

Abstract
Tobacco smoking is the most common cause of lung cancer, but approximately 10–25% of patients with lung cancer are life-long never smokers. The cause of lung cancer in never smokers is unknown, although tobacco-smoke exposure may play a role in some of these patients. Lung cancer that develops in the absence of significant tobacco-smoke exposure appears to be a unique disease entity with novel genomic and epigenomic alterations and activation of molecular pathways that are not generally seen in tobacco-smoke-induced lung cancer. These molecular alterations are very likely responsible for the unique clinico-pathological features of lung cancer in never smokers (LCINS), and some of these molecular alterations – such as the activating EGFR TK mutations and EML4–ALK fusion – significantly influence therapeutic choices and treatment outcomes. In the last few years there has been a number of studies exploring the molecular characteristics of LCINS, and some of them have reported new and significant findings. Here we review the key findings from these studies and discuss their potential therapeutic implications.
1. Introduction
Globally, over a million patients are diagnosed with lung cancer each year, making it the most common type of cancer in the world [1]. Even though tobacco smoking is considered to be the most common cause of lung cancer, it is estimated that 10–25% of all patients diagnosed with lung cancer are never smokers [2]. Never smokers with lung cancer are more likely to be women, have adenocarcinoma histology and are of East Asian ethnicity when compared to tobacco smokers with lung cancer [3–5] Apart from these now well-established epidemiological differences, recent research has uncovered several key molecular alterations that are more frequently detected in never smokers with lung cancer. Some of these molecular alterations – such as activating mutations in the tyrosine kinase (TK) domain of the epidermal growth factor receptor (EGFR) gene and the EML4–ALK fusion – have therapeutic relevance in the treatment of patients with advanced-stage lung cancer [6–10]. Comprehensive genomic analysis by whole genome sequencing has also identified significant differences between the tumour genome of lung cancer in never smokers (LCINS) and tobacco smokers with lung cancer [11] (Table 1). In this review we will discuss the genomic and epigenomic findings that characterise LCINS.
Table 1.
Characteristic molecular variations in lung cancer in never smokers (LCINS).
| Markers | Lung cancer in tobacco smokers | LCINS | |
|---|---|---|---|
| Genomic changes | Point mutations in protein coding regions | Primarily G → T transversions | Primarily G → A transitions |
| KRAS mutation | Common, 30–43% | Rare, 0–7% | |
| EGFR mutation | Rare, 0–7% | Common, 45% | |
| TP53 mutation; G → T to G → A ratio | Ratio = 1.5 | Ratio = 0.23 | |
| STK11 mutations | 14% | 3% | |
| EML4-ALK fusion | 2–3% | 5–11% | |
| ROS fusion | <1% | 1.5–6% | |
| RET fusion | <1% | 2% | |
| Epigenomic changes | Methylation index (MI) | High MI | Low MI |
| p16 and APC methylation | Common | Rare | |
| Loss of protein expression in hMSH2 | Less common, 10% | More common, 40% |
2. Inherited susceptibility to LCINS
Despite the fact that tobacco smoking is the primary cause of lung cancer, identification of familial clustering of patients with lung cancer is suggestive of an inherited risk factor. Several studies have reported that patients with LCINS are more likely to have a family member diagnosed with lung cancer than a tobacco smoker with the same disease [12–15]. A systematic review of 11 studies identified that a positive family history of lung cancer increases the risk of developing lung cancer by 1.5-fold in never smokers [16]. A linkage study of 52 families with two or more members diagnosed with lung cancer identified the 6q23–25 region to be a major susceptibility locus for lung cancer [17]. In addition, three large genome-wide association studies (GWASs) identified the 15q24–25.1 locus as the site harbouring genetic polymorphisms associated with lung cancer risk [18–20]. However, a pooled analysis of data from all three studies did not find the 15q24–25.1 locus to be associated with increased risk for LCINS [21].
Studies have also examined whether polymorphisms of genes involved in carcinogen metabolism, DNA repair and inflammation are associated with increased risk for developing LCINS. Pooled analysis of studies evaluating CYP1A1 and GSTM1 polymorphisms identified that CYP1A1-I462V polymorphism was associated with two- to three-fold increased risk for developing LCINS. Interestingly, the CYP1A1-I462V polymorphism was associated with increased risk for LCINS only in Caucasians, not in Asians [22]. However, these findings are limited by the small sample size of patients with LCINS in each individual study, and they were focused on a limited number of molecular alterations. Individual studies have shown specific polymorphisms involving DNA repair genes (XRCC1 and ERCC2) and genes involved in interleukin production (IL1, IL6 and IL10) to be associated with increased risk for LCINS [23–25]. These studies are limited by their relatively small sample size and require independent validation to ascertain that these polymorphisms are associated with increased risk for LCINS.
3. Markers of tobacco exposure
Significant differences have been reported in the frequency and patterns of gene mutations between LCINS and lung cancer in tobacco smokers (reviewed in [26]). Some of the earliest studies identified that mutations in the tumour suppressor gene TP53 were less frequent in LCINS (8–47%) when compared with tobacco smokers with lung cancer (26–71%) [27–29]. Also a significant dose–response relationship between tobacco smoke and TP53 mutations has been reported in patients with non-small-cell lung cancer (NSCLC) [27]. In a sample of 30 resected NSCLC tumor samples the odds of having TP53 mutations in a patient smoking 20 cigarettes per day for 30 years were 5.3 when compared with a patient with LCINS. Tobacco-smoke exposure was also associated with a distinct mutational spectrum in the TP53 gene, with increased frequency of G → T transversion mutations when compared to LCINS [30,31].
Mutations involving the KRAS oncogene are rare in patients with LCINS and are more frequently reported in tobacco smokers with lung cancer [32–36]. In a sample of 106 patients with adenocarcinoma, the incidence of KRAS mutations was significantly higher in the smokers cohort versus the never smokers (43% versus 0%, P = 0.001) [35]. Similarly KRAS mutations are more frequently identified in tobacco smokers and are predominantly G → T transversion mutations [31].
4. Fusions and mutations involving kinase genes
Analyses of tumor samples from patients with excellent response to treatment with EGFR TK inhibitors led to the discovery of activating mutations involving the EGFR TK gene [6,7]. At around the same time it was also discovered that patients with LCINS had a better response to EGFR TK inhibitors such as gefitinib [37]. Several retrospective studies subsequently established that patients with LCINS were more likely to harbour the EGFR TK mutation than tobacco smokers with lung cancer [8,38,39]. One of the largest studies (n = 1082) confirmed that activating EGFR TK mutations were more frequent in patients with LCINS than in tobacco smokers with lung cancer: 54% versus 16% [40]. The higher incidence of EGFR TK mutations in LCINS has been a consistent finding across different ethnic and geographical divisions. In addition, the frequency of EGFR TK mutations is inversely related to tobacco-smoke exposure. The proportion of EGFR TK mutations in patients with less than 20 pack year exposure was 55% versus 27% for 20–50 pack years and 22% for >50 pack years (P < 0.001) [38]. Pham and colleagues reported similar findings: decreasing incidence of EGFR TK mutations with increasing pack years [39]. The difference was significant when exposure was >15 pack years (9%) versus never smokers (51%); P < 0.005. In addition, EGFR TK mutations were not detected in tobacco smokers with more than 75 pack year exposure.
The EGFR TK inhibitor erlotinib was initially approved for the treatment of all patients with advanced NSCLC in the second- and third-line settings. The discovery of activating EGFR TK mutations led to several randomised trials comparing EGFR TK inhibitors with chemotherapy in the front-line setting in patients with EGFR TK mutations [41–43]. Results from these trials have now established EGFR TK inhibitors as the standard front-line treatment for patients with advanced-stage NSCLC that is positive for EGFR TK mutation.
Mutations involving the HER2 gene have been shown to be more frequent in never smokers with adenocarcinoma [9]. In a sample of 671 NSCLC tumours, the overall frequency of HER2 mutations was low at 1.6% (11/671), but they were more frequently identified in never or light smokers (8 of 248, 3.2%; P = 0.02). The HER2 mutations were not detected in tumours harbouring either the activating EGFR-TK or KRAS mutations.
The STK11 gene encodes a serine–threonine kinase and plays an important role in cell proliferation and survival. Mutations involving the STK11 gene have been reported in 8% of all patients with lung cancer. In addition, they are more frequently present in tobacco smokers with lung cancer than in patients with LCINS (14% versus 3%; P = 0.007) [44].
EML4–ALK is a novel fusion gene present in approximately 5% of patients with NSCLC and is associated with an excellent therapeutic response to treatment with an Alk kinase inhibitor [10,45,46]. The fusion gene was more frequently identified in never smokers and younger patients with lung cancer. In addition, it appears to be mutually exclusive to EGFR TK and KRAS mutations.
Two new transforming fusions involving the RET and ROS1 kinase genes at the 3′ end have been identified in patients with lung cancer [47]. In one study, tumour samples from 936 patients with surgically resected NSCLC were tested for RET fusion genes by the reverse transcriptase polymerase chain reaction (PCR). The RET fusion was detected in 13 patients (1.4%), and these patients predominantly had adenocarcinoma histology (84.6%), were never smokers (82%) and many of them were younger: age ⩽ 60 years at the time of diagnosis (73%) [48]. RET fusions have been shown to promote cell proliferation, and treatment with vandetanib, a multi kinase inhibitor with activity against RET kinase, was able to inhibit RET-induced cell proliferation [47]. Fusions involving the ROS1 gene in lung cancer were first reported in 2007 [49] and in a subsequent study, a fluorescent in situ hybridisation (FISH) based assay of 1000 NSCLC tumour samples identified ROS1 fusions in 18 (1.7%) samples [50]. Similar to patients with ALK or RET fusions, ROS fusions were found primarily in younger patients who were never smokers and had adenocarcinoma histology. Cell lines expressing ROS fusion were sensitive to treatment with the ALK inhibitor crizotinib. Overall, fusion genes involving the ALK, RET and ROS kinases are relatively rare molecular events in patients with NSCLC. These patients have similar clinico-pathological features, including that of being a never smoker. In addition, these fusions appear to be mutually exclusive to each other and to other known driver mutations in lung cancer, such as EGFR TK and KRAS mutations.
5. Epigenetic alterations
Methylation of tumour suppressor genes – including p16INK4a, DAPK, RASSF1A, RARβ, APC, CDH13, MGMT, hMLH1, hMSH2 and GSTP1 – leading to epigenetic silencing has been reported in lung cancer (reviewed in [51,52]). Studies have reported that methylation of the tumour suppressor gene p16 is less frequent in LCINS in comparison to lung cancer in tobacco smokers [53–57]. In a sample of 514 NSCLC tumours, which included 112 never smokers with adenocarcinoma, p16 (P = 0.007) and APC (P = 0.0007) methylation rates were significantly lower in never smokers than tobacco smokers with adenocarcinoma [54]. There was no significant difference in the methylation rate of the other tumour suppressor genes RASSF1A, RARβ, CDH13, MGMT and GSTP1 between the two groups. The methylation index (total number of genes methylated/total number of genes examined) was significantly higher in tobacco smokers with lung cancer when compared to LCINS. In a follow-up study of 383 NSCLC tumours, the authors confirmed that the p16 methylation rate and the methylation index were significantly lower in LCINS (P < 0.0001) [55]. The methylation rate for APC was significantly lower (P < 0.0001) in never smokers when the analysis was restricted to adenocarcinoma. Subsequent studies have also reported a low p16 methylation rate in never smokers with adenocarcinoma [56,58]. There was no significant difference in the methylation rates of RASSF1A and DAPK between tobacco smokers with lung cancer and LCINS [56].
The loss of protein expression in protein mismatch repair genes hMLH1 and hMSH2 was reported to be more frequent in LCINS than in lung cancer in tobacco smokers [59]. In a sample of 77 resected NSCLC tumours, the loss of protein expression for hMLH1 (70% versus 46%) and hMSH2 (40% versus 10%) was more frequent in LCINS. The authors also reported that promoter methylation was the predominant mechanism for the loss of protein expression in both genes.
6. Next-generation sequencing in LCINS
The advent of next-generation sequencing technologies now allows us unprecedented access to the tumour genome. Recently, next-generation sequencing of several tumour–normal pairs from patients with NSCLC was reported, and some of these patients were never smokers. Whole genome and transcriptome sequencing was performed in 17 patients with NSCLC, including five never smokers and 12 tobacco smokers [11]. The total number of mutations involving genes in protein coding regions was significantly higher in smokers than in never smokers; median 209 versus 18. In addition, the mutations in tobacco smokers were primarily G → T transversions, whereas in LCINS they were G → A transitions. For the first time this study identified that the G → A transition point mutations in never smokers is a genome-wide phenomenon and is not restricted to KRAS and TP53 genes.
Genomic and epigenomic profiling of tumour–normal pairs from six Korean patients with LCINS with exome seq, RNA seq, micro RNA seq and methylated DNA immunoprecipitation-sequencing (MeDIP-seq) confirmed the low mutation rate in LCINS [60]. They reported a total of 47 somatic mutations from the six LCINS tumour samples. In addition, they identified several novel fusion genes, including CCDC6–RET fusion which has been previously reported and could be a potential therapeutic target. Pathway analysis identified that genes involved in cell cycle regulation – particularly in G2/M transition – are very likely to have played a significant role in the development of these tumours.
7. Conclusion
Cancer is a disease that is characterised by genomic and epigenomic alterations that result in malignant transformation of normal tissue. Such transforming genomic and epigenomic alterations are considered the drivers of the malignant disease and determine the clinical behaviour of the disease. In the case of lung cancer, tobacco-smoke exposure appears to be an important factor in determining the type of oncogenic drivers associated with the disease. This is well exemplified by findings from several studies showing that mutations involving TP53 and KRAS genes are more frequent in tobacco smokers with lung cancer, whereas LCINS is characterised by EGFR TK mutations, ALK, RET and ROS fusions. The differences between LCINS and lung cancer in tobacco smokers are not restricted to a few genes. Recent next-generation sequencing studies have found that the genome of LCINS is significantly different from the tumour genome of a tobacco smoker with lung cancer (Fig. 1). Overall, the number of mutations is significantly lower in LCINS, and the point mutations are primarily G → A transitions.
Fig. 1.
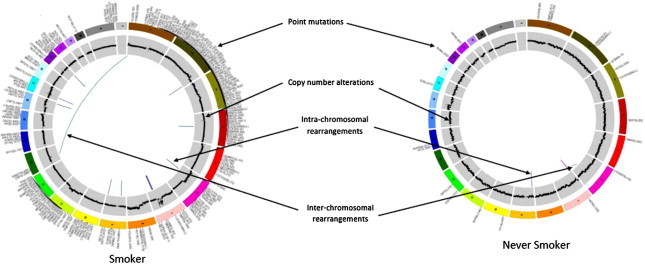
Circos plots of tumour genome from a never smoker with lung cancer and a tobacco smoker with lung cancer. Adapted from Govindan et al [11].
The higher number of genomic alterations seen in smokers with lung cancer is very likely due to the mutagenic field effect of tobacco-smoke exposure. The vast majority of these genomic alterations in tobacco smokers with lung cancer are believed to be passengers that do not have any role in the malignant transformation or progression. In contrast, in LCINS the absence of tobacco-smoke exposure and the relatively smaller number of identified genomic alterations suggest that most if not all of them play a role in its malignant transformation. Hence the LCINS genome may provide us with a relatively enriched and easily identifiable set of oncogenic drivers for lung cancer. In addition, the relatively small number of genomic alterations in LCINS also presents better opportunities for the development of targeted therapies against LCINS. With the advances in sequencing technology and decreasing costs it is possible that, in the near future, advanced-stage LCINS may be primarily treated with molecularly targeted therapy, and it would be possible to achieve prolonged periods of disease control similar to the treatment of chronic myeloid leukaemia (CML) and gastrointestinal stromal tumour (GIST).
Conflict of interest statement
The author is not a government employee. For the last 2 years, he has been a consultant for Pfizer, Roche Genentech, Bristol-Myers Squibb, Merck, Boehringer-Ingelheim, Abbott Oncology and Covidien.
References
- 1.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J., Shin H.R., Bray F. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2011;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 3.Nordquist L.T., Simon G.R., Cantor A., Alberts W.M., Bepler G. Improved survival in never-smokers vs current smokers with primary adenocarcinoma of the lung. Chest. 2004;126:347–351. doi: 10.1378/chest.126.2.347. [DOI] [PubMed] [Google Scholar]
- 4.Subramanian J., Velcheti V., Gao F., Govindan R. Presentation and stage-specific outcomes of lifelong never-smokers with non-small cell lung cancer (NSCLC) J Thorac Oncol. 2007;2:827–830. doi: 10.1097/JTO.0b013e318145af79. [DOI] [PubMed] [Google Scholar]
- 5.Toh C.K., Gao F., Lim W.T. Never-smokers with lung cancer: epidemiologic evidence of a distinct disease entity. J Clin Oncol. 2006;24:2245–2251. doi: 10.1200/JCO.2005.04.8033. [DOI] [PubMed] [Google Scholar]
- 6.Lynch T.J., Bell D.W., Sordella R. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 7.Paez J.G., Janne P.A., Lee J.C. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 8.Shigematsu H., Lin L., Takahashi T. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 9.Shigematsu H., Takahashi T., Nomura M. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642–1646. doi: 10.1158/0008-5472.CAN-04-4235. [DOI] [PubMed] [Google Scholar]
- 10.Soda M., Choi Y.L., Enomoto M. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 11.Govindan R., Ding L., Griffith M. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tokuhata G.K., Lilienfeld A.M. Familial aggregation of lung cancer in humans. J Natl Cancer Inst. 1963;30:289–312. [PubMed] [Google Scholar]
- 13.Kreuzer M., Heinrich J., Kreienbrock L. Risk factors for lung cancer among nonsmoking women. Int J Cancer. 2002;100:706–713. doi: 10.1002/ijc.10549. [DOI] [PubMed] [Google Scholar]
- 14.Wu A.H., Fontham E.T., Reynolds P. Family history of cancer and risk of lung cancer among lifetime nonsmoking women in the United States. Am J Epidemiol. 1996;143:535–542. doi: 10.1093/oxfordjournals.aje.a008783. [DOI] [PubMed] [Google Scholar]
- 15.Brownson R.C., Alavanja M.C., Caporaso N., Berger E., Chang J.C. Family history of cancer and risk of lung cancer in lifetime non-smokers and long-term ex-smokers. Int J Epidemiol. 1997;26:256–263. doi: 10.1093/ije/26.2.256. [DOI] [PubMed] [Google Scholar]
- 16.Matakidou A., Eisen T., Houlston R.S. Systematic review of the relationship between family history and lung cancer risk. Br J Cancer. 2005;93:825–833. doi: 10.1038/sj.bjc.6602769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey-Wilson J.E., Amos C.I., Pinney S.M. A major lung cancer susceptibility locus maps to chromosome 6q23-25. Am J Hum Genet. 2004;75:460–474. doi: 10.1086/423857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amos C.I., Wu X., Broderick P. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hung R.J., McKay J.D., Gaborieau V. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. doi: 10.1038/nature06885. [DOI] [PubMed] [Google Scholar]
- 20.Thorgeirsson T.E., Geller F., Sulem P. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spitz M.R., Amos C.I., Dong Q., Lin J., Wu X. The CHRNA5-A3 region on chromosome 15q24-25.1 is a risk factor both for nicotine dependence and for lung cancer. J Natl Cancer Inst. 2008;100:1552–1556. doi: 10.1093/jnci/djn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hung R.J., Boffetta P., Brockmoller J. CYP1A1 and GSTM1 genetic polymorphisms and lung cancer risk in Caucasian non-smokers: a pooled analysis. Carcinogenesis. 2003;24:875–882. doi: 10.1093/carcin/bgg026. [DOI] [PubMed] [Google Scholar]
- 23.Zhou W., Liu G., Miller D.P. Polymorphisms in the DNA repair genes XRCC1 and ERCC2, smoking, and lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2003;12:359–365. [PubMed] [Google Scholar]
- 24.Kiyohara C., Takayama K., Nakanishi Y. Lung cancer risk and genetic polymorphisms in DNA repair pathways: a meta-analysis. J Nucleic Acids. 2010;2010:701760. doi: 10.4061/2010/701760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian B., Zhang H., Zhang L., Zhou X., Yu H., Chen K. Association of genetic polymorphisms in DNA repair pathway genes with non-small cell lung cancer risk. Lung Cancer. 2011;73:138–146. doi: 10.1016/j.lungcan.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 26.Subramanian J., Govindan R. Molecular genetics of lung cancer in people who have never smoked. Lancet Oncol. 2008;9:676–682. doi: 10.1016/S1470-2045(08)70174-8. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki H., Takahashi T., Kuroishi T. P53 mutations in non-small cell lung cancer in Japan: association between mutations and smoking. Cancer Res. 1992;52:734–736. [PubMed] [Google Scholar]
- 28.Hainaut P., Pfeifer G.P. Patterns of p53 G{->}T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22:367–374. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 29.Husgafvel-Pursiainen K., Boffetta P., Kannio A. P53 mutations and exposure to environmental tobacco smoke in a multicenter study on lung cancer. Cancer Res. 2000;60:2906–2911. [PubMed] [Google Scholar]
- 30.Toyooka S.T.T., Gazdar Adi F. The TP53 gene, tobacco exposure, and lung cancer. Hum Mutat. 2003;21:229–239. doi: 10.1002/humu.10177. [DOI] [PubMed] [Google Scholar]
- 31.Vahakangas K.H., Bennett W.P., Castren K. P53 and K-ras Mutations in lung cancers from former and never-smoking women. Cancer Res. 2001;61:4350–4356. [PubMed] [Google Scholar]
- 32.Slebos R.J.H.R., Dalesio O., Mooi W.J., Offerhaus G.J., Rodenhuis S. Relationship between K-ras oncogene activation and smoking in adenocarcinoma of the human lung. J Natl Cancer Inst. 1991;83:1024–1027. doi: 10.1093/jnci/83.14.1024. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y.C., Lee H.S., Chen S.K., Yang S.C., Chen C.Y. Analysis of K-ras gene mutations in lung carcinomas: correlation with gender, histological subtypes, and clinical outcome. J Cancer Res Clin Oncol. 1998;124:517–522. doi: 10.1007/s004320050208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gealy R., Zhang L., Siegfried J.M., Luketich J.D., Keohavong P. Comparison of mutations in the p53 and K-ras genes in lung carcinomas from smoking and nonsmoking women 1. Cancer Epidemiol Biomarkers Prev. 1999;8:297–302. [PubMed] [Google Scholar]
- 35.Ahrendt S.A., Decker P.A., Alawi E.A. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92:1525–1530. doi: 10.1002/1097-0142(20010915)92:6<1525::aid-cncr1478>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 36.Le Calvez F., Mukeria A., Hunt J.D. TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res. 2005;65:5076–5083. doi: 10.1158/0008-5472.CAN-05-0551. [DOI] [PubMed] [Google Scholar]
- 37.Miller V.A., Kris M.G., Shah N. Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non-small-cell lung cancer. J Clin Oncol. 2004;22:1103–1109. doi: 10.1200/JCO.2004.08.158. [DOI] [PubMed] [Google Scholar]
- 38.Kosaka T., Yatabe Y., Endoh H. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919–8923. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 39.Pham D., Kris M.G., Riely G.J. Use of cigarette-smoking history to estimate the likelihood of mutations in epidermal growth factor receptor gene exons 19 and 21 in lung adenocarcinomas. J Clin Oncol. 2006;24:1700–1704. doi: 10.1200/JCO.2005.04.3224. [DOI] [PubMed] [Google Scholar]
- 40.Shigematsu H., Gazdar A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:257–262. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- 41.Mok T.S., Wu Y.L., Thongprasert S. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 42.Maemondo M., Inoue A., Kobayashi K. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 43.Rosell R., Carcereny E., Gervais R. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 44.Koivunen J.P., Kim J., Lee J. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br J Cancer. 2008;99:245–252. doi: 10.1038/sj.bjc.6604469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw A.T., Yeap B.Y., Mino-Kenudson M. Clinical features and outcome of patients with non–small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwak E.L., Bang Y.-J., Camidge D.R. Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeuchi K., Soda M., Togashi Y. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 48.Wang R., Hu H., Pan Y. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012;30:4352–4359. doi: 10.1200/JCO.2012.44.1477. [DOI] [PubMed] [Google Scholar]
- 49.Rikova K., Guo A., Zeng Q. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 50.Bergethon K., Shaw A.T., Ignatius Ou S.-H. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–870. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Digel W., Lubbert M. DNA methylation disturbances as novel therapeutic target in lung cancer: preclinical and clinical results. Crit Rev Oncol Hematol. 2005;55:1–11. doi: 10.1016/j.critrevonc.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 52.Bowman R.V., Yang I.A., Semmler A.B., Fong K.M. Epigenetics of lung cancer. Respirology. 2006;11:355–365. doi: 10.1111/j.1440-1843.2006.00859.x. [DOI] [PubMed] [Google Scholar]
- 53.Kim D.H., Nelson H.H., Wiencke J.K. P16(INK4a) and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res. 2001;61:3419–3424. [PubMed] [Google Scholar]
- 54.Toyooka S., Maruyama R., Toyooka K.O. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. Int J Cancer. 2003;103:153–160. doi: 10.1002/ijc.10787. [DOI] [PubMed] [Google Scholar]
- 55.Toyooka S., Suzuki M., Tsuda T. Dose effect of smoking on aberrant methylation in non-small cell lung cancers. Int J Cancer. 2004;110:462–464. doi: 10.1002/ijc.20125. [DOI] [PubMed] [Google Scholar]
- 56.Divine K.K.P.L., Marron-Terada P.G., Liechty K.C. Multiplicity of abnormal promoter methylation in lung adenocarcinomas from smokers and never smokers. Int J Cancer. 2005;114:400–405. doi: 10.1002/ijc.20761. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y., Lan Q., Siegfried J.M., Luketich J.D., Keohavong P. Aberrant promoter methylation of p16 and MGMT genes in lung tumors from smoking and never-smoking lung cancer patients. Neoplasia. 2006;8:46–51. doi: 10.1593/neo.05586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scesnaite A., Jarmalaite S., Mutanen P. Similar DNA methylation pattern in lung tumours from smokers and never-smokers with second-hand tobacco smoke exposure. Mutagenesis. 2012;27:423–429. doi: 10.1093/mutage/ger092. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y.-C., Lu Y.-P., Tseng R.-C. Inactivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched sputum samples. J Clin Invest. 2003;111:887–895. doi: 10.1172/JCI15475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim S.C., Jung Y., Park J. A high-dimensional, deep-sequencing study of lung adenocarcinoma in female never-smokers. PLoS One. 2013;8:e55596. doi: 10.1371/journal.pone.0055596. [DOI] [PMC free article] [PubMed] [Google Scholar]