Northern Hemisphere climatic cooling? Mol Phylogenet Evol. 2010;55:1139–1145. doi: 10.1016/j.ympev.2010.01.020. [DOI] [PubMed] [Google Scholar]
- Slotte T, Foxe JP, Hazzouri KM, Wright SI. Genome-wide evidence for efficient positive and purifying selection in Capsella grandiflora, a plant species with a large effective population size. Mol Biol Evol. 2010;27:1813–1821. doi: 10.1093/molbev/msq062. [DOI] [PubMed] [Google Scholar]
- Slotte T, et al. Genomic determinants of protein evolution and polymorphism in Arabidopsis. Gen Biol Evol. 2011;3:1210–1219. doi: 10.1093/gbe/evr094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotte T, et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nature Genet. 2013;45:831–835. doi: 10.1038/ng.2669. [DOI] [PubMed] [Google Scholar]
- Smith NGC. Are radical and conservative substitution rates useful statistics in molecular evolution? J Mol Evol. 2003;57:467–478. doi: 10.1007/s00239-003-2500-z. [DOI] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. Biometry: the principles and practice of statistics in biological research. 4th ed. New York: W. H. Freeman and Co; 2012. [Google Scholar]
- Stenøien HK, Såstad S. Genetic variability in bryophytes: does mating system really matter? J Bryol. 2001;4:313–318. [Google Scholar]
- Subramanian S. Significance of population size on the fixation of nonsynonymous mutations in genes under varying levels of selection pressure. Genetics. 2013;193:995–1002. doi: 10.1534/genetics.112.147900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:609–612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szövényi P, Rensing SA, Lang D, Shaw AJ, Wray G. Generation-biased gene expression in a bryophyte model system. Mol Biol Evol. 2011;28:803–812. doi: 10.1093/molbev/msq254. [DOI] [PubMed] [Google Scholar]
- Szövényi P, Ricca M, Shaw AJ. Multiple paternity and sporophytic inbreeding depression in a dioicous moss species. Heredity. 2009;103:394–403. doi: 10.1038/hdy.2009.82. [DOI] [PubMed] [Google Scholar]
- Szövényi P, Terracciano S, Ricca M, Giordano S, Shaw AJ. Recent divergence, intercontinental dispersal and shared polymorphism are shaping the genetic structure of amphi-Atlantic peatmoss populations. Mol Ecol. 2008;17:5364–5377. doi: 10.1111/j.1365-294X.2008.04003.x. [DOI] [PubMed] [Google Scholar]
- Szövényi P, et al. Selection is no more efficient in haploid than in diploid life stages of an angiosperm and a moss. Mol Biol Evol. 2013;30:1929–1939. doi: 10.1093/molbev/mst095. [DOI] [PubMed] [Google Scholar]
- Taylor PJ, Eppley SM, Jesson LK. Sporophytic inbreeding depression in mosses occurs in a species with separate sexes but not in a species with combined sexes. Am J Bot. 2007;94:1853–1859. doi: 10.3732/ajb.94.11.1853. [DOI] [PubMed] [Google Scholar]
- Vijay N, Poelstra JW, Künstner A, Wolf JBW. Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments. Mol Ecol. 2013;22:620–634. doi: 10.1111/mec.12014. [DOI] [PubMed] [Google Scholar]
- Wang J, Hill WG, Charlesworth D, Charlesworth B. Dynamics of inbreeding depression due to deleterious mutations in small populations: mutation parameters and inbreeding rate. Genet Res (Camb). 1999;74:165–178. doi: 10.1017/s0016672399003900. [DOI] [PubMed] [Google Scholar]
- Wernegreen JJ. Reduced selective constraint in endosymbionts: elevation in radical amino acid replacements occurs genome-wide. PLoS One. 2011;6:e28905. doi: 10.1371/journal.pone.0028905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JBW, Künstner A, Nam K, Jakobsson M, Ellegren H. Nonlinear dynamics of nonsynonymous (dN) and synonymous (dS) substitution rates affects inference of selection. Genome Biol Evol. 2009;1:308–319. doi: 10.1093/gbe/evp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Andolfatto P. The impact of natural selection on the genome: emerging patterns in Drosophila and Arabidopsis. Annu Rev Ecol Evol Syst. 2008;39:193–213. [Google Scholar]
- Wright SI, Kalisz S, Slotte T. Evolutionary consequences of self-fertilization in plants. Proc R Soc B. 2013;280:20130133. doi: 10.1098/rspb.2013.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Ness RW, Foxe JP, Barrett SCH. Genomic consequences of selfing and outcrossing in plants. Int J Plant Sci. 2008;169:105–118. [Google Scholar]
- Yang L, Gaut BS. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol Biol Evol. 2011;28:2359–2369. doi: 10.1093/molbev/msr058. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- Zhang J. Rates of conservative and radical nonsynonymous nucleotide substitutions in mammalian nuclear genes. J Mol Evol. 2000;50:56–68. doi: 10.1007/s002399910007. [DOI] [PubMed] [Google Scholar]
- Zhang Z, et al. KaKs Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics. 2006;4:259–263. doi: 10.1016/S1672-0229(07)60007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zörgö E, et al. Ancient evolutionary trade-offs between yeast ploidy states. PLoS Genet. 2013;9:e1003388. doi: 10.1371/journal.pgen.1003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.