

Abstract
B cell development involves rapid cellular proliferation, gene rearrangements, selection and differentiation, and provides a powerful model to study DNA repair processes in vivo. Analysis of the contribution of the base excision repair (BER) pathway in lymphocyte development has been lacking primarily due to the essential nature of this repair pathway. However, mice deficient for the BER enzyme, apurinic/apyrimidinic (AP) endonuclease 2 (APE2) protein develop relatively normally, but display defects in lymphopoiesis. Here we present an extensive analysis of bone marrow hematopoiesis in mice nullizygous for APE2 and find an inhibition of the pro-B to pre-B cell transition. We find that APE2 is not required for V(D)J-recombination, and that the turnover rate of APE2-deficient progenitor B cells is nearly normal. However, the production rate of pro- and pre-B cells is reduced due to a p53-dependent DNA damage response. FACS-purified progenitors from APE2-deficient mice differentiate normally in response to IL-7 in in vitro stromal cell co-cultures, but pro-B cells show defective expansion. Interestingly, APE2-deficient mice show a delay in recovery of B lymphocyte progenitors following bone marrow depletion by 5-fluorouracil, with the pro-B and pre-B cell pools still markedly decreased two weeks after a single treatment. Our data demonstrate that APE2 has an important role in providing protection from DNA damage during lymphoid development, which is independent from its ubiquitous and essential homolog APE1.
Introduction
The base excision repair (BER) pathway is one of the major cellular defenses against oxidative damage to DNA, and is essential to protect the genomic stability of all cells (1, 2). However, protection from reactive oxygen species (ROS) may be especially important for the expansion of hematopoietic cells, since the generation of new blood cells (either to maintain daily numbers or in response to infection or bone marrow injury) involves many rounds of rapid proliferation and differentiation, and the associated metabolic activity generates intracellular ROS. Developing lymphocytes require highly efficient DNA repair mechanisms not only because they undergo several rounds of replication, but also because DNA repair is essential for the recombination of immunoglobulin genes (3). Cells that have succeeded at V(D)J-recombination are positively selected based on the integrity of the expressed immunoglobulin or T cell receptor encoded by the DNA rearrangement. This makes lymphopoiesis a powerful model to study DNA repair processes in vivo (4). For instance, the non-homologous end joining (NHEJ) repair pathway has been extensively studied in lymphoid cells (5), as deficiency for most of the NHEJ constituents almost uniformly results in failure of lymphocyte development and immunodeficiencies in both humans and mice (6).
BER is essential for both cell viability and embryogenesis. In somatic cells, BER is responsible for the repair of over 10,000 lesions per cell per day (1, 2). DNA bases damaged by oxidation are removed by glycosylases, generating an abasic site that is recognized by AP-endonuclease (APE). APE cleaves the DNA backbone, allowing DNA Polβ to insert a new nucleotide, after which the nick is sealed by DNA ligase. This repair pathway is essential during development, and deficiency in APE1, Polβ or Ligase 1 causes embryonic lethality in mice (7-9). This has made the role of BER in vivo difficult to examine.
Very little is known about APE2-mediated DNA repair processes, as opposed to the well-studied homolog APE1, which is ubiquitously expressed and highly efficient at cleaving abasic sites. APE1 and APE2 have nearly identical active sites, but in contrast to APE1, the AP endonuclease activity of APE2 is very inefficient (10). On the contrary, APE2 has very efficient 3′ to 5′ exonuclease activity and 3′-phosphodiesterase activities, while APE1 is weaker at these activities (11-13). Recent biochemical experiments are consistent with a role for APE2 in the repair of oxidative damage to DNA, although this has not been demonstrated in vivo. The exonuclease activity of APE2 can remove mismatched bases (12), and preferentially removes A opposite the oxidatively damaged and highly mutagenic base 8-oxoG (13). Replicative polymerases insert A opposite 8-oxoG, creating G:C to A:T transversions. The addition of PCNA, shown to interact with APE2, enhances this activity, and treatment of cells with H2O2 causes redistribution of APE2 into discrete nuclear foci and co-localization of APE2 with PCNA.
In addition to its exo activity, APE2 has efficient 3′ phosphodiesterase activity (10, 13). DNA strand breaks with 3′-phosphoglycolate termini can be generated from attack by ROS (14), and the 3′ phosphodiesterase activity of APE2 that removes these blocking groups is much more efficient than its endonuclease activity, and more efficient than the 3′ phosphodiesterase activity of APE1 (10, 12). Similarities between APE2 and the L. major AP endonuclease, LMAP, in an amino acid residue shown to impart increased 3′ phosphodiesterase activity also suggest the importance of this enzymatic activity in protection from oxidative damage since LMAP, but not APE1, was able to revert the hypersentitivity of xth nfo repair-deficient E. coli to H202 (15). All combined, these data suggest that, while APE1 is the essential AP-endonuclease, the enhanced exonuclease and 3′ phosphodiesterase activities of APE2 may provide cells with additional protection from oxidative damage.
APE1 is known to be essential to repair the tens of thousands of abasic sites generated in cells per day by glycosylase activity and spontaneous base hydrolysis, but the importance of APE2 in vivo is not clear. In a previous study it was shown that mice deficient in APE2 display thymic atrophy and have reduced numbers of peripheral blood B and T cells (16), but the reason for this defect is unknown. As oxidative DNA damage by ROS is associated with aging phenotypes and cancer (1, 17, 18), it is important to understand how this damage is repaired at the biochemical level as well as which processes in vivo are most sensitive to this type of damage.
APE2-deficiency provides a unique model in that it allows us to study the impact of physiologically-acquired oxidative damage during early hematopoiesis and subsequent stages of B-cell development. We analyzed B cell development in the bone marrow and found that APE2-deficiency results in a partial block in the generation of pre-B cells, which is not related to defective V(D)J recombination. Our results are consistent with a loss of pre-B cells due to a p53-dependent DNA damage response. Furthermore, we demonstrate that APE2 is necessary for the rapid recovery of lymphoid progenitors following chemotherapeutic depletion of the bone marrow. Our data demonstrate that APE2 is an important enzyme for the maintenance and regeneration of B cell precursor pools.
Materials and Methods
Mice
All mouse strains were backcrossed to C57BL/6 for more than 8 generations. apex1+/−mice were obtained from E. Friedberg (19) (University of Texas Southwestern Medical Center, Dallas, TX). apex2+/− mice (20) and H2-SVEX transgenic mice (21) were previously described. p53 +/− mice were obtained from Stephen Jones, and were previously described (22). (apex2+/− and p53+/−) and (apex2+/− and apex1+/− ) mice were crossed to create doubly-deficient mice or apex1+/− apex2Y/− mice. Because apex2 is on the X chromosome, we used male apex2Y/− mice and apex2Y/+ littermates in all experiments. B6.SJL mice originally purchased from Jackson Labs (Bar Harbor, ME) were bred in our facility. Mice were housed in the Institutional Animal Care and Use Committee-approved specific pathogen-free facility at the University of Massachusetts Medical School. The mice were bred and used according to the guidelines from the University of Massachusetts Animal Care and Use Committee.
Flow cytometry
For FACS analysis, bone marrow cells were flushed from the tibias and femurs of the hind leg, filtered through 70 μ nylon mesh and re-suspended in staining media containing biotin-, flavin-, and phenol red-deficient RPMI 1640 (Irvine Scientific), 10 mM HEPES, pH 7.2, 0.02% sodium azide, 1 mM EDTA, and 2% fetal bovine serum. Cells were incubated with anti-CD16/32 antibody (eBioscience) for 10 min. on ice to block Fc receptors, primary antibodies for 30 min., washed before the addition of streptavidin-pacific blue (Invitrogen) secondary reagent for 15 min., and then washed three more times. After the final wash, cells were resuspended in 1 ug/ml propidium iodide to exclude dead cells. Primary antibodies included CD93-Cy7PE (AA4.1), B220-APC (RA3-6B2), CD19-PE-TR (6D5), CD43-PE (S7), IgM-biotin or -FITC (II/41), CD49bbiotin or -FITC (DX5), Ly6C-biotin or -FITC (AL-21), Sca-1-FITC (E13-161.7), c-kit-Cy5.5-PE (2B8), CD135-PE (Flt3), CD127-APC (A7R34), and biotin-labeled CD3 (145-2C11), TER119, GR-1 (8C5), and CD11b/Mac-1 (M1/70). Antibodies were purchased from BD Biosciences, eBioscience, or Invitrogen. All cell populations were gated on viable cells by propidium iodide exclusion and singlets by forward scatter area and height. Multi-potent progenitors (MPP) and a population highly enriched in hematopoietic stem cells, hereafter referred to as hematopoietic stem and progenitor cells (HSPC), were identified as Lineage-negative (Lin−: IgM−, B220−, CD19−, CD3−, TER119−, GR-1−, CD11b−) and CD127-negative (IL-7Rα−), c-kit+ and Sca-1+ (LSK cells) that are flt3+ (MPP) and flt3− (HSPC). Common lymphoid progenitors (CLP) were gated as Lin−CD127+AA4.1+Sca1+/LOflt3+ckitINT. Other bone marrow subsets were defined as follows: pre-pro B cells, IgM−Ly6C−Dx5−B220+CD43+CD19−AA4.1+; pro-B cells, IgM−Ly6CDx5−B220+CD43+CD19+AA4.1+; pre-B cells, IgM−Ly6C−Dx5−B220+CD43−/LOCD19+AA4.1+; immature B cells, Ly6C−Dx5−B220+IgM+CD43−/LOCD19+AA4.1+ and recirculating B cells, Ly6C−Dx5−B220+IgM+CD43−/LOCD19+AA4.1−. Vex was detected using 407-nM excitation. Flow cytometry was performed on a 3 laser, 9 detector LSRII (Becton Dickinson). Data were analyzed with FlowJo software (Tree Star).
Bone marrow transfers
2×106 bone marrow cells from WT or APE2-deficient mice (C57BL/6, CD45.2+) were transferred i.v. into B6.SJL (CD45.1+) recipients that had been sub-lethally irradiated (650 rad) 24 h previously. Recipient mice were maintained on antibiotics and reconstitution of their bone marrow was analyzed by flow cytometry 7 weeks later, as described above, with the addition of anti-CD45.2-FITC (ebioscience). Pro-B and pre-B cells are defined as in Fig. 1, here and throughout the manuscript. The percent of CD45.2+ donor cells in the bone marrow was 63.8 +/− 3.1% in mice that received WT (n = 3 recipients) and 62 +/− 2.2% in mice that received APE2 (n = 2 recipients) bone marrow. For competitive bone marrow transplants, APE2-deficient (C57BL/6, CD45.2+) and WT (B6.SJL, CD45.1+) bone marrow cells were mixed at the indicated ratios and a total of 2×106 cells were transplanted i.v. into lethally irradiated (900 rad) F1 C57BL/6 × B6.SJL recipients. Peripheral blood from the tail vein was analyzed by flow cytometry 8 weeks after transplant. Two-tailed one-sample t-test showed significance (p < 0.05) relative to the theoretical mean (the expected reconstitution rate), except where noted in the figure legend.
Figure 1.

Partial block in pro-B to pre-B cell transition in APE2-deficient mice. (A) Shown are the total cell numbers of the indicated progenitors in two hind limbs of WT and APE2-deficient mice. n = 3 for HSPC, MPP, CLP and pre-pro-B cells, and n = 5 for pro-B and pre-B cells. (B) Representative FACS plot of pro-B (B220+CD43+) and pre-B cells (B220+CD43×/LO). Numbers shown are the percentage of cells in the viable, IgMDX5−Ly6C− gate that are pre-B cells, IgM− B220+ cells, and pro-B cells. (C) Total cell numbers in mice that are haploinsufficient for APE1 (APE1+/−; n=3), deficient in APE2 (n=9), both (APE1+/− APE2Y/−; n=8), or their WT littermates (n=17), combined from many flow cytometry experiments. Data are from one hind limb. (A,C) Results of t test (A) and non-parametric one-way ANOVA (C) show significance vs. WT (* p <0.05, ** p<0.01). In addition, APE1+/− x APE2 immature B cells are significantly reduced relative to APE1+/− (p <0.05).
Analysis of DNA Synthesis and Cell Cycle
Cell cycle status was determined in pro-B and pre-B cells that were purified by FACS, and then fixed in 70% ethanol overnight prior to incubation with RNaseA and propidium iodide (50 ug/ml each, Sigma) for 20 min. at 37°C. Cells were then analyzed by flow cytometry and the Watson Pragmatic model was applied to fit the curves to the stages of the cell cycle. Cell turnover and production rates were determined by continuous BrdU labeling of in vivo DNA synthesis. Mice were injected i.p. with 1 mg BrdU (Sigma) in 0.2 ml PBS every 12 hours for 1 or 2 days and 6 hours prior to sacrifice. Bone marrow cells were harvested and surface stained for pro-B and pre-B cell markers, as above, except that B220-AF647 (Invitrogen) was used. Cells were permeabilized in 70% ethanol overnight, washed, fixed in 1% paraformaldehyde in PBS with 0.2% Tween-20 for 1 h, washed, and treated with 5 U/ml DNase1 (Roche) in 0.15 M NaCl, 4.2 mM MgCl2 for 30 min. at 37°C. After one wash with deficient RPMI/5% FBS/0.1% Tween-20, cells were incubated with anti-BrdU-AF594 (Invitrogen) for 30 min. at room temperature and washed twice more before analysis by flow cytometry. Renewal rates (turnover) and production rates were calculated as described (23).
Stromal Cell Co-cultures
2,000 FACS-purified pro-B cells were plated onto nearly confluent cultures of GFP+ OP9 stromal cells in 6-well plates with RPMI 1640 with 2 mM L-glutamine, non-essential amino acids, 100 U/ml penicillin, 100 ug/ml streptomycin (all from Gibco), 5% fetal bovine serum (Atlanta Biologicals, Atlanta, GA), 5 × 10×5 M β-mercaptoethanol (Sigma), 10 ng/ml IL-7 (Peprotech Inc., Rocky Hill, NJ) and 10 ng/ml Flt3-ligand (Bio X Cell, West Lebanon, NH). Culture medium and cytokines were replaced every three days. Wells were harvested after 6 or 9 days by pipetting, and OP9 cells were excluded from FACS analysis on the basis of GFP expression and forward and side scatter.
Results
APE2-deficient mice have a partial block at the pro-B to pre-B cell transition
To assess the role of APE2 in B cell development, we used multiparametric flow cytometry to determine the numbers of pro-B, pre-B, and immature B cells, as well as their precursor populations including hematopoietic stem and progenitor cells (HSPC), multi-potent progenitors (MPP), common lymphoid progenitors (CLP) and pre-pro-B cells in the bone marrow. In adult APE2-deficient mice, HSPCs, MPPs, CLPs, pre-pro-B cells and pro-B cells are present in normal numbers, whereas there is a two-fold reduction in the number of pre-B cells (Fig. 1 A-C). This finding is consistent with decreased numbers of B220+IgM− B cells in the bone marrow of APE2-deficient mice, as reported by Ide et al (16). In addition, we analyzed apex1+/− mice, which are viable but haploinsufficient, and exhibit reduced APE1-mediated DNA repair compared to WT littermates (19). The number of B cell progenitors was not significantly altered from WT littermate mice in apex1+/−. Also, we did not find a further reduction in pre-B cells in apex1+/− apex2Y/− compared to apex2 Y/− mice (Fig. 1C). Immature B cells, which develop from the pre-B cell pool, were slightly, but not significantly reduced in APE2-deficient mice, but this population was further reduced in apex1+/− apex2Y/− mice, suggesting that APE2 may be more important when levels of APE1 are reduced. We then asked whether the defect in B cell development in APE2-deficient mice is intrinsic to the hematopoietic system by transplanting CD45.2+ bone marrow from WT or APE2-deficient mice into irradiated CD45.1+ recipients. After 7 weeks, the number of pro-B cells was not different in recipients injected with APE2-deficient relative to WT bone marrow, but the number of pre-B cells was reduced three-fold (3.02 ± 0.27 × 106, WT; 0.99 ± 0.05 × 106, APE2.) These data show that APE2 has a unique, intrinsic role in B cell development, and that its deficiency results in reduced numbers of pre-B and immature B cells.
APE2 is not required for V(D)J-recombination
Our finding of a partial block in development in APE2-deficient mice at the pre-B cell stage led to the question of whether APE2 might be important for V(D)J-recombination. Toward this end, we crossed APE2-deficient mice to a V(D)J-recombination-reporter transgenic mouse (21). The transgene consists of the coding sequence for violet-excited green fluorescent protein (Vex-GFP) in the antisense orientation, flanked by recombination signal sequences. Cells that express RAG1 and RAG2 and are capable of performing V(D)J-recombination can be assessed by flow cytometry based on Vex-GFP expression resulting from inversional recombination (21, 24). In two independent experiments, we found that APE2-deficient pro- and pre-B cells recombine this substrate just as efficiently as WT cells. APE2-deficient pro-B cells were 88.7 ± 0.0% Vex+ (average of two mice ± range) compared to 87.8 ± 0.5% Vex+ WT pro-B cells. Pre-B cells were 86.2 ± 0.1% (APE2-deficient) and 87.0 ± 2% (WT) Vex+. Although we cannot rule out a subtle contribution, we conclude that APE2 is not required for V(D)J-recombination.
Turnover and production rates of APE2-deficient B cell progenitors
To determine if the reduced pre-B cell numbers in APE2-deficient bone marrow are related to decreased proliferation rates in APE2-deficient pro-B and pre-B cells, the rate of incorporation of the thymidine analog, BrdU, into DNA was measured. Bone marrow was harvested and analyzed by FACS to assess the turnover rate (percent of the pool labeled per day) and the production rate (number of cells labeled per day) of pro-B and pre-B cells. APE2-deficiency did not significantly affect the turnover rate of pro-B or pre-B cells (Fig. 2A and 2B, left). However, the production rate of both pro-B and pre-B cells was decreased in APE2-deficient mice, where pro-B cell production was down 3-fold (p <0.0001) and pre-B cell production was down 3.5-fold (p <0.004). The latter difference reflects the decreased pre-B cell numbers shown in Fig. 1. However, since APE2-deficient pro-B cell numbers are normal in the steady-state (Fig. 1A), but 3-fold fewer are produced per day, these data demonstrate that homeostatic pressures are acting to maintain cell numbers in the pro-B cell compartment. We conclude that APE2-deficiency causes a decrease in the number of pro-B and pre-B cells produced per day, but does not affect the proliferation rates of these cells.
Figure 2.

Turnover and production rates of pro-B and pre-B cells determined by BrdU incorporation. (A) Representative FACS plot gated on pre-B cells from the bone marrow of mice injected with BrdU every 12 hours for 1 or 2 days, and 6 hours before harvest. The background staining in a control PBS-injected mouse is shown (light gray histogram). (B) Data shown is representative of two experiments, each having 3 WT (solid squares) and 3 APE2-deficient (open circles) mice for each time point. Gray/shaded lines depict 95% confidence intervals for linear regression plots.
Decreased pre-B cell production is p53-dependent
The accumulation of DNA damage, resulting in a perturbation of the cell cycle and/or cell death, could be the basis of the lower production rate of pro- and pre-B cells in the bone marrow of APE2-deficient mice. We observed only a slight increase in apoptosis in APE2-deficient pre-B cells by intracellular staining of cleaved caspases (not shown). The difference was not statistically significant, but dead cells are removed very efficiently in vivo by macrophages. However, we did find that the loss of pre-B cells in APE2-deficient mice is p53-dependent. When APE2-deficient mice were bred onto a p53-deficient background, the number of APE2-deficient pre-B cells was not significantly diminished relative to the numbers in WT or p53-deficient mice (Fig. 3). Activation of the p53 pathway strongly suggests that APE2-deficient pre-B cells sustain DNA damage, as was previously shown for APE2-deficient thymocytes (25), and also suggests that p53 inhibits their further development.
Figure 3.

The reduction in pre-B cell numbers in APE-deficient mice is p53-dependent. The number of pre-B cells in the BM of mice deficient in p53, APE2, or both, and their WT littermates was determined by flow cytometry. n = 5 for all groups. APE2 is significantly different from all other groups (t test: * p <0.05). APE2 x p53 doubly deficient mice are not different from WT or p53-deficient mice.
After the pro-B to pre-B cell transition, the pre-B II stage is characterized by exit from the cell cycle, decrease in cell size and initiation of recombination at the Ig light chain locus (26). To further analyze this transition, we determined the cell cycle status of FACS purified pro- and pre-B cells by propidium iodide (PI) staining. We found no difference between WT and APE2-deficient pro-B cells, but fewer APE2-deficient pre-B cells have exited the cell cycle and decreased in size (Fig. 4). Unlike WT, the APE2-deficient pre-B cell pool has the same proportion of cells in S and G2/M phases as the pro-B cell pool. Therefore, not only is the number of pre-B cells reduced in APE2-deficient mice, but of these B220+ CD43×/lo cells, a smaller percent have succeeded at differentiation to the small, non-cycling pre-B II stage. From these combined data, we conclude that APE2-deficient progenitors are partially blocked at the transition between the cycling pro-B/preB I stage and the small resting pre-B II stage by a p53-dependent pathway.
Figure 4.

Reduced transition to small non-cycling pre-B II cell stage in APE2-deficient mice. The cell cycle stage of FACS-purified pro-B and pre-B cells was determined by propidium iodide staining. A representative FACS plot is shown depicting cell size (FSC, y axis) vs. DNA content (PI, x axis) with hand-drawn gates as example. Below are the results (mean ± SEM) from three independent FACS sorts, where the percent in each stage of cell cycle was determined using the Watson Pragmatic model. (* APE2 relative to WT: p <0.02).
Defects at earlier stages of hematopoiesis are revealed during recovery from chemotherapeutic treatment in APE2-deficient mice
It is not surprising that developing APE2-deficient lymphoid cells are lost at the pre-B cell stage, since checkpoints for genomic integrity following V(D)J-recombination occur at this stage. Decreased expression of the pro-survival genes Bcl-XL and Bcl-2 makes these cells highly sensitive to apoptosis (27-30). However, this does not mean that APE2 is only important at the pro-B to pre-B cell transition. Defects in hematopoiesis can be masked by homeostatic compensation, which maintains the size of precursor populations at each stage of development (31). This is demonstrated by our finding of a reduced production rate of pro-B cells in APE2-deficient mice by BrdU labeling (Fig. 2), despite finding normal numbers of pro-B cells in untreated mice (Fig. 1). To determine if APE2-deficient mice have defects in stages of hematopoiesis other than the pro- to pre-B cell transition, we perturbed bone marrow homeostasis by injecting mice with the chemotherapeutic agent 5-fluorouracil (5FU). 5FU gets incorporated into both DNA and RNA of dividing cells, and results in bone marrow depletion, sparing only quiescent HSCs and a few mature, re-circulating IgM+ cells (32). The efficiency of repopulation of the bone marrow by HSCs was monitored over time, harvesting bone marrow 5, 10 and 14 days after 5FU treatment, and analyzing early lymphoid progenitor cells by flow cytometry (Fig. 5 A-D). The HSPC compartment in both WT and APE2-deficient mice expanded three to four fold in response to bone marrow depletion (not shown), but the recovery of APE2-deficient downstream progenitors on day 10 after 5FU was substantially reduced (Fig. 5C, black bars) relative to WT littermates (compare to line at 100%), as well as relative to untreated APE2-deficient mice (gray bars). By 14 days after 5FU treatment, the earliest progenitors had recovered to levels seen in untreated mice, but pro-B and pre-B cell numbers in APE2 deficient mice were still greatly reduced (Fig. 5D). Even after one month, APE2-deficient pro-B and pre-B cells still showed a trend toward reduced numbers, although these values were not significantly different from the numbers in untreated mice (Fig. 5E). Altogether, these results demonstrate that the defect in expansion of APE2-deficient B cell progenitors is even greater when under pressure to regenerate the empty niche after bone marrow depletion.
Figure 5.

Delayed recovery of APE2-deficient B cell progenitors from 5FU challenge. APE2-deficient mice and their WT littermates were injected once i.p. with PBS (day 0) or 5FU (150 mg/g body weight) 5, 10, 14 or 28 days before analysis of bone marrow by flow cytometry. (A,B) Representative FACS plot showing gating strategy for HSPC and MPP (A) and CLP (B) in bone marrow of PBS-treated control mice (“day 0”), or mice 5 or 10 days after 5FU injection. (C-D) The number of APE2-deficient progenitors on day 10 (C) and day 14 (D) after injection (black bars) is shown as a percent of that in identically treated WT mice. The same populations in untreated APE2-deficient mice are shown as a percent of those in untreated WT mice for comparison (gray bars). Each experiment was a paired observation between one APE2-deficient and one WT littermate male mouse per time point and the average of independent experiments (n = 3 for HSPC through pre-pro B cells and n = 7 for pro-B and pre-B cells) ± SEM is shown. (E) Number of pro- and pre-B cells 28 days after 5FU injection (data are combined from two flow cytometry experiments, total n = 4 WT and 3 APE2-deficient mice). (F) Number of total mononuclear cells in the bone marrow as a function of time after 5FU injection; data was combined from all of the experiments shown in C-G (n = 4-8 for WT mice and 3-6 for APE2-deficient mice, depending on the time point). (G) Number of viable lymphoid (B220+) or myeloid (either Mac-1+ or Gr-1+) cells in the bone marrow of untreated mice (n = 3) or mice 14 days (n = 5 WT and 4 APE2-deficient) or 28 days (n = 4 WT and 3 APE2-deficient) after 5FU injection. (C-G) t test results vs. WT: * p <0.05, ** p<0.01, *** p<0.002).
14 days after 5FU injection, we observed that the total number of mononuclear cells in the bone marrow of APE2-deficient mice had recovered to almost normal levels (Fig. 5F) while the number of lymphocyte progenitors was still greatly reduced. As bone marrow consists mostly of two major hematopoietic lineages, lymphoid and myeloid, we measured the percent of bone marrow cells positive for the myeloid markers Mac-1 and Gr-1, compared to B220+ lymphoid cells, 14 days after 5FU injection. Indeed, we found that myeloid cells were not significantly reduced in APE2-deficient relative to WT mice, whereas B220+ cells were still five-fold reduced at this time point (Fig. 5G). In order to further evaluate the multi-lineage repopulation potential of APE2-deficient bone marrow, we performed competitive bone marrow transplants. APE2-deficient (CD45.2+) and WT (CD45.1+) bone marrow cells were competitively transplanted at the indicated ratios into lethally irradiated (CD45.2 × CD45.1) F1 recipients (Fig. 6). Analysis of peripheral blood after 8 weeks by flow cytometry showed that APE2-deficient bone marrow did not compete effectively against WT bone marrow, and did not achieve the expected rates of reconstitution. Reconstitution of B, T and myeloid lineages were all impaired.
Figure 6.
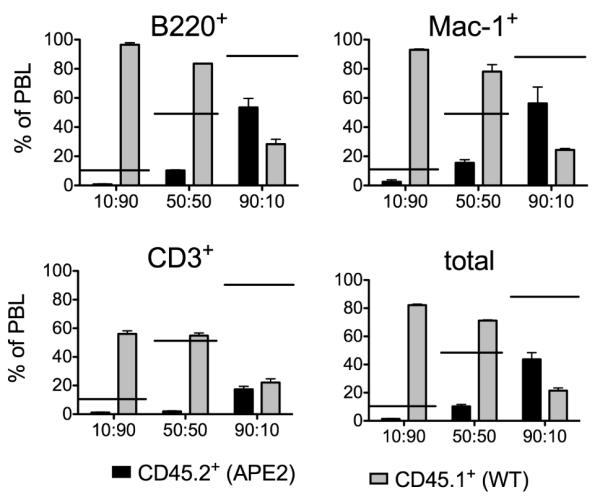
Competitive bone marrow transplants. Peripheral blood (PBL) analysis 8 weeks after transfer. x axis shows ratio of APE2-deficient:WT bone marrow transplanted. The level of reconstitution by APE2-deficient cells is significantly different from the expected value (indicated by horizontal bars) in all cases (p <0.05) with the exception of Mac-1+ cells in the 90:10 transplant.
Defective expansion of IL-7-responsive progenitors in vitro
We next assessed the ability of APE2-deficient progenitors from untreated mice to expand and differentiate in vitro by co-culture with OP9 stromal cells, IL-7 and Flt3L. This culture system allows us to distinguish between potential defects in the expansion of APE2-deficient IL-7R+ cells (CLP, pre-pro, and pro-B cells), and the ability of these progenitors to differentiate along the lymphoid lineage. FACS-purified HSPC, MPP, CLP and pro-B cells from untreated WT and APE2-deficient mice were co-cultured for 7 days and then analyzed by flow cytometry for lymphoid (CD19) and myeloid (Mac-1/GR-1) markers. As shown in Fig. 7A, HSPCs almost exclusively generated myeloid cells within the timeframe of this experiment, but MPPs were able to differentiate into both lymphoid and myeloid cells, while CLPs generated only lymphoid cells, and pro-B cells expanded. We conclude from this that APE2-deficient progenitors were able to differentiate normally in vitro. Pro-B cell expansion was then analyzed quantitatively after 6 and 9 days. WT pro-B cells expanded 60-fold after 6 days, while APE2-deficient pro-B cells expanded only 20-fold (p <0.005) and failed to expand further. After 9 days in culture, approximately ten-fold fewer APE2-deficient pro-B cells were recovered compared to WT pro-B cells (p <0.01)(Fig. 7B). Consistent with our results in vivo, the decreased expansion of these cells in vitro was dependent on p53, since pro-B cells purified from apex2Y/-p53−/− mice proliferated exponentially, similar to p53−/− pro-B cells on day 6 and only slightly (1.4-fold) slower on day 9 (p <0.05).
Figure 7.

APE2-deficient B cell progenitors differentiate normally but expand poorly in vitro. (A) The indicated progenitor populations were purified by FACS and co-cultured with stromal cells, IL-7 and Flt3L. Sorted HSPC, MPP and CLP input cells were negative for CD19 and Mac-1. The percent of non-stromal cells expressing CD19 or Mac-1/Gr-1 (myeloid) was determined by flow cytometric analysis of cultures after 7 days. FACS plots are representative of results from two independent sorts. (B) 2,000 FACS-purified pro-B cells from WT, apex2Y/− (APE2), p53−/− (p53) or apex2Y/− p53−/−(DKO) bone marrow were cultured as in (A) and the number of viable cells was counted after 6 and 9 days. CD19 and CD43 expression on the expanded cells was confirmed by flow cytometry. The average of 3 replicate wells is shown. Data on WT and APE2-deficient pro-B cells are representative of 2 independent sorting experiments. Data on p53−/− and DKO pro-B cells are from one sort.
Altogether, these data demonstrate that APE2-deficient lymphoid progenitors can respond to IL-7, differentiate normally and initiate proliferation, but have a reduced capacity for expansion. In vivo, these cells are predominantly lost at the small, non cycling pre-B II cell stage due to checkpoints that activate a p53-dependent damage response. We conclude that APE2 is required for the efficient expansion of B cell progenitors in response to IL-7, both in vitro and in vivo.
Discussion
The weak AP endonuclease activity of APE2 and the overlap in biochemical properties between APE1 and APE2 have hampered experiments to determine the biological function of APE2. We previously discovered that APE2 has a role in class switch recombination (CSR), as APE2-deficient cells have reduced CSR and reduced ability to generate the DNA breaks required for CSR (33). In this study we demonstrate that APE2 is important during the expansion of lymphoid progenitors in the bone marrow. We uncovered a role for APE2 in B cell development under homeostatic conditions, where there is a two-fold decrease in pre-B cells numbers in the absence of APE2. APE2-deficient pro-B cells also show a defective expansion in vitro in response to IL-7. Both the decreased proliferation in vitro and the loss of pre-B cells in vivo is dependent on p53, supporting the conclusion that APE2-deficiency activates a p53-dependent DNA damage response. In addition, we observed a significant increase in p21 mRNA (transcriptional target gene of p53) in APE2-deficient pre-B cells (data not shown). This result is consistent with the increased p53 phosphorylation, γH2AX foci and DNA breaks observed in the thymus of APE2-deficient mice (25). Many lymphocytes are lost due to checkpoints that control for DNA integrity following V(D)J-recombination. These checkpoints are active at the pro-B to pre-B cell transition, which might explain why APE2-deficiency results in defects during this stage of B cell development. For instance, the anti-apoptotic genes Bcl-XL and Bcl-2 that protect pro-B cells during gene rearrangements are down-regulated at this transition, causing pre-B cells to be prone to apoptosis in the absence of positive selection signals (27-30).
In addition, we found that APE2 is especially important in the recovery of lymphoid development after chemotherapeutic challenge. APE2-deficient B cell progenitors fail to expand rapidly in vivo after 5FU injection. Defective repopulation of the bone marrow upon transplantation has been observed in mice deficient in several DNA repair pathways including non-homologous end joining (Ku80), nucleotide excision repair (XPD), telomerase (mTR) (34), Brca2 (35) and mismatch repair (Msh2) (36). Although the HSCs in DNA repair-deficient mice accumulate DNA damage with age, the frequency of HSCs is not affected (34). Rather, it seems that the ability of these cells and their downstream progenitors to expand and repopulate the bone marrow upon transfer into irradiated recipients is compromised by the DNA damage. This is similar to our results with APE2-deficient lymphoid progenitors, which identifies APE2 as an important factor in hematopoiesis. Defects in granulopoiesis as well as lymphopoiesis are observed in other DNA repair deficient mice (Ku80×/−, mTR×/−, and XPDTTD) (34). We found that myeloid progenitors are able to recover rapidly following 5FU challenge in the absence of APE2, but upon competitive bone marrow transplantation, the myeloid lineage was also impaired. It is possible that APE2-deficiency might impair lymphopoiesis to a greater extent than granulopoiesis, although this requires further investigation. Genomic stability may be of greater significance to lymphocytes, since they can be very long-lived and will undergo further rounds of proliferation and differentiation if activated by antigen. Granulocytes are short-lived and need continued stem cell activity to be regenerated, and are thus considered a good correlate to stem cell function (37). It will be interesting to test the effect of APE2-deficiency on long-term bone marrow repopulation by an enriched stem cell population and serial transplantation to determine if APE2 is important for maintaining HSC function. DNA repair mechanisms in HSCs and the consequences of mutations arising from aberrant repair are only beginning to be understood (38-40). It was recently shown that the preferential use of the error-prone NHEJ pathway in quiescent HSCs can lead to genomic rearrangements (40). The contribution of BER to genomic stability in HSC has not been examined, but seems likely to be important, as BER is an accurate repair pathway that is active in non-dividing cells.
DNA damage caused by 5FU can be repaired by both APE1 and the DNA mismatch repair pathway (41-43). However, there are also some data consistent with the idea that APE2 can repair these lesions. In yeast, mutants of the homologs of APE1 and APE2, apn1 and apn2, show only mild sensitivity to 5FU, but the double mutant apn1apn2 is very sensitive (42). In human studies, a recent gene expression profiling study found that decreased apex2 expression was associated with a favorable outcome in glioblastoma patients treated with radiation and capecitabine, a compound that is converted into 5FU in vivo (44). These results suggest that both APEs can repair 5FU damage; however, we believe it is unlikely that residual 5FU (or 5FU damage) remains to contribute to the bone marrow recovery phenotype we observe up to one month after 5-FU injection in APE2-deficient mice. It has been shown that quiescent HSCs are protected from 5FU damage (45), and that 5FU is cleared from the body by the liver within 4 hours of injection (46). Furthermore, our finding that purified APE2-deficient pro-B cells from untreated mice have defects in expansion in vitro confirms the need for APE2 in the expansion of B cell precursors, even in the absence of exogenous DNA damage. We propose that APE2 is important for the rapid expansion of hematopoietic precursors following any bone marrow-depleting treatment, such as radiation or chemotherapy. Furthermore, APE2 could participate in repair of DNA lesions caused by these treatments and thus be a factor in resistance to such therapies.
The accumulation of oxidative damage is associated with aging phenotypes (1, 17), and our findings suggest that APE2-deficiency accelerates the changes known to occur in the bone marrow during aging. This phenotype includes defects in the pro-B to pre-B cell transition, decreased IL-7 responsiveness of B cell progenitors, skewing in the bone marrow toward the myeloid lineage, impaired recovery from bone marrow injury, increased marginal zone B cells (33), and thymic atrophy (16). Decreased lymphocyte production and skewing toward the myeloid lineage is observed in the bone marrow of both mice and humans with age (47, 48), although this is not thought to be due to DNA damage (48, 49). Although many factors contribute to the decline of lymphocyte production in the bone marrow with age, including a decrease in hormones and stromal cell function (49-52), our data support that the accumulation of DNA damage might also be a contributing factor. The lymphocyte compartment is somewhat unique in the body, since these cells undergo many rounds of rapid proliferation and can be very long lived. As such, they are especially dependent upon efficient DNA repair for survival and to prevent cell transformation and malignancy. This is true not only during development, but also during the proliferative response of B cells to antigen. Consistent with this, we have also found defects in the expansion of APE2-deficient germinal center B cells (manuscript in preparation).
Acknowledgements
The authors wish to thank Michael Cancro and William Quinn (U. of Pennsylvania, Philadelphia, PA) for helpful advice on BrdU labeling, Juan Carlos Zúñiga-Pflücker (Department of Immunology, University of Toronto, and Sunnybrook Research Institute, Toronto, Ontario, Canada) for OP9 stromal cells, Anna Ucher and the UMMS Flow Cytometry Facility for excellent technical assistance and Janet Stavnezer for helpful comments on the manuscript.
This work was supported by National Institutes of Health Grants RO1 AI065639 (to C.E.S.) and RO1 AI043534 (to R.M.G.). J.E.J.G. was supported by the Irvington Institute Fellowship Program of the Cancer Research Institute. R.M.G. is a member of the UMass DERC (DK32520) and as such acknowledges that the project described was supported by Grant Number 5 P30 DK32520 from the National Institute of Diabetes and Digestive and Kidney Diseases.
2. Abbreviations used in this paper
- 5FU
5-fluorouracil
- APE
apurinic/apyrimidicendonuclease
- BER
base excision repair
- BSO
buthionine sulfoxamine
- CLP
common lymphoid progenitor
- HSC
hematopoietic stem cell
- HSPC
hematopoietic stem and progenitor cell
- MPP
multi-potent progenitor
- ROS
reactive oxygen species
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annual review of genetics. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 2.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu.Rev.Immunol. 2006;24:541–570. doi: 10.1146/annurev.immunol.23.021704.115830. [DOI] [PubMed] [Google Scholar]
- 4.Mills KD, Ferguson DO, Alt FW. The role of DNA breaks in genomic instability and tumorigenesis. Immunol.Rev. 2003;194:77–95. doi: 10.1034/j.1600-065x.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 5.Jolly CJ, Cook AJ, Manis JP. Fixing DNA breaks during class switch recombination. The Journal of experimental medicine. 2008;205:509–513. doi: 10.1084/jem.20080356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Villartay JP. V(D)J recombination deficiencies. Advances in experimental medicine and biology. 2009;650:46–58. doi: 10.1007/978-1-4419-0296-2_4. [DOI] [PubMed] [Google Scholar]
- 7.Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc.Natl.Acad.Sci.U.S.A. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bentley D, Selfridge J, Millar JK, Samuel K, Hole N, Ansell JD, Melton DW. DNA ligase I is required for fetal liver erythropoiesis but is not essential for mammalian cell viability. Nature genetics. 1996;13:489–491. doi: 10.1038/ng0896-489. [DOI] [PubMed] [Google Scholar]
- 9.Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. The EMBO journal. 2000;19:1397–1404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadi MZ, Ginalski K, Nguyen LH, Wilson DM., III Determinants in nuclease specificity of Ape1 and Ape2, human homologues of Escherichia coli exonuclease III. J.Mol.Biol. 2002;316:853–866. doi: 10.1006/jmbi.2001.5382. [DOI] [PubMed] [Google Scholar]
- 11.Hadi MZ, Wilson DM., III Second human protein with homology to the Escherichia coli abasic endonuclease exonuclease III. Environ.Mol.Mutagen. 2000;36:312–324. [PubMed] [Google Scholar]
- 12.Burkovics P, Szukacsov V, Unk I, Haracska L. Human Ape2 protein has a 3′-5′ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006;34:2508–2515. doi: 10.1093/nar/gkl259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burkovics P, Hajdu I, Szukacsov V, Unk I, Haracska L. Role of PCNA-dependent stimulation of 3′-phosphodiesterase and 3′-5′ exonuclease activities of human Ape2 in repair of oxidative DNA damage. Nucleic acids research. 2009;37:4247–4255. doi: 10.1093/nar/gkp357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertoncini CR, Meneghini R. DNA strand breaks produced by oxidative stress in mammalian cells exhibit 3′-phosphoglycolate termini. Nucleic acids research. 1995;23:2995–3002. doi: 10.1093/nar/23.15.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castillo-Acosta VM, Ruiz-Perez LM, Yang W, Gonzalez-Pacanowska D, Vidal AE. Identification of a residue critical for the excision of 3′-blocking ends in apurinic/apyrimidinic endonucleases of the Xth family. Nucleic acids research. 2009 doi: 10.1093/nar/gkp021. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ide Y, Tsuchimoto D, Tominaga Y, Nakashima M, Watanabe T, Sakumi K, Ohno M, Nakabeppu Y. Growth retardation and dyslymphopoiesis accompanied by G2/M arrest in APEX2-null mice. Blood. 2004;104:4097–4103. doi: 10.1182/blood-2004-04-1476. [DOI] [PubMed] [Google Scholar]
- 17.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 18.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Meira LB, Devaraj S, Kisby GE, Burns DK, Daniel RL, Hammer RE, Grundy S, Jialal I, Friedberg EC. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 2001;61:5552–5557. [PubMed] [Google Scholar]
- 20.Ide Y, Tsuchimoto D, Tominaga Y, Iwamoto Y, Nakabeppu Y. Characterization of the genomic structure and expression of the mouse Apex2 gene. Genomics. 2003;81:47–57. doi: 10.1016/s0888-7543(02)00009-5. [DOI] [PubMed] [Google Scholar]
- 21.Borghesi L, Gerstein RM. Developmental separation of V(D)J recombinase expression and initiation of IgH recombination in B lineage progenitors in vivo. The Journal of experimental medicine. 2004;199:483–489. doi: 10.1084/jem.20031802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 23.Cancro MP, Sah AP, Levy SL, Allman DM, Schmidt MR, Woodland RT. xid mice reveal the interplay of homeostasis and Bruton’s tyrosine kinase-mediated selection at multiple stages of B cell development. International immunology. 2001;13:1501–1514. doi: 10.1093/intimm/13.12.1501. [DOI] [PubMed] [Google Scholar]
- 24.Borghesi L, Hsu LY, Miller JP, Anderson M, Herzenberg L, Herzenberg L, Schlissel MS, Allman D, Gerstein RM. B lineage-specific regulation of V(D)J recombinase activity is established in common lymphoid progenitors. The Journal of experimental medicine. 2004;199:491–502. doi: 10.1084/jem.20031800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dan Y, Ohta Y, Tsuchimoto D, Ohno M, Ide Y, Sami M, Kanda T, Sakumi K, Nakabeppu Y. Altered gene expression profiles and higher frequency of spontaneous DNA strand breaks in APEX2-null thymus. DNA Repair (Amst) 2008;7:1437–1454. doi: 10.1016/j.dnarep.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Johnson K, Hashimshony T, Sawai CM, Pongubala JM, Skok JA, Aifantis I, Singh H. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28:335–345. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 27.Nunez G, Merino R, Grillot D, Gonzalez-Garcia M. Bcl-2 and Bcl-x: regulatory switches for lymphoid death and survival. Immunology today. 1994;15:582–588. doi: 10.1016/0167-5699(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 28.Lu L, Osmond DG. Apoptosis and its modulation during B lymphopoiesis in mouse bone marrow. Immunological reviews. 2000;175:158–174. [PubMed] [Google Scholar]
- 29.Merino R, Ding L, Veis DJ, Korsmeyer SJ, Nunez G. Developmental regulation of the Bcl-2 protein and susceptibility to cell death in B lymphocytes. The EMBO journal. 1994;13:683–691. doi: 10.1002/j.1460-2075.1994.tb06307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li YS, Hayakawa K, Hardy RR. The regulated expression of B lineage associated genes during B cell differentiation in bone marrow and fetal liver. The Journal of experimental medicine. 1993;178:951–960. doi: 10.1084/jem.178.3.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancro MP, Smith SH. Peripheral B cell selection and homeostasis. Immunologic research. 2003;27:141–148. doi: 10.1385/IR:27:2-3:141. [DOI] [PubMed] [Google Scholar]
- 32.Tsuboi I, Hirabayashi Y, Harada T, Koshinaga M, Kawamata T, Kanno J, Inoue T, Aizawa S. Role of hematopoietic microenvironment in prolonged impairment of B cell regeneration in age-related stromal-cell-impaired SAMP1 mouse: effects of a single dose of 5-fluorouracil. J Appl Toxicol. 2008;28:797–805. doi: 10.1002/jat.1341. [DOI] [PubMed] [Google Scholar]
- 33.Guikema JE, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, Schrader CE. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J.Exp.Med. 2007;204:3017–3026. doi: 10.1084/jem.20071289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 35.Navarro S, Meza NW, Quintana-Bustamante O, Casado JA, Jacome A, McAllister K, Puerto S, Surralles J, Segovia JC, Bueren JA. Hematopoietic dysfunction in a mouse model for Fanconi anemia group D1. Mol Ther. 2006;14:525–535. doi: 10.1016/j.ymthe.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 36.Reese JS, Liu L, Gerson SL. Repopulating defect of mismatch repair-deficient hematopoietic stem cells. Blood. 2003;102:1626–1633. doi: 10.1182/blood-2002-10-3035. [DOI] [PubMed] [Google Scholar]
- 37.Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells: Overexpression of Bcl-2 increases both their number and repopulation potential. The Journal of experimental medicine. 2000;191:253–264. doi: 10.1084/jem.191.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milyavsky M, Gan OI, Trottier M, Komosa M, Tabach O, Notta F, Lechman E, Hermans KG, Eppert K, Konovalova Z, Ornatsky O, Domany E, Meyn MS, Dick JE. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell stem cell. 7:186–197. doi: 10.1016/j.stem.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 39.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell stem cell. 6:309–322. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, Morrison CG, Passegue E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell stem cell. 7:174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNeill DR, Wilson DM., 3rd A dominant-negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA-damaging agents. Mol Cancer Res. 2007;5:61–70. doi: 10.1158/1541-7786.MCR-06-0329. [DOI] [PubMed] [Google Scholar]
- 42.Matuo R, Sousa FG, Escargueil AE, Soares DG, Grivicich I, Saffi J, Larsen AK, Henriques JA. DNA repair pathways involved in repair of lesions induced by 5-fluorouracil and its active metabolite FdUMP. Biochemical pharmacology. 2010;79:147–153. doi: 10.1016/j.bcp.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 43.Li LS, Morales JC, Veigl M, Sedwick D, Greer S, Meyers M, Wagner M, Fishel R, Boothman DA. DNA mismatch repair (MMR)-dependent 5-fluorouracil cytotoxicity and the potential for new therapeutic targets. British journal of pharmacology. 2009;158:679–692. doi: 10.1111/j.1476-5381.2009.00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grunda JM, Fiveash J, Palmer CA, Cantor A, Fathallah-Shaykh HM, Nabors LB, Johnson MR. Rationally designed pharmacogenomic treatment using concurrent capecitabine and radiotherapy for glioblastoma; gene expression profiles associated with outcome. Clin Cancer Res. 2010;16:2890–2898. doi: 10.1158/1078-0432.CCR-09-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berardi AC, Wang A, Levine JD, Lopez P, Scadden DT. Functional isolation and characterization of human hematopoietic stem cells. Science. 1995;267:104–108. doi: 10.1126/science.7528940. [DOI] [PubMed] [Google Scholar]
- 46.Bocci G, Danesi R, Di Paolo AD, Innocenti F, Allegrini G, Falcone A, Melosi A, Battistoni M, Barsanti G, Conte PF, Del Tacca M. Comparative pharmacokinetic analysis of 5-fluorouracil and its major metabolite 5-fluoro-5,6-dihydrouracil after conventional and reduced test dose in cancer patients. Clin Cancer Res. 2000;6:3032–3037. [PubMed] [Google Scholar]
- 47.Dykstra B, de Haan G. Hematopoietic stem cell aging and self-renewal. Cell and tissue research. 2008;331:91–101. doi: 10.1007/s00441-007-0529-9. [DOI] [PubMed] [Google Scholar]
- 48.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephan RP, Reilly CR, Witte PL. Impaired ability of bone marrow stromal cells to support B-lymphopoiesis with age. Blood. 1998;91:75–88. [PubMed] [Google Scholar]
- 50.Lamberts SW, van den Beld AW, van der Lely AJ. The endocrinology of aging. Science. 1997;278:419–424. doi: 10.1126/science.278.5337.419. [DOI] [PubMed] [Google Scholar]
- 51.Labrie JE, 3rd, Sah AP, Allman DM, Cancro MP, Gerstein RM. Bone marrow microenvironmental changes underlie reduced RAG-mediated recombination and B cell generation in aged mice. The Journal of experimental medicine. 2004;200:411–423. doi: 10.1084/jem.20040845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayack SR, Shadrach JL, Kim FS, Wagers AJ. Systemic signals regulate ageing and rejuvenation of blood stem cell niches. Nature. 463:495–500. doi: 10.1038/nature08749. [DOI] [PubMed] [Google Scholar]