

Abstract
Peroxiredoxin (Prx)II belongs to a family of redox-active proteins that use redox-sensitive cysteine in the active site to reduce peroxides. PrxII is induced by various oxidative stimuli and plays an important protective role against oxidative radical damage by reactive oxygen species. PrxII expression levels are correlated with resistance to radiation therapy or certain anti-cancer drugs in radioresistant breast cancer cells, glioblastomas, and head and neck cancer cells as well as in tissue isolated from head and neck patients who do not respond to radiation therapy. Small interfering RNA (siRNA) that inhibits the PrxII gene expression has been shown to partially reverse the radioresistant phenotype in radiation resistant breast cancer cells and sensitizes glioma cells to oxidative stress, highlighting the potential clinical importance of PrxII in radiation resistance in cancer. This article focuses on the role that PrxII may play in chemoresistant breast cancer cells.
Keywords: siRNA, redox, cysteine disulfide bridges, targeted therapy, reactive oxygen species
Mechanisms of chemoresistance overview
Breast cancer is the leading cause of gynecologic cancer deaths, and >70% of patients in advanced stages develop a recurrence within 5 years.1 Many patients with advanced disease become refractory to conventional chemotherapeutic agents, resulting in recurrence and resistance to the treatment.2 Resistant cancer cells acquire abilities that sensitive cells do not have, including survival mechanisms which enable them to evade eradication of chemotherapeutic agent effects. Despite the development of new therapies, breast cancer is still the second leading cause of death in Western countries. Emerging evidence suggests that cancer stem cells (CSCs) represent the most tumorigenic and chemotherapeutic resistant cells within a heterogeneous tumor mass.3 The population of breast cancer stem cells is increased after chemotherapy.4,5 Although the standard combination of surgery and chemotherapy can effectively reduce tumor mass, most patients with residual breast cancer stem cells eventually acquired chemoresistance. Hence, recurrent cancer is inevitable in the vast majority of cases.6,7 Thus understanding the molecular dysregulation underlying chemoresistance is important for enhancing therapeutic outcome.
One of many mechanisms of tumor cells to survive against anticancer treatments is related to the activation of antioxidative systems including catalase, superoxide dismutase, glutaredoxin and peroxiredoxins. Peroxiredoxin (Prx)II is a member of the peroxiredoxin family in the complex antioxidant networks. It was previously known as a natural killer-enhancing factor B.8 PrxII is highly inducible by therapeutic radiation and drugs in cancer cells. Several resistant tumor cells have shown upregulated PrxII levels including breast cancer, head and neck cancer and glioma cells. PrxII has been known to play an important protective role against oxidative radical damage by reactive oxygen species (ROS).9–11 ROS generation by chemotherapeutic agents is one of the major mechanisms to eliminate cancer cells. However, the level of ROS generation is a determinant for ROS’s role as a cellular toxic component or as a cellular signaling molecule or both. In general, high concentration of ROS that exceed the cellular repair capacity can destroy cellular antioxidant defense systems, impair cellular redox balance and cause damage to DNA, protein, lipids and other cellular components. As a result, high concentration of ROS will lead to cell death. While low concentration of ROS may serve as a signaling molecule to activate cellular repair mechanisms, and turn on cellular proliferation and apoptosis pathways.
Ionizing radiation (IR) and chemotherapeutic agents work to eradicate cancer cells by producing a high level of ROS, recognition of DNA damage, activation of specific kinases, transcription factors and their downstream pathways, and finally execution of cytotoxic programs. An enhanced cellular defense system in cancer cells is one of the important ways for the cells to sustain the cytotoxic effect of the chemotherapeutic agents and exhibit chemoresistant phenotype. There is a group of antioxidant defense systems in cells including thioredoxins (Trxs),12 glutaredoxins (Grxs), superoxide dismutase (SOD) and peroxiredoxins (Prxs).13–17 They are found to be upregulated in many human cancer cells correlating with tumor resistance (Table 1). Other mechanisms of resistance are related to factors including DNA repair, cell cycle status, survival pathways, and altered immunoresponse.18 Resistance cells apply these systems to maintain themselves in a stable condition upon radiation and drug treatment.
Table 1.
Activation of antioxidant systems in drug and radio-resistant tumors
| Sources of oxidative stress | Antioxidant systems | Resistant cells |
|---|---|---|
| Dox | MnSOD | Erythroleukemia |
| Cisplatin, Dox, etoposide, IR | GSH | Lung, breast colon cancer, melanoma, leukemia, brain tumor |
| NO/peroxynitrite | Catalase | Gastric carcinoma cells sarcoma |
| Dox | GSTpi Glutathione transferase Peroxiredoxin II |
Acute myeloid leukemia cells |
| Dox | MnSOD | MCF-7 breast cancer cells |
| IR | PrxII | Head and neck cancer |
| Cisplatin | Trx | Lung cancer cells |
| CDDP | Grx | Pancreatic ductal carcinoma Acute myeloid leukemia cells |
Abbreviations: CDDP, Cis-diamminedichloroplatinum; Dox, doxorubicin; Grx, glutaredoxin; GSH, glutathione; GSTpi, Glutathione S-transferase P1-1; IR, ionizing radiation; MCF-7, Michigan Cancer Foundation-7; MnSOD, mitochondrial superoxide dismutase 2; NO, nitrogen oxide; PrxII, peroxiredoxin II; Trx, thioredoxin.
PrxII has emerged as an antioxidant molecule playing a crucial role in MCF+ fraction ionizing 3 (FIR3) radioresistant cells that are enriched with a subpopulation of breast cancer stem cells. Thus, PrxII plays an important role to reduce the deleterious effect of ROS, restore the cellular redox balance, protect cells from oxidative damage and increase the survival rate of cells. As a consequence, it leads to the resistance to IR or other chemotherapeutic treatments. The role of PrxII in radioresistance of MCF-7 breast cancer cells could be through antioxidant defenses, affecting properties such as redox regulation, proliferative signaling, damage repair and enabling cells to survive with sustained levels of elevated ROS produced by chemotherapeutic treatment. Therefore, alteration in PrxII levels, determination of their mechanisms of action and targeting its modulation by chemical or biological means may lead to new strategies that can be developed to improve radiation therapeutic ratios.
This article focuses on evidence highlighting the role of PrxII as a regulator of elevated ROS production during chemotherapy and its potential as a target for cancer therapy. The paper is structured into three main sections with regard to different aspects of PrxII function. First, we focus on the structure and functions of PrxII and its importance in antioxidant defense. Second, we consider how PrxII activates these processes by antioxidant removal through oxidase activity and altering redox potential in cellular gene expression to bring about a common set of changes in function and activity in cancer cells. And third, we focus on the available evidence that membrane association of PrxII in protecting cells from ROS damage is one of the important mechanisms for cancer chemoresistance. The relevance of these findings, their importance and significance for novel targeted approaches to anti-cancer therapy will be addressed.
The structure and functions of PrxII and its importance in antioxidant defense
Prxs represent a major group of detoxification enzymes that use redox-sensitive cysteine, the active site peroxidatic cysteine (Cys), to reduce peroxides.19,20 Prxs are divided into two subgroups: 2-Cys Prxs and 1-Cys Prxs.21 2-Cys Prx proteins contain both the N-and C-terminal Cys residues and 1-Cys Prx proteins contain only the N-terminal conserved Cys residues. All six Prxs contain at least one essential Cys per 21 to 25 kDa subunit. PrxII shares structural homology with other peroxiredoxins exhibiting a thioredoxin fold and a conserved catalytic Cys residue at the N-terminus of the protein.22 They do not contain metal cofactors or other non-Cys redox centers, but do share the same basic catalytic mechanism, in which an active-site Cys is oxidized to a dimer with a disulfide linkage and sulfenic acid by the peroxide substrate. PrxII isozyme is a typical 2Cys Prx and contains two conserved Cys residues, the peroxidatic Cys (generally near residue 50), and the resolving Cys (near residue 170). The resolving Cys can form chemical bonds with peroxidative Cys through internal and external disulfide bridges.
PrxII like all other members in the family is capable of serving as a peroxidase and involves thioredoxin and/or glutathione as the electron donor. Cys is one of the most rarely used amino acids in the proteins of most organisms studied so far. Therefore, when highly conserved in proteins, it usually plays crucial roles in the structure, function, or regulation of the protein. Cys residues form coordinate compounds with metals in many zinc or iron-sulfur proteins, undergo nucleophilic reaction as the active site of the enzymes and form disulfide bridges with other Cys. This is due to the ability of thiol groups to stabilize protein structures by forming covalent bonds, as well as due to their high reactivity and redox properties.
The formation of excessive amounts of ROS produced by chemotherapeutic reagents, including peroxide (O22−) and superoxide anions (O2−*) poses an oxidation threat to cells when they accumulate to a high concentration. Chronic exposure of MCF-7 breast cancer cells to IR upregulates the PrxII and induces an adaptive response that results in enhanced tolerance to the subsequent cytotoxicity of IR. This upr egulation of PrxII was shown to be one of the means acquired by MCF + FIR3 radioresistant breast cancer cells that MCF + fraction ionizing radiation sensitive 4 (FIS4) sensitive cells do not have. The catalytic mechanism of Prx in ROS scavenging, involved nucleophilic attack by the peroxidatic Cys at the Prx enzyme active site. The resulting Cys sulfenic intermediate can form a dimer with another Prx subunit. The subsequent reduction of the dimer by a cellular reducing agent will regenerate the sulfhydryl group of Cys in the Prx active site (Figure 1). The peroxidatic Cys can also be oxidized to sulfinic acid (SO2H) and sulfonic acid (SO3H) depending upon the severity of oxidative stress.
Figure 1.
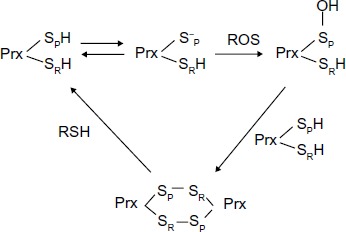
Cysteine oxidation states of peroxiredoxin II in response to oxidative stress.
Abbreviations: Cys, cysteine, Prx, 2-Cys peroxiredoxin; ROS, reactive oxygen species; SpH, peroxidative Cys thiol group; SRH, resolving Cys thiol group in Prx; RSH, sulfhydryl groups.
Like other antioxidant molecules, the role of PrxII in cellular survival is closely associated with the metabolizing and scavenging systems that actively engage in removing ROS. The detrimental effects of ROS can be eliminated. This is one of the survival mechanisms acquired by resistant cells to counteract the toxic effect of ROS, which enable them to evade chemotherapeutic agents’ purge. Down-regulation of PrxII by small interfering (si)RNA in MCF + FIR3 radioresistant breast cancer cells compromises cell viability in several contexts, including lowering intracellular protein thiol and glutathione levels, impairing the cellular membrane Ca2+ pump, increasing cellular toxicity to IR and sensitizing radioresistant MCF + FIR3 cells to IR. By using siRNA to inhibit the PrxII gene expression, PrxII was also shown to be essential in protecting against ROS-induced toxicity in glioma cells and head and neck cancer cells.23 The results clearly implicated the functionality of PrxII antioxidant systems in cancer development and progression.11,23 Chemo- or radiotherapy are still the most commonly used therapeutic methods for the treatment of recurrent tumors. The drawback of the methods are their lack of specificity in targeting cancer cells. Thus, targeting PrxII antioxidant defense systems may be a promising approach to the selective killing of cancer cells. Mechanisms based on combinations of cancer inhibitors with free-radical-producing agents may have clinical applications.24
Peroxide-linked redox signaling
Due to its ability to diffuse over significant distances within and between cells, ROS serves a signaling function in cellular communication.25–27 This dual role of ROS as a potentially damaging compound and as a messenger demands a balanced defense system. ROS generation by chemotherapeutic agents exerts its impact on cellular signaling pathways depending upon concentration levels. At physiological low levels, ROS can function as “redox messengers” in intracellular signaling and regulation, whereas a high level of ROS induces oxidative modification of cellular macromolecules, inhibits protein function, and promotes cell death. In addition to its role as a peroxidase in antioxidant defense, PrxII also serves divergent functions which are associated with only by their toxicity generated by ROS or DNA damaging properties, but also by activating signaling pathways that regulate cellular proliferation, apoptosis and metastasis, making redox-regulated disulfide bond formation an important alternative post-translational control mechanism in addition to the phosphorylation/dephosphorylation mechanism.
Prx proteins also detoxify alkyl hydroperoxides and peroxynitrite, despite the fact that significant differences exist in substrate specificity and kinetic properties. Through this activity Prx is also likely to modulate ROS and reactive nitrogen species (RNS) related signaling. Thus, PrxII serves as an upstream sensor of ROS to regulate the function of its downstream signal proteins in a redox-dependent manner. The sensor function of PrxII is attributed to its ability to react with low levels of ROS and to a major conformational change induced by disulfide formation between two distantly located peroxidative and resolving Cys residues. The switch between two conformations and subsequent relocalization was suggested to have a regulatory function and to mediate redox-dependent signaling.28,29 It is likely that PrxII plays a role in redox-dependent regulation by its association with cellular redox status such as the intracellular levels of glutathione and other reducing reagents and not by catalytic peroxide detoxification. This redox reaction originated from ROS involving cysteine thiol-disulfide exchange is critical for the intracellular monitoring of oxidation status. Changes in the ratio of the reduced and disulfide form in cells can affect signaling pathways that participate in a broad array of physiological responses from cell proliferation, apoptosis, to gene expression that involve ROS as a second messenger.30,31 Redox signaling has been shown to be an important mechanism in cell survival and cell death.32
The mechanisms by which redox status can change the proliferative activity of cancer cells are related to transcriptional and post-transcriptional modifications of proteins that play a critical role in cell cycle control. Binding sites of the redox-regulated transcription factors such as NFκB, AP1, Kelch-like erythroid cell-derived protein1 (Keap1) are located in the promoter region of a large variety of genes that are directly involved in the apoptosis and proliferation of cellular events (Figure 2). Oxidant and antioxidant homeostasis, especially thiol-disulfide balance is critical for many signal events in the adaptation for cells to survive under oxidative stress and conferring chemoresistance of cancer cells.33–36 Modification of proteins through oxidative thiol has long been discussed as a concept for regulating cellular events. In recent years, convincing evidence has shown that redox switches play significant roles in many proteins for regulation of their activities in response to ROS and RNS.37 NFκB is known to be a redox-sensitive transcription factor. It plays an important role in cell signaling transduction pathways through regulation of the gene expression in radioresistant breast cancer cells.38
Figure 2.
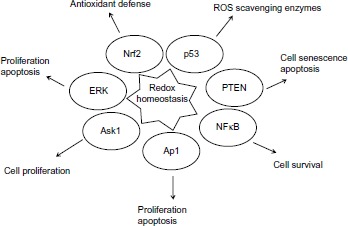
Redox sensitive transcription factors and signaling pathways regulated by redox homeostasis.
Abbreviations: Ask1, apoptosis signaling kinase 1; ERK, extracellular signal-regulated kinase; NFκB, nuclear factor kappalight-chain-enhancer of activated B cells; Nrf2, nuclear factor like 2; PTEN, phosphatase and tensin homologue; ROS, reactive oxygen species.
Cellular redox status can also impact the expression of a number of oncogenes and tumor suppressors. One of those representatives is the p53 tumor suppressor protein. The function of p53 is associated with proliferation, apoptosis, senescence, sensing and repairing damaged DNA. p53 plays a role in facilitating the function of anticancer drugs and its activation is sufficient to induce cell death. Oxidation modification of the cysteine residues in p53 results in inactivation of p53 and renders cells resistant to anticancer treatment.39 In addition, p53 also shows a gain of resistance phenotype, in which tumor cells harboring wild-type p53 were found resistant to a large variety of antitumor agents including IR and several anticancer drugs.40 The mechanisms of resistance involving p53 include molecular defects in post-translational modification, deregulation of downstream signaling pathways, and increased proteasomal degradation. Suppression of tumor suppressor protein phosphatase and tensin homologue (PTEN) function by oxidative conditions increases breast cancer chemotherapeutic resistance.41 Nuclear factor like 2 (Nrf2) is a redox-sensitive transcription factor. Under normal conditions, Nrf2 forms a complex with Keap1. The redox sensor in Keap1 under the oxidative stimuli can turn on the signals for nuclear translocation of Nrf2 and results in activation of the Keap1-Nrf2 pathway. Stable overexpression of Nrf2, a major transcription factor that promotes the synthesis of glutathione, resulted in enhanced resistance of cancer cells to chemotherapeutic agents including cisplatin, DOX (doxorubicin), and etoposide.42 Redox regulation mediated apoptosis signaling kinase 1 (Ask1) multimerization and its reduction, and facilitating DNA binding by binding of reduced thioredoxin to Ask1. The changes in the cellular redox balance have been shown to activate the extracellular signal-regulated kinase (ERK), MAP kinase and c-Jun N-terminal kinase (JNK) proliferation and apoptosis signaling pathways.43,44
In summary, oxidative inactivation of PrxII turned out to be one of the possible ways to interfere with ROS signaling.6,11,23 Activation of downstream genes by the redox-sensitive transcription factors leads to the promotion of cell survival by increasing the capacity to maintain redox homeostasis, inhibiting apoptotic execution, and enhancing DNA repair.28–32 It is clear that ROS and PrxII antioxidant defense systems play a role in modulating cell proliferation and apoptosis through regulation of cellular redox states. MCF + FIR3 radioresistant cells with upregulation of PrxII may be more susceptible to abnormal levels of certain redox-sensitive transcription factors or dysregulation of tumor suppressor proteins that promote cell survival, a cellular adaptation to acquire radioresistance (Figure 3).6,15
Figure 3.
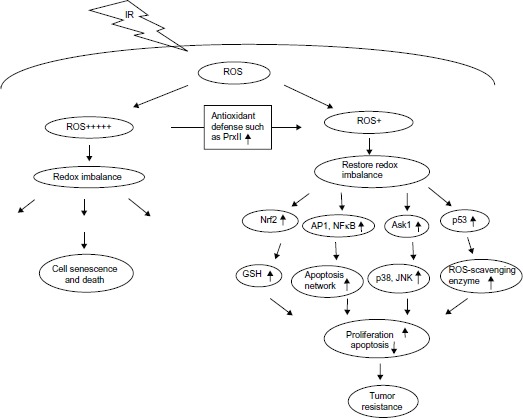
Possible roles of peroxiredoxin II in radiation and drug response through redox signaling.
Abbreviations: AP1, activator protein; Ask1, apoptosis signaling kinase 1; GSH, glutathione; IR, ionizing radiation; JNK, c-Jun N-terminal kinase; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; Nrf2, nuclear factor like 2; PrxII, peroxiredoxin II; ROS, reactive oxygen species.
Notes: + indicates level of ROS; +++++ indicates high level of ROS.
Membrane association of PrxII
Evidence indicates that members of the Prx family can not only scavenge peroxides but can also reduce peroxidized membrane phospholipids, suggesting their role in membrane protection against oxidative stress. Peroxiredoxin VI (PrxVI), a member of the Prx family, has been shown to play an important role in protecting against the damaging effects of ROS, or in a signaling role, by controlling levels of H2O2 in neutrophils. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is composed of multiple components of phagocyte oxidase (phox). Neutrophils provide the first line of defense against microbial invasion through activation of NADPH oxidase. PrxVI binds to phox complex assembled on the plasma membrane to enhance NADPH oxidase activity. During this process, PrxVI translocates to the membrane in stimulated cells and participates in assembly and activation of the NADPH oxidase.45 Possible membrane association of PrxIV was also observed in spermiogenesis. The localization of PrxIV was found to be associated with the membrane of the acrosomal vesicle in the elongated spermatid.46
PrxII was initially characterized as a membrane-associated protein whose reversible binding to the membrane was linked to the regulation of the Ca2+- activated K+ channel in erythrocytes.47 The association of PrxII to the membrane rose with increasing H2O2 concentration and was only observed when the oxidized form of the enzyme was present in the cytosol. PrxII reduces both hydrogen peroxide and organic peroxides, protects the membrane against lipid peroxidation and derives its reducing power from the thioredoxin/thioredoxin reductase and NADP+/NADPH systems. This membrane association of PrxII is believed to be involved in mechanisms of defense against lipid peroxidation.48–50 In a mouse model of sickle cell disease, interaction of PrxII with a membrane was further shown to be modulated during hypoxia-induced acute sickle cell related vaso-occlusive crises.51 Binding of PrxII to the red cell membrane has been reported in patients with hereditary spherocytosis. Progressive association of PrxII to the membrane detected in aged red blood cells (RBCs) suggests a potential RBC oxidative stress marker.52
PrxII is a cytosolic protein and is associated with the membrane, once activated. A previous study has shown that the N-terminal cytoplasmic domain of band 3 protein facilitates the PrxII membrane association to protect the membrane from oxidation damage in red blood cells.53 Band 3 is not only an ion channel but also acts as a membrane anchor site to the red cell skeleton and binds a number of cytoplasmic proteins to mediate the signal transduction of cellular pathways. Prx II was recruited to the membrane in response to oxidative stress. PrxII has also been shown to protect membrane from IR damage in radioresistant breast cancer cells.6 However, how PrxII redox transformations are linked to membrane association and ion transportation is not yet established.
PrxII as a novel target in breast cancer treatment
The pro-oxidant effect of chemotherapeutic agents in connection with activation of cellular defense systems has been part of a continuous effort to unlock the key for cancer resistance mechanisms. PrxII is one of the most overexpressed proteins in radioresistant breast cancer cells and it is upregulated in head and neck cancer cells. It has been shown that silencing PrxII expression by siRNA in resistant breast cancer cells and glioma cells leads to a significant reduction in cell proliferation and radioresistance. Furthermore, the activities and properties of some Prxs are regulated by over-oxidation, phosphorylation and nitrosylation. Despite research progress in structure and functions of PrxII, it is far from being understood comprehensively. Several lines of research suggested exploring further the role of PrxII in cancer chemoresistance; identifying sensitive targets and the signaling pathways involved could provide meaningful and useful information. It is yet to be established that Prxs interact with various target proteins, since change in intracellular PrxII levels will alter the cellular protein expression profile. These screens are not complete yet. Prxs are likely to play an important role in modulating the ROS/RNS regulation networks and the intensity of the systems’ response. All these data implicate PrxII in cancer radioresistance and suggest that PrxII may play an important role in chemoresistance and can be a potential target for therapeutic development.6,8,11,15,18,20,23
In developing approaches for managing resistance, most effort has focused on development of drugs to target the proteins for counter-resistance. Specific modulation of oxidative stress levels or regulation of specific production of certain reactive oxygen species in tumor cells could be a powerful tool to enhance therapeutic outcome. However, it is impossible to cure patients with advanced breast cancer in all cases. One of the reasons is the heterogeneity of breast CSCs, which leads to distinctive sensitivities to the therapy used for the subset of CSCs. It may be useful to use combination therapy by inhibition of the signaling pathway customized to tumor type that needs to be targeted and combined with chemotherapy or radiotherapy. Our recent investigations6 of radiation response of radioresistant breast cancer cells by inhibition of PrxII expression using PrxII-siRNA in MCF + FIR3 radioresistant breast cancer cells suggest that an alternate management strategy may be effective in some cases, for instance, therapeutic IR combined with siRNA that targets certain genes overexpressed in resistant cancer cells. This combination method has shown that it enhances radiation induced cell death by combining the gene targeting property of siRNA and the cytotoxicity of radiation therapy. This type of combination therapy could be the major direction for breast cancer treatment in the future. Next-generation DNA sequencing and mass spectrometry based proteomics would facilitate implementation of personalized medicine. The establishment of comprehensive gene/protein networks from cancer patients could provide more accurate platforms for clinical prognoses.54,55 We hope that this strategy will lead to a new generation of radiotherapy as cancer chemotherapeutics.
Acknowledgments
We would like to thank California State University, Dominguez Hills for the Research, Scholarly and Creative Activity (RSCA) grant support and Dr Robin Roth for proofreading this manuscript.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Breast Cancer Facts and Figures 2012–2013 [webpage on the Internet] Atlanta: American Cancer Society Inc; [Accessed August 9, 2013]. Available from: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-033876.pdf. [Google Scholar]
- 2.Menaa C, Li JJ. The role of radiotherapy-resistant stem cells in breast cancer recurrence. Breast Cancer Manage Journal. 2013;2(2):89–92. doi: 10.2217/bmt.13.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdullah LN, Chow EKH. Mechanisms of chemoresistance in cancer stem cells. Clin Trans Med. 2013;2(2):1–9. doi: 10.1186/2001-1326-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillips TM, McBride WH, Pajonk F. The response of CD24(–/low)/CD44 breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 6.Diaz AJG, Tamae D, Yen Y, Li JJ, Wang T. Enhanced radiation response in MCF-7 radioresistant breast cancer cells by targeting peroxiredoxin II. Breast Cancer. 2013;5:87–101. doi: 10.2147/BCTT.S51378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duru N, Fan M, Candas D, et al. Her2-associated radioresistance of breast cancer stem cells isolated from her2-negative breast cancer cells. Clin Cancer Res. 2012;18:6634–6647. doi: 10.1158/1078-0432.CCR-12-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shau H, Kim A. Identification of natural killer enhancing factor as a major antioxidant in human red blood cells. Biochem Biophys Res Commun. 1994;199(1):83–88. doi: 10.1006/bbrc.1994.1197. [DOI] [PubMed] [Google Scholar]
- 9.Rhee S, Kang SW, Chang TS, Jeong W, Kim K. Peroxiredoxin, a novel family of peroxidase. IUBMB Life. 2001;52(1–2):35–41. doi: 10.1080/15216540252774748. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann B, Hecht HJ, Flohe l. Peroxiredoxins. Biol Chem. 2002;383(3–4):347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 11.Smith-Pearson PS, Kooshki M, Spitz DR, Poole LB, Zhao W, Robbins ME. Decreasing peroxiredoxin II expression decreases glu-tathione, alters cell cycle distribution, and sensitizes glioma cells to ionizing radiation and H(2)O(2) Free Radic Biol Med. 2008;45(8):1178–1189. doi: 10.1016/j.freeradbiomed.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66(8):75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 13.Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780(11):1304–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Eldridge A, Fan M, Woloschak G, Grdina DJ, Chromy BA, Li JJ. Manganese superoxide dismutase interacts with a large scale of cellular and mitochondrial proteins in low-dose radiation-induced adaptive radioprotection. Free Radic Biol Med. 2012;53(10):1838–1847. doi: 10.1016/j.freeradbiomed.2012.08.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang T, Tamae D, LeBon T, Shively JE, Yen Y, Li JJ. The role of peroxiredoxin II in radiation-resistant MCF-7 breast cancer cells. Cancer Res. 2005;65(22):10,338–10,346. doi: 10.1158/0008-5472.CAN-04-4614. [DOI] [PubMed] [Google Scholar]
- 16.Wangpaichitr M, Sullivan EJ, Theodoropoulos G, et al. The relationship of thioredoxin-1 and cisplatin resistance: its impact on ROS and oxidative metabolism in lung cancer cells. Mol Cancer Ther. 2012;11(3):604–615. doi: 10.1158/1535-7163.MCT-11-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Backos DS, Frianklin CC, Reigan P. The role of glutathione in brain tumor drug resistance. Biochem Pharmacol. 2012;83(8):1005–1012. doi: 10.1016/j.bcp.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 18.Yu D, Hung MC, editors. Breast Cancer Chemosensitivity. 608. Landes Bioscience and Springer Science; 2007. (AEMB). [Google Scholar]
- 19.Bae Y, Oh H, Rhee SG, Yoo YD. Regulation of reactive oxygen species generation in cell signaling. Mol Cell. 2011;32(6):491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poynton RA, Hampton MB. Peroxiredoxins as biomarkers of oxidative stress. Biochim Biophys Acta. 2014;1840(2):906–912. doi: 10.1016/j.bbagen.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Wood ZA, Schroder E, Robin Harris J, Pool LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28(1):32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 22.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300(5619):650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 23.Park SH, Chung YM, Lee YS, et al. Antisense of human Peroxiredoxin II enhances radiation-induced cell death. Clin Cancer Res. 2000;6:4915–4920. [PubMed] [Google Scholar]
- 24.Trachootham D, Zhang H, Zhang W, et al. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood. 2008;112(5):1912–1922. doi: 10.1182/blood-2008-04-149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pitzschke A, Forzani C, Hirt H. Reactive oxygen species signaling in plants. Antioxid Redox Signal. 2006;8(9–10):1757–1764. doi: 10.1089/ars.2006.8.1757. [DOI] [PubMed] [Google Scholar]
- 26.Halliwell B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006;141(2):312–322. doi: 10.1104/pp.106.077073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noctor G, Veljovic-Jovanovic S, Foyer CH. Peroxide processing in photosynthesis: antioxidant coupling and redox signalling. Philos Trans R Soc Lond B Biol Sci. 2000;355(1402):1465–1475. doi: 10.1098/rstb.2000.0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konig J, Lotte K, Plessow R, Brockhinke A, Baier M, Dietz KJ. Reaction mechanism of plant 2-Cys peroxiredoxin. Role of the C terminus and the quaternary structure. J Biol Chem. 2003;278(27):24,409–24,420. doi: 10.1074/jbc.M301145200. [DOI] [PubMed] [Google Scholar]
- 29.Dietz KJ, Jacob S, Oelze ML, et al. The function of peroxiredoxins in plant organelle redox metabolism. J Exp Bot. 2006;57(8):1697–1709. doi: 10.1093/jxb/erj160. [DOI] [PubMed] [Google Scholar]
- 30.Xue L, Zhou B, Liu X, et al. Structurally Dependent Redox Property of Ribonucleotide Reductase Subunit p53R2. Cancer Research. 2006;66(4):1900–1905. doi: 10.1158/0008-5472.CAN-05-2656. [DOI] [PubMed] [Google Scholar]
- 31.Shao J, Zhou B, Di Bilio AJ, et al. A Ferrous-triapine complex mediates formation of reactive oxygen species that inactivate human ribonucleotide reductase. Mol Cancer Ther. 2006;5:586–592. doi: 10.1158/1535-7163.MCT-05-0384. [DOI] [PubMed] [Google Scholar]
- 32.Wang K, Fang H, Xiao D, et al. Converting redox signaling to apoptotic activities by stress-responsive regulators HSF1 and NRF2 in fenretinide treated cancer cells. PLoS One. 2009;4(10):e7538. doi: 10.1371/journal.pone.0007538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maillet A, Pervaiz S. Redox regulation of p53, redox effectors regulated by p53: A subtle balance. Antioxidants And Redox Signaling. 2012;16(11):1285–1294. doi: 10.1089/ars.2011.4434. [DOI] [PubMed] [Google Scholar]
- 34.Gloire G, Piette J. Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid Redox Signal. 2009;11(9):2209–2222. doi: 10.1089/ars.2009.2463. [DOI] [PubMed] [Google Scholar]
- 35.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biology. 2013;1(1):45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Yau C, Gray JW, et al. Enhanced NFkB and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer. 2007;7(59):1–15. doi: 10.1186/1471-2407-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brandes N, Schmitt S, Jacob U. Thiol-based Redox Switches in Eukaryotic Proteins. Antioxid Redox Signal. 2008;11(5):997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo G, Wang T, Gao Q, et al. Expression of ErbB2 enhances radiation-induced NF-kappaB activation. Oncogene. 2004;23(2):535–545. doi: 10.1038/sj.onc.1207149. [DOI] [PubMed] [Google Scholar]
- 39.Wosikowski K, Regis JT, Robey RW, et al. Normal p53 status and function despite the development of drug resistance in human breast cancer cells. Cell Growth Differ. 1995;6:1395–1403. [PubMed] [Google Scholar]
- 40.Martinez-Rivera M, Siddik ZH. Resistance and gain-of-resistance phenotypes in cancers harboring wild-type p53. Biochemical Pharmacology. 2012;83:1049–1062. doi: 10.1016/j.bcp.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steelman LS, Navolanic PM, Sokolosky ML, et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene. 2008;27(29):4086–4095. doi: 10.1038/onc.2008.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang XJ, Sun Z, Villeneuve NF, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29(6):1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:1–4. 287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 44.Filomeni G, Auilano K, Rotilio G, Ciriolo MR, et al. Reactive oxygen species-dependent c-Jun NH2-terminal kinase/c-Jun signaling cascade mediates neuroblastoma cell death induced by diallyl disulfide. Cancer Res. 2003;63(18):5940–5949. [PubMed] [Google Scholar]
- 45.Ambruso DR. Peroxiredoxin-6 and NADPH oxidase activity. Methods Enzymol. 2013;527:145–167. doi: 10.1016/B978-0-12-405882-8.00008-8. [DOI] [PubMed] [Google Scholar]
- 46.Sasagawa I, Matsuki S, Suzuki Y, et al. Possible involvement of the membrane-bound form of peroxiredoxin 4 in acrosome formation during spermiogenesis of rats. Eur J Biochem. 2001;268(10):3053–3061. doi: 10.1046/j.1432-1327.2001.02200.x. [DOI] [PubMed] [Google Scholar]
- 47.Moore RB, Mankad MV, Shriver SK, Mankad VN, Plishker GA. Reconstitution of Ca(2+)-dependent K+ transport in erythrocyte membrane vesicles requires a cytoplasmic protein. J Biol Chem. 1991;266(28):18,964–18,968. [PubMed] [Google Scholar]
- 48.Johnson RM, goyetter G, Jr, Ravindranath Y, Ho YS. Oxidation of glutathione peroxidase-deficient red cells by organic peroxides. Blood. 2002;100(4):1515–1516. doi: 10.1182/blood-2002-04-1124. [DOI] [PubMed] [Google Scholar]
- 49.Rocha S, Vitorino RM, Lemos-Amado FM, et al. Presence of cytosolic peroxiredoxin 2 in the erythrocyte membrane of patients with hereditary spherocytosis. Blood Cells Mol Dis. 2008;41(1):5–9. doi: 10.1016/j.bcmd.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Low TY, Seow TK, Chung MC. Separation of human erythrocyte membrane associated proteins with one-dimensional and two-dimensional gel electrophoresis followed by identification with matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Proteomics. 2002;2(9):1229–1239. doi: 10.1002/1615-9861(200209)2:9<1229::AID-PROT1229>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 51.De Franceschi L, Franco RS, Bertoldi C, et al. Pharmacological inhibition of calpain-1 prevents red cell dehydration and reduces Gardos channel activity in a mouse model of sickle cell disease. FASEB. 2013;27(2):750–759. doi: 10.1096/fj.12-217836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rinalducci S, D’Amici GM, Blasi B, Vaglio S, Grazzini G, Zolla L. Peroxiredoxin-2 as a candidate biomarker to test oxidative stress levels of stored red blood cells under blood bank conditions. Transfusion. 2011;51(7):1439–1449. doi: 10.1111/j.1537-2995.2010.03032.x. [DOI] [PubMed] [Google Scholar]
- 53.Matte A, Bertoldi M, Mohanda N, An X, et al. Membrane association of peroxiredoxin 2 in red cells is mediated by the N-terminal cytoplasmic domain of band 3. Free Radic Biol med. 2013;55:27–35. doi: 10.1016/j.freeradbiomed.2012.10.543. [DOI] [PubMed] [Google Scholar]
- 54.Williamson AJ, Whetton AD. The requirement for proteomics to unravel stem cell regulatory mechanisms. J Cell Physiol. 2011;226(10):2478–2483. doi: 10.1002/jcp.22610. [DOI] [PubMed] [Google Scholar]
- 55.Fendler B, Atwal G. Systematic deciphering of cancer genome networks. Yale J Biol Med. 2012;85(3):339–345. [PMC free article] [PubMed] [Google Scholar]