

Abstract
Alveolar type II epithelial cells (ATIIs) are one of the primary targets for influenza A pneumonia. The lack of a culture system for maintaining differentiated ATIIs hinders our understanding of pulmonary innate immunity during viral infection. We studied influenza A virus (IAV)-induced innate immune responses in differentiated primary human ATIIs and alveolar macrophages (AMs). Our results indicate that ATIIs, but not AMs, support productive IAV infection. Viral infection elicited strong inflammatory chemokine and cytokine responses in ATIIs, including secretion of IL-8, IL-6, MCP-1, RANTES, and MIP-1β, but not TNF-α, whereas AMs secreted TNF-α as well as other cytokines in response to infection. Wild-type virus A/PR/8/34 induced a greater cytokine response than reassortant PR/8 virus, A/Phil/82, despite similar levels of replication. IAV infection increased mRNA expression of IFN genes IFN-β, IL-29 (IFN-λ1), and IL-28A (IFN-λ2). The major IFN protein secreted by type II cells was IL-29 and ATIIs appear to be a major resource for production of IL-29. Administration of IL-29 and IFN-β before infection significantly reduced the release of infectious viral particles and CXC and CC chemokines. IL-29 treatment of type II cells induced mRNA expression of antiviral genes MX1, OAS, and ISG56 but not IFN-β. IL-29 induced a dose-dependent decrease of viral nucleoprotein and an increase of antiviral genes but not IFN-β. These results suggest that IL-29 exerts IFN-β-independent protection in type II cells through direct activation of antiviral genes during IAV infection.
Influenza A virus (IAV)3 is a common pathogen that can cause severe viral pneumonia with alveolar inflammation and an infiltration of neutrophils and mononuclear cells (1). The main antigenic determinants of influenza A viruses are the hemagglutinin (H or HA) and neuraminidase (N) transmembrane glycoproteins. Based on the antigenicity of these glycoproteins, influenza A viruses are further subdivided into 16 H (H1–H16) and 9 N (N1–N9) subtypes. H1N1 and H3N2 subtypes are the main isolates currently circulating in the human population. The original H1N1 strain or Spanish flu caused a worldwide pandemic, with an estimated 20–50 million deaths (2, 3). This virus has recently been reconstituted by reverse genetics and shown to be extremely virulent (2). Avian influenza is a recent, emerging infection that has a mortality rate of ~50% (4). In comparison to human H1N1 viruses, high pathogenic strains of avian influenza virus, H5N1, can produce severe viral pneumonia and a strong proinflammatory response or cytokine storm (5, 6). Although influenza can produce devastating pneumonia, studies on primary human alveolar epithelial cells are limited.
The alveolar epithelium lines the gas exchange portion of the lung and is the main target for influenza A pneumonia. The alveolar epithelium is composed of two main cell types: type I and type II cells. Alveolar type II cells (ATIIs) synthesize and secrete pulmonary surfactant, secrete chemokines and cytokines, and participate in the innate immune response of the lung (7). Recent studies have shown that avian flu receptor α2,3-linked sialic acid is predominantly expressed by ATIIs (8–12). Hence, the response of differentiated ATIIs to influenza A infection is very important for understanding the pathogenesis of viral pneumonia. We have developed a culture system for maintaining differentiated ATIIs that is suitable for studying viral infections (13, 14).
Type I IFNs and the recently discovered type III IFNs (15, 16), are well known for their antiviral activities. In humans, the type I IFN family consists of 14 IFN-α genes, single IFN-β, IFN-ω, limitin, and IFN-κ genes (17, 18). These genes are all localized to chromosome 9 and use the same receptor, the IFN-αβR (19). The type III IFNs (IFN-λ) identified in 2003 include IL-29 (IFN-λ1), IL-28A (IFN-λ2), and IL-28B (IFN-λ3), are located on chromosome 19, and bind to a different receptor (IFN-λR) (15, 16, 20). The type III IFN receptor IFN-λR is made up of two subunits: IFNLR1 and IL-10R2 (21). IFNLR1 is IFN-λ specific and under stringent expression regulation; it is not detected in fibroblasts or endothelial cells (15). The other unit, IL-10R2, is ubiquitously expressed and shared with IL-10, IL-22, and IL-26. Type I and type III IFNs have similar functions and are induced by a number of viral infections including IAV, encephalomyocarditis virus, and vesicular stomatitis virus (16, 22, 23). Although these two types of IFNs use different receptors (IFN-αβR and IFN-λR), their initial signaling is through JAK1 and STAT1 and both have significant antiviral activity (24).
The purpose of this study is to explore the innate immune response elicited by differentiated human ATIIs infected with well-characterized influenza viruses. We investigated the inflammatory response and IFN production in ATIIs and alveolar macrophages (AMs) infected by a H1N1 wild-type virus (A/PR/8/34) and a H3N2 reassortant PR/8 virus (A/Phil/82). To our knowledge, this is the first report to investigate influenza infection in differentiated ATIIs and AMs from the same donor and should help us to better understand the alveolar response to IAV infection.
Materials and Methods
Donor information
We obtained human lungs from deidentified organ donors whose lungs were not suitable for transplantation but donated for medical research. In general, we selected donors with reasonable lung function with a PaO2: FIO2 ratio of >250, a clean clinical history and chest radiograph that does not indicate infection, and a limited time on the ventilator. The gender, age, and smoking history are variable and have not been selection criteria. The human donors used in this article included 8 males and 16 females with age ranges from 18 to 87, and there were 12 current smokers, 2 exsmokers, and 10 nonsmokers. Hence, there is great deal of variability among the donors, as expected.
Isolation and culture of human alveolar type II and macrophage cells
Human ATIIs were isolated from deidentified human lungs that were not suitable for transplantation and donated for medical research as described previously (13, 14). The committee for the Protection of Human Subjects at National Jewish Medical and Research Center approved this research. In brief, the middle lobe was perfused, lavaged, and then instilled with elastase (12.9 U/ml; Roche Diagnostics) for 50 min at 37°C. The lung was minced and the cells were isolated by filtration and partially purified by centrifugation on a discontinuous density gradient made of Optiprep (Accurate Chemical Scientific) with densities of 1.080 and 1.040 and by negative selection with CD14-coated magnetic beads (Dynal Biotech) and binding to IgG-coated petri dishes (Sigma-Aldrich). The cells were counted and cytocentrifuge cell preparations were made to assess cell purity by staining for cytokeratin (CAM 5.2; DakoCytomation). The yield of ATIIs was ~300 × 106 per isolation and the purity was ~80% before plating and over 95% after adherence in culture (13). The purity of isolated AMs is ~90% before plating and close to 100% after adherence in culture.
Isolated cells were plated on millicell inserts (Millipore) coated with a mixture of rat tail collagen and Matrigel (BD Biosciences) in 10% FBS in DMEM and then cultured with 1% charcoal-stripped FBS along with keratinocyte growth factor, isobutylmethylxanthine, 8-bromo-cAMP, dexamethasone, and antibiotics to achieve their differentiated phenotype as described in detail previously (13, 14).
AM cells were isolated from the same donor from which corresponding ATIIs were obtained. Briefly, the lung was lavaged with HEPES-buffered saline and 2 mM EDTA and the lavage fluid was centrifuged at 4°C for 10 min. If there were a significant number of RBC, the RBC were lysed with Pharm Lyse (BD Biosciences). The macrophages were resuspended in DMEM supplemented with 10% FBS, 2 mM glutamine, 2.5 μg/ml amphotericin B, 100 μg/ml streptomycin, 100 U/ml penicillin G, and 10 μg/ml gentamicin (Life Technologies) and plated on tissue culture plates. After a 4-h or overnight adherence, the cells were washed with DMEM to remove the nonadherent cells and then were cultured for 2 more days before infection. The purity of the AM preparations was confirmed by immunostaining for CD68 (clone KP1; DakoCytomation).
Virus preparation
IAV was grown in the chorioallantoic fluid of 10-day-old chicken eggs and purified on a discontinuous sucrose gradient as previously described (25). The virus was dialyzed against PBS without added calcium or magnesium to remove sucrose, aliquoted, and stored at −80°C until needed. The wild-type H1N1 strain A/PR/8/34 was a gift from Dr. J. Abramson (Bowman Gray School of Medicine, Winston-Salem, NC). The HA titer of each virus preparation was determined by titration of virus samples in PBS with thoroughly washed human type O, Rh− RBC as described previously (25). The A/Phil/82 was originally provided by Dr. E. Margot Anders (University of Melbourne, Melbourne, Australia); this virus is a reassortant PR/8 virus, the HA, N, and NP genes are from Philippines/2/82 (H3N2) and the PB1, PB2, M, and NS genes are from PR/8. After thawing, the viral stocks contained 1 × 109 PFU/ml.
Influenza viral infection of ATIIs and AMs
Primary ATIIs and AMs were cultured as described above and inoculated with MOCK (PBS), A/PR/8/34, or A/Phil/82 at a MOI of 0.5–1; after a 1 h adsorption, the inoculum was aspirated, cells were washed two times with DMEM, and refed with cell-type appropriate medium and incubated at 37°C in an atmosphere of 10% CO2. The MOI was a calculated MOI based on DNA measurements for plating efficiency. At designated times after inoculation, triplicate samples of culture supernatant were collected for virus titration and cytokine analysis. RNA and protein were extracted from cells for gene and protein expression as described previously (13).
Double immunostaining
At 24 h postinoculation (hpi), ATIIs and AMs were fixed in methanol and stained for viral Ags and cell markers. Macrophages were stained with goat anti-human influenza A viral Ag and mouse anti-CD68. The type II cells were stained for goat anti-human surfactant protein A (Santa Cruz Biotechnology) and mouse anti-influenza A viral Ag (Millipore) according to standard protocols as described previously (13).
Infectious viral release curve
The culture supernatant from infected AMs and ATIIs was used for evaluation of viral release. For type II cells, only the apical medium was evaluated. The virus in the medium was titrated in Madin-Darby canine kidney cells (MDCK) with a fluorescent focus assay as prescribed previously (26). Briefly, confluent MDCK cells were infected with diluted IAV-infected cell supernatant for 45 min at 37°C in PBS, followed by washing in serum-free DMEM. The monolayers were then incubated for 7 h at 37°C with 5% CO2 in DMEM. After washing, the monolayers were fixed with 80% acetone and 20% H2O2 (v/v) for 10 min at 4°C and then stained with mouse anti-influenza A viral nucleoprotein (NP). The foci were counted with fluorescent microscopy. Dilutions of virus were used to find the dose yielding 50 fluorescent foci per high-powered (magnification, ×40) field. The results from three independent donors were averaged, and the viral titers were expressed as log10 fluorescent focus unit/ml. Additional studies of viral release were done to confirm these findings with a TCID50 assay.
Real-time RT-PCR
Real-time RT-PCR was used to measure the mRNA expression of IFN and antiviral gene expression as previously described (13). The specific primers and probes for IFNs that we designed used in this study are IFN-β (forward: 5′-CTTACAGGTTACCTCCGAAACTGAA-3′; reverse: 5′-TTGAAGAATGCTTGAAGCAATTGT-3′, probe: 5′-ATCTCCTAGCCTGTGCCTCTGGGACTG-3′); IL-29 (forward: 5′-GGGAACCTGTGTCTGAGAACGT-3′; reverse: 5′-GAGTAGGGCTCAGCGCATAAATA-3′; probe: 5′-CTGAGTCCACCTGACACCCCACACC-3′); and IL-28A (forward: 5′-GGCCTCTGTCACCTTCAACCT-3′; reverse, 5′-CCCCACTGGCAACACAATTC-3′; probe: 5′-TCCGCCTCCTCACGCGAGACC-3′). 36B4 expression was used to normalize equal mRNA copy for each sample (13). The specific probes for MX1, OAS, and ISG56 were purchased from Applied Biosystems) and designed by the manufacturer.
Western blotting
Protein expression of influenza A viral NP and GAPDH was detected by Western blotting modified slightly from the standard protocol (13). Different from previous Western blotting, fluorescent-labeled secondary Abs were used instead of HRP-conjugated secondary Abs (13). The final development was conducted using the Odyssey system (L1-COR Biosciences). Mouse anti-GAPDH was purchased from Promega, mouse anti-influenza A NP Ab was acquired through Millipore, and secondary Abs were obtained through L1-COR Biosciences.
Multiplex bead-based cytokine analysis
A human multiplex Ab bead kit (Invitrogen) was used to measure chemokine and cytokine secretion from infected cells. The method allows simultaneous evaluation of 25 human cytokines, chemokines, and growth factors: IL-1α, IL-1ß, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p40, IL-13, IL-15, IL-17, IFN-α, IFN-γ, TNF-α, GM-CSF, MCP-1, MIP-1α, MIP-1ß, IFN-induced protein 10 (IP-10), eotaxin, RANTES, and monokine induced by IFN-γ (monokine induced by IFN-γ (MIG)). The sensitivity of each cytokine varied. The multiplexed assay was performed at the National Jewish Luminex Core Facility according to the manufacturer’s instructions. To generate a standard curve, 2-fold serial dilutions of appropriate standards were prepared in DMEM. Standards and supernatant samples were pipetted at 50 μl/well and mixed with 50 μl of the bead mixture. After a 1-h incubation, wells were washed three times with washing buffer using a vacuum manifold. PE-conjugated secondary Ab was added for 45 min, the wells were washed twice, and samples were analyzed using the Bio-Plex suspension array system, which includes a fluorescent reader and Bio-Plex Manager analytic software (Bio-Rad). One hundred beads were counted for each analyte per well. Values (pg/ml) were calculated using Bio-Rad software.
IFN treatment of type II cells
Human IFN-β (PBL InterferonSource) and IL-29 (R&D Systems) were used to treat ATIIs preinfection. ATIIs were cultured for 7 days and treated with IFN-β and IL-29 for 18 h, the cells were then washed two times and subjected to viral inoculation. For assay of viral protein expression, viral release, and chemokine secretion, the cells were harvested 24 hpi. In addition, total RNA was harvested at 4 hpi for evaluation of antiviral gene expression by real-time RT-PCR.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 4. One-way ANOVA was used to compare the difference between two or more groups. Appropriate post hoc tests were selected for multiple comparisons. A value of p < 0.05 was considered significant with all methods.
Results
Differentiated human ATIIs support a productive IAV infection
Human ATIIs were isolated and cultured in vitro for 8 days to achieve the differentiated phenotype (13). The differentiated ATIIs were then infected with influenza A/PR/8/34 or A/Phil/82. Fig. 1, top panel, indicates both viruses infected ATIIs similarly and maximal titer was reached by 24 hpi and maintained through 48 hpi (Fig. 1, lower panel). There was obvious detachment in wild-type A/PR/8/34-infected cells at 48 hpi, whereas no significant cytopathic effect was observed in reassortant PR/8 virus (A/Phil/82)-infected cells. We also examined the infectious viral release in IAV-infected A549 cells, but we did not detect a productive infection in the MDCK assay without exogenous proteases (data not shown).
FIGURE 1.

Influenza A viruses induce a productive infection in differentiated human ATIIs. Top panels, Influenza infection in ATIIs. Primary cultured differentiated human ATIIs were inoculated with influenza A viruses for 1 h at a MOI of 0.5 and harvested at 24 hpi for immunofluorescent stain of viral Ag (red) and surfactant protein A (green). Bottom panel, Infective viral release from virus-infected ATIIs. Supernatants from infected cells at designated times after inoculation (MOI = 0.5) were collected for quantitation of infectious virus by fluorescent focus assay. Data represent results from three independent donors.
IAV infection induces a strong inflammatory response in ATIIs
The IAV-infected cells were evaluated for the secretion of 25 chemokines and cytokines by Luminex multiplex technology at 24 hpi. As shown in Table I, influenza greatly stimulated the production of IL-6, IP-10, MIP-1α, MIP-1β, and RANTES by type II cells. To confirm the observed differences between virus- and MOCK-infected cells, culture supernatants were further evaluated for chemokine release by independent ELISAs. The level of IL-8 after infection was out of range in the Multiplex assay, but it was measured by ELISA. As shown in Fig. 2, top panel, both viruses induced a significant secretion of proinflammatory cytokines such as IL-6, IL-8, RANTES, MCP-1, and MIP-1β. In general, wild-type A/PR/8/34 was a much stronger inducer of cytokines than A/Phil/82. Additional studies showed a time- and dose-dependent secretion of cytokines in several different individuals (data not shown). In addition, heat inactivation of virus almost abolished the increase of IL-8 and MCP-1 (Fig. 2, bottom panel).
Table I.
Influenza A increases secretion of inflammatory cytokines in type II cellsa
| Cytokine | MOCK (pg/ml) | A/PR/8/34 (pg/ml) | A/Phil/82 (pg/ml) |
|---|---|---|---|
| IL-1β | 3.2 ± 2.5 | 33.4 ± 4.2 | 15.6 ± 8.4 |
| IL-1Ra | 29.2 ± 4.7 | 226.5 ± 57.3 | 96.8 ± 18.2 |
| IL-6 | 71.8 ± 6.8 | 5376 ± 1388.8 | 4553 ± 1417 |
| IP-10 | 0 | 4496 ± 2027 | 584.1 ± 148.5 |
| TNF-αb | 0 | 3.5 ± 2.6 | 0.1 ± 0.06 |
| MIP-1α | 1.1 ± 1.1 | 258.7 ± 142.6 | 54.5 ± 9.9 |
| MIP-1βb | 0.4 ± 0.2 | 721.7 ± 366.1 | 50.2 ± 6.3 |
| RANTESb | 18.4 ± 7.4 | 3583.7 ± 2550.4 | 362.6 ± 12.1 |
| MCP-1b | 434.5 ± 78.0 | 7865.3 ± 4267.3 | 4154.2 ± 946.2 |
Culture supernatants from influenza virus-infected human ATII collected at 24 hpi were tested by Luminex multiplex technology. The data represent mean and SE of four independent donors.
Indicates result verified by independent ELISA.
FIGURE 2.

Influenza A viral infection induces secretion of inflammatory cytokines. Primary cultured human type II cells were exposed to influenza A viruses or heat-inactivated virus at a MOI of 0.5 for 1 h. Culture supernatants were collected at 24 hpi for measurement of selected cytokines by ELISA. Top panels, Verification of multiplex data by ELISA in different donors. Bottom panels, Heat inactivation of virus abolishes the virus-induced chemokine response. The heat-inactivated viruses are designated as I-PR/8 and I-Phil/82. Data represent results from three independent experiments from different individuals.
IL-29 (IFN-λ1) is the main IFN secreted by virus-infected ATIIs
IFN production in response to viral infection was measured by real-time RT-PCR and ELISA. Both influenza A viruses induced early increases of mRNA synthesis of IFN genes IFN-β, IL-28A (IFN-λ2), and IL-29 (IFN-λ1) at 4 and 24 hpi (Fig. 3, top panel), we did not detect any induction of mRNA of IFN-α and IFN-γ (data not shown). To confirm that the observed differences of mRNA are reflected in levels of protein secretion, secretion of IFN-β and IL-29 (IFN-λ1) was measured by ELISA. Consistent with RNA data, significant secretion of IL-29 was detected in medium of virus-infected cells, as shown in Fig. 3, bottom panel. Both viruses induced a time- and dose-dependent IL-29 response; again, the A/PR/8/34 virus was a much stronger inducer of IL-29 than A/Phil/82. However, we failed to detect any IFN-β protein secreted from the supernatant of ATIIs after influenza viral infection at the limit of detection level of ELISA, 25 pg/ml (data not shown). We further compared the release of IL-29 in viral-infected ATIIs and AMs by independent ELISA and found ATIIs produced more IL-29 than AMs from the same donors (Fig. 4, top panel). These results suggest that IL-29 is the main IFN protein induced by IAV infection in ATIIs.
FIGURE 3.

IAV infection induces IFN production in ATIIs. Top panels, Influenza infection increases mRNA level of IFN genes. Human type II cells were infected by IAV and total RNA were extracted for evaluation of IFN gene expression by real-time RT-PCR. The data show the relative level of corresponding gene to a constitutive probe, 36B4. Bottom panels, IAV infection induces a dose- and time-dependent IL-29 (IFN-λ1) release from differentiated type II cells. Left panel, Dose-dependent IL-29 secretion; right panel, time-dependent IL-29 secretion. Human type II cells were infected with influenza A viruses and culture supernatants from infected cells were evaluated for IL-29 protein by ELISA at 24 hpi. One-way ANOVA was used to compare the differences between two or more groups. Appropriate post hoc tests were used for multiple comparisons. *, Indicates there is a difference between the virus and control group; #, indicates there is difference between PR/8 and Phil/82.
FIGURE 4.

Type II cells secrete IL-29 and macrophages secrete TNF-α in response to IAV infection. Cultured human ATIIs and macrophages from the same donor were inoculated with IAV at the same MOI. Supernatant medium was collected for quantitation of IL-29 and TNF-α and protein by ELISA.
IL-29 plays a direct antiviral role in ATIIs in response to IAV infection through activation of antiviral genes
Because wild-type virus A/PR/8/34 was the main chemokine and cytokine inducer, it was chosen for evaluation of the antiviral effect of IL-29. Differentiated type II cells were treated with IL-29 (IFN-λ1) and IFN-β before infection and examined for influenza A NP expression and antiviral gene expression at designated times after infection. At 4 hpi, IL-29 alone induced the mRNA expression of MX1 and OAS and ISG56 in human type II cells. In virus-infected type II cells, IL-29 pretreatment induced a dose-dependent mRNA increase of antiviral genes MX1, OAS, and ISG56 but not IFN-β in virus-infected cells (Fig. 5). At 24 hpi, a significant decrease of NP expression was detected by Western blotting in IL-29- and IFN-β-treated cells (Fig. 6). To investigate whether IL-29 decreases the infectious viral release and inflammatory response, independent experiments were performed with IL-29. As shown in Fig. 7, pretreatment with IL-29 or IFN-β significantly reduced viral release and infection-induced secretion of MIP-1β, RANTES, MCP-1, and IL-8.
FIGURE 5.

IL-29 treatment increases antiviral gene expression in A/PR/8/34 virus-infected ATIIs. Type II cells were treated with IL-29 and IFN-β 18 h before infection. The mRNA expression of antiviral genes was evaluated by real-time RT-PCR at 4 hpi. Data represent one experiment of three independent experiments with different donors that produced the same results.
FIGURE 6.
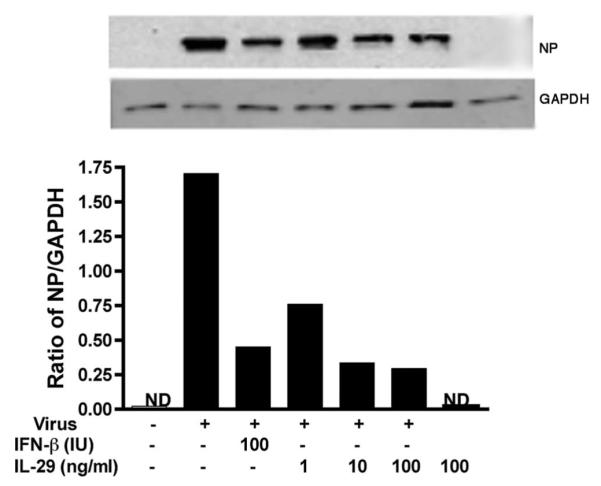
IL-29 pretreatment decreases viral NP expression in A/PR/8/34 virus-infected ATIIs. Type II cells were treated with IL-29 and IFN-β 18 h before infection. The protein was harvested at 24 hpi for detection of NP expression by Western blotting and normalized to the level of GAPDH. Data represent one experiment of three independent experiments with different donors that produced the same results.
FIGURE 7.

IL-29 and IFN-β pretreatment reduces viral release and chemokine secretion in ATIIs in response to influenza A infection. Type II cells were treated with IL-29 and IFN-β before infection. Top panel, IFN treatment significantly reduced viral release. Bottom panels, IFN treatment significantly decreased A/PR/8/34-induced inflammatory chemokine secretion. The infectious viral release and chemokine secretion were measured by plaque assay and ELISA, respectively. Data represent one experiment from three independent experiments with different donors that produced the same results.
AMs secrete significantly increased TNF-α in response to IAV infection
AMs from the same donors as ATIIs were also investigated for IAV infection and viral release. Immunofluorescent studies demonstrated that >90% of the cells were infected by virus at 24 hpi in AMs at same MOI used in the ATII infections (Fig. 8, top panel). However, there was also no increased release of virus above the level of residual inoculum even at 48 hpi with the absence of exogenous protease (Fig. 8, bottom panel). There was no significant cytopathic effect observed at this time. In addition, there was no increase in viral release by a TCID50 assay in the presence or absence of exogenous trypsin (data not shown). Cytokine secretion from AMs from the same donors as ATIIs was also evaluated by the Luminex multiplex assay. As compared with ATIIs, AMs are a rich source of chemokines and cytokines, secrete a higher level of cytokines than ATIIs under basal adherent conditions, and secrete significant levels of TNF-α, IFN-α, MIP-1α, MIP-1β, IP-10 (CXCL10), and MIG (CXCL9) after viral infection (Table II). Notably, AMs secrete TNF-α in response to viral infection, whereas ATIIs do not (Fig. 4, bottom panel). In addition, heat inactivation of the virus partially decreased the secretion of IL-8, MCP-1, RANTES, and MIP-1β, and almost completely inhibited the secretion of TNF-α in virus-treated AMs (Fig. 9).
FIGURE 8.

Influenza A viruses induce a nonproductive infection in human AMs. Top panels, Influenza infection in AMs. Primary cultured AMs were inoculated with influenza A viruses for 1 h at a MOI of 0.5 and harvested at 24 hpi for the immunofluorescent stain of viral Ag (green) and CD68 (red). Bottom panel, Infective viral release from virus-infected AMs. Primary cultured AMs were inoculated with influenza A viruses for 1 h at a MOI of 0.5 and supernatants from infected cells were collected at designated times after inoculation (MOI = 0.5) for quantitation of infectious virus by fluorescent focus assay. Data represent results from three independent donors.
Table II.
Influenza A increases the secretion of inflammatory cytokines in AMsa
| Cytokine | MOCK Mean (pg/ml) |
A/PR/8/34 Mean (pg/ml) |
A/Phil/82 Mean (pg/ml) |
|---|---|---|---|
| IL-1β | 2.6 | 41.0 | 38.5 |
| IL-1Ra | 345 | 1,505 | 861 |
| IL-6b | 402 | 1,931 | 1,216 |
| TNF-αb | 121 | 2,018 | 2,582 |
| IP-10 | 328 | 12,001 | 8,266 |
| MIP-1α | 5,514 | 55,714 | 81,268 |
| MIP-1βb | 4,333 | 33,652 | 13,793 |
| RANTESb | 381 | 10,094 | 2,191 |
| MCP-1b | 19,994 | 23,651 | 26,857 |
Culture supernatants from influenza virus-infected human AMs collected at 24 hpi were tested by Luminex multiplex technology. The data represent the mean from two independent donors.
Indicates data verified by ELISA.
FIGURE 9.
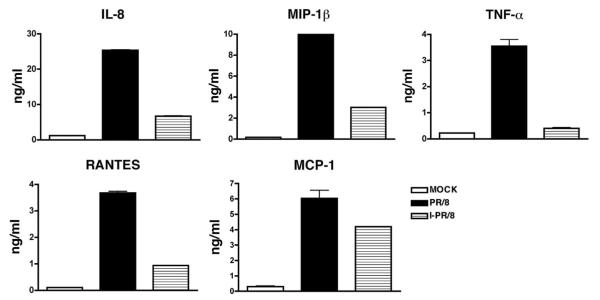
Heat inactivation decreases the cytokine response induced by A/PR/8/34 in AMs. Cultured AMs were exposed to alive and heat-inactivated A/PR/8/34 virus. Secretion of IL-8, MCP-1, MIP-1β, RANTES, and TNF-α was measured by ELISA at 24 hpi. Data represent one experiment from five independent experiments with different donors.
Discussion
In this study, we compared the innate immune response induced by IAV virus A/PR/8/34 (wild type) and A/Phil/82 (reassortant PR/8 virus). Although both viruses have similar ability to infect and replicate in ATIIs, the wild-type strain A/PR/8/34 is a stronger inducer of chemokines and cytokines. The result from the fluorescent focus assay without exogenous trypsin indicates that IAV infection induces a productive viral release in differentiated ATIIs (Fig. 1) but not in A549 cells (data not shown), an extensively used tumor-derived pulmonary epithelial cell line. Thus, study of the IAV infection in A549 cells might not reflect the in vivo type II situation. In this study, we also investigated IAV infection in AMs from the same donor. Consistent with the previous report (27), there is no productive viral release from IAV-infected AMs even though a similar amount of virus causes higher infection rates in AMs than that in ATIIs (Fig. 7). This suggests that A549 cells and AMs might lack certain cellular proteases that are often required to cleave viral proteins to form the mature infectious virus particles (28).
Recently, Bottcher et al. (29) reported trypsin-like protease TMPRSS2 (transmembrane protease, serine 2) and HAT (human airway trypsin-like protease) proteolytically activate influenza A virus in A549 and MDCK cells to induce a productive infection (29). Interestingly, we found that differentiated ATIIs but not AMs express a high level of TMPRSS2 mRNA by microarray experiments (data not shown) that might help to explain why AMs do not support a productive infection but ATIIs do. Because secondary bacterial infection is a common complication in severe influenza infection, further study is required to investigate the effect of viral infection on the phagocytic function of human AMs.
A robust cytokine response is the first line of lung defense against external challenges. However, excessive innate immune responses may also cause additional tissue damage and inflammation. This scenario may be especially true with pandemic and avian flu infections in which it is proposed that a cytokine storm is a major reason for the increased levels of mortality. Recently, numerous studies suggest an important role for alveolar epithelial cells in the regulation of the pulmonary inflammatory response to environmental insults such as LPS and respiratory viruses (30–32). As shown in Table I and Fig. 2, virus-infected differentiated human ATIIs secrete abundant IL-8 (CXCL8) and MCP-1 (CCL2) that are thought to be main stimulants for neutrophil and monocyte migration in acute lung inflammation (32, 33). IAV infection also induces other CXCL and CC chemokines such as IP-10 (CXCL10), RANTES (CCL5), MIP-1α (CCL3), and MIP-1β (CCL4). In addition, chemokine production by ATIIs is dependent on viral replication (Fig. 2, bottom panel). Our results provide strong evidence that distal lung epithelial cells directly contribute to IAV-induced lung inflammation and would be a critical target to control the avian or pandemic flu.
Production of type I IFNs is a classic way the host combats viral infection. However, cell types differ with respect to the amount and types of IFN produced during infection. As reported by Kumagai et al. (34), AMs are thought to be major IFN-α producers in pulmonary infection with RNA viruses, whereas IFN-β is produced mainly by epithelial cells, dendritic cells, or fibroblasts during the infection (35). We are showing here that IL-29 (IFN-λ1) might be the main IFN protein secreted by alveolar type II epithelial cells in response to IAV infection. By comparison in wild-type A/PR/8 virus-infected AMs and ATIIs from the same donors, we found that epithelial cells not macrophages are the main resource for IFN-λ production. In an effort to determine whether IL-29 plays a protective role in the IAV infection in ATIIs, we found that IL-29 alone increased mRNA of antiviral genes MX1, OAS1, and ISG56. Human MX1, also named MxA, is an IFN-induced gene. The MxA protein belongs to the GTPase family and targets the viral NP to inhibit the replication of the IAV (36, 37). Recent evidence suggests that the antiviral activity of MxA differs markedly with the strains of influenza virus and the PR/8 virus has a very high sensitivity to human MxA protein (37, 38)). The OAS1 gene encodes a member of the 2′,5′-oligoadenylate synthetase (2′,5′OAS) family, which is involved in the innate immune response to viral infection. It also functions on the inhibition of viral replication through activation of latent RNase (RnaseL) to degrade viral RNA (39). ISG56 is also an IFN-induced gene. Different from the induction of MxA, which is dependent on IFN signaling and Stat1, ISG56 can be directly induced by the influenza virus (40, 41). IL-29 treatment before viral infection induced a dose-dependent increase of antiviral gene expression and a decrease of infectious virus release and virus-induced cytokine response. These data indicate that IL-29 has a direct antiviral function in human ATIIs in response to influenza infection through activation of antiviral genes (22, 42).
Different from type I IFN genes, IFN-λ genes have several introns and are located on chromosome 19 instead of 9. They use different receptors. The IFN-λ-specific receptor IFNLR1 (IL-28RA) has much more limited expression in different cell types and organs (19, 43). It has been shown that, in vitro, cell response to IFN-λ is closely dependent on the expression of the IL-28RA receptor subunit (44). We compared the IL-28RA mRNA expression in cell lines from T cells, B cells, monocytes, and epithelial cells and found that IL-28RA is specifically expressed by epithelial cells (data not shown). This suggests that IL-29 might have special role in protection of the epithelium. The response to IFN-λ is also more restricted to epithelial cells than type I IFNs (19, 43). In contrast, the response to IFN-α/β is observed in various cell types in these organs and is prominent in endothelial cells (45). Although type I IFN has stronger antiviral effects than IFN-λ (46), side effects such as high fever hinder its clinical use, presumably because of the widespread expression of the type I receptor. In our study, we found that IFN-β treatment also protected type II cells in response to viral infection. However, an increase of IFN-β mRNA was not observed in IL-29-treated type II cells compared with viral infection alone which suggests that the antiviral effect of IL-29 is independent of IFN-β. This is supported by the very recent in vivo study that demonstrated administration of IFN-λ readily induced the antiviral factor of MX1 in mouse lungs and efficiently protected IFNAR1 knockout mice from lethal influenza virus infection. This study also concluded that IFN-λ contributes to resistance against viral pathogens infecting the lung but not the liver (47).
Contoli et al. (48) reported that treatment with IFN-λ decreases the viral titer and IL-8 secretion in rhinovirus-infected asthma patients (48). Deficient induction of IFN-λ was highly correlated to the severity of virus-induced asthma exacerbation. It is interesting that IL-29 alone stimulates production of IL-6, IL-8, and IL-10 production but not TNF-α and IL-1, the known proinflammatory cytokines in PBMC (49). IL-29 increases mRNA of CXC chemokine MIG/CXCL9, IP-10/CXCL10, and I-TAC/CXCL11 in PBMC (50). As shown in Fig. 7, the secretion of IL-8, RANTES, MCP-1, and MIP-1β are decreased by IL-29 pretreatment accompanying a decrease of infectious viral release. Additional studies are needed to determine whether IL-29 also regulates the immune response in virus-infected type II cells besides its direct antiviral activity.
Although in humans the replication of the influenza virus is generally restricted to the epithelial cells of the upper and lower respiratory tracts, AMs also play an important role in viral infection through their function as the pivotal phagocytic defense cells in the lung and secrete proinflammatory cytokines. We and others found that AMs produce a higher level of cytokines under basal conditions than ATIIs (Table II). Influenza A induces a strong CXCL and CC response in both ATIIs and AMs when compared with the basal condition (Tables I and II). This indicates that both cell types likely play a significant role in the regulation of neutrophil and monocyte recruitment into the alveolar air space during IAV infection. As shown in Fig. 2, bottom panel, the chemokine response in ATIIs is mainly virus replication dependent, whereas, heat inactivation of A/PR/8 virus did not completely abolish the virus-induced chemokine secretion in AMs (Fig. 9), which suggests other factors other than virus also contribute to the chemokine response in AMs. As shown in Table II and Fig. 4, AMs specifically secrete TNF-α, which is a well-known proinflammatory cytokine and has been extensively documented in viral infection-induced inflammation including IAV; therefore, the TNF-α-induced second wave of chemokine secretion might explain the extra chemokine response besides viral infection in AMs (51–53).
The current studies involved well-characterized laboratory strains of influenza virus, and the observations will need to be confirmed with circulating strains of influenza and expanded into more virulent strains, such as H5N1 strains. It is interesting to note that the cytopathic and cytokine response was less with the recombinant virus in which different HA, NA, and NP genes were present, although infectivity was not altered.
In conclusion, influenza A virus induces an robust innate immune response in both human ATIIs and AMs. These two cell types differ in the amount and types of cytokines produced, especially TNF-α. Differentiated ATIIs are the main source for production of IL-29 in response to influenza A infection. IL-29 exerts an antiviral effect during influenza A infection through activation of antiviral genes. The molecular mechanism of influenza A-induced IL-29 production in type II cells, the interaction of ATIIs and resident AMs in virus-induced lung inflammation, and the signaling pathway involved in IL-29- induced antiviral response require further investigation.
Acknowledgments
We thank Dr. Hong Wei Chu for support with the project. We also thank Rizwan Manzer, Karen Edeen, Emily Travanty, Yoko Ito, Beata Kosmider, and Xueni Chen for assistance with human type II cell isolations. Finally, we thank Teneke M. Warren and Caroline Cook for assistance with manuscript preparation.
Footnotes
This work was supported by National Institutes of Health Grant HL-029891, the Parker B. Francis Fellowship Foundation, U.S. Army Medical Research and Material Command Grant W81XWH-07-1-550; and the ExxonMobil Foundation.
Abbreviations used in this paper: IAV, influenza A virus; HA, hemagglutinin; N, neuraminidase; ATII, alveolar type II cell; AM, alveolar macrophage; MOI, multiplicity of infection; hpi, hour postinoculation; MDCK, Madin-Darby canine kidney cell; NP, nucleoprotein; IP-10, IFN-induced protein 10.
Disclosures The authors have no financial conflict of interest.
References
- 1.Yeldandi AV, Colby TV. Pathologic features of lung biopsy specimens from influenza pneumonia cases. Hum. Pathol. 1994;25:47–53. doi: 10.1016/0046-8177(94)90170-8. [DOI] [PubMed] [Google Scholar]
- 2.Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, Cox NJ, Katz JM, Taubenberger JK, Palese P, Garcia-Sastre A. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 3.Luk J, Gross P, Thompson WW. Observations on mortality during the 1918 influenza pandemic. Clin. Infect. Dis. 2001;33:1375–1378. doi: 10.1086/322662. [DOI] [PubMed] [Google Scholar]
- 4.Thitithanyanont A, Engering A, Ekchariyawat P, Wiboon-ut S, Limsalakpetch A, Yongvanitchit K, Kum-Arb U, Kanchongkittiphon W, Utaisincharoen P, Sirisinha S, et al. High susceptibility of human dendritic cells to avian influenza H5N1 virus infection and protection by IFN-α and TLR ligands. J. Immunol. 2007;179:5220–5227. doi: 10.4049/jimmunol.179.8.5220. [DOI] [PubMed] [Google Scholar]
- 5.Tran TH, Nguyen TL, Nguyen TD, Luong TS, Pham PM, Nguyen VC, Pham TS, Vo CD, Le TQ, Ngo TT, et al. Avian influenza A (H5N1) in 10 patients in Vietnam. N. Engl. J. Med. 2004;350:1179–1188. doi: 10.1056/NEJMoa040419. [DOI] [PubMed] [Google Scholar]
- 6.Chan MC, Cheung CY, Chui WH, Tsao SW, Nicholls JM, Chan YO, Chan RW, Long HT, Poon LL, Guan Y, Peiris JS. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 2005;6:135. doi: 10.1186/1465-9921-6-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mason RJ. Biology of alveolar type II cells. Respirology. 2006;11(Suppl.):S12–S15. doi: 10.1111/j.1440-1843.2006.00800.x. [DOI] [PubMed] [Google Scholar]
- 8.Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, Holtzman MJ, Brody SL. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 2006;80:7469–7480. doi: 10.1128/JVI.02677-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, Kuiken T. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am. J. Pathol. 2007;171:1215–1223. doi: 10.2353/ajpath.2007.070248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinya K, Ebina M, Yamada S, Ono M, Kasai N, Kawaoka Y. Avian flu: influenza virus receptors in the human airway. Nature. 2006;440:435–436. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 11.Gu J, Xie Z, Gao Z, Liu J, Korteweg C, Ye J, Lau LT, Lu J, Gao Z, Zhang B, et al. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet. 2007;370:1137–1145. doi: 10.1016/S0140-6736(07)61515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholls JM, Chan MC, Chan WY, Wong HK, Cheung CY, Kwong DL, Wong MP, Chui WH, Poon LL, Tsao SW, et al. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat. Med. 2007;13:147–149. doi: 10.1038/nm1529. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Edeen K, Manzer R, Chang Y, Wang S, Chen X, Funk CJ, Cosgrove GP, Fang X, Mason RJ. Differentiated human alveolar epithelial cells and reversibility of their phenotype in vitro. Am. J. Respir. Cell Mol. Biol. 2007;36:661–668. doi: 10.1165/rcmb.2006-0410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mossel EC, Wang J, Jeffers S, Edeen KE, Wang S, Cosgrove GP, Funk CJ, Manzer R, Miura TA, Pearson LD, et al. SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology. 2008;372:127–135. doi: 10.1016/j.virol.2007.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 16.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-λ mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 17.LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, Taylor K, Buergin M, Chinchilla D, Roshke V, et al. Interferon-κ, a novel type I interferon expressed in human keratinocytes. J. Biol. Chem. 2001;276:39765–39771. doi: 10.1074/jbc.M102502200. [DOI] [PubMed] [Google Scholar]
- 18.Oritani K, Medina KL, Tomiyama Y, Ishikawa J, Okajima Y, Ogawa M, Yokota T, Aoyama K, Takahashi I, Kincade PW, Matsuzawa Y. Limitin: an interferon-like cytokine that preferentially influences B-lymphocyte precursors. Nat. Med. 2000;6:659–666. doi: 10.1038/76233. [DOI] [PubMed] [Google Scholar]
- 19.Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annu. Rev. Biochem. 1987;56:727–777. doi: 10.1146/annurev.bi.56.070187.003455. [DOI] [PubMed] [Google Scholar]
- 20.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 21.Commins S, Steinke JW, Borish L. The extended IL-10 superfamily: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. J. Allergy Clin. Immunol. 2008;121:1108–1111. doi: 10.1016/j.jaci.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 22.Osterlund P, Veckman V, Siren J, Klucher KM, Hiscott J, Matikainen S, Julkunen I. Gene expression and antiviral activity of α/β interferons and interleukin-29 in virus-infected human myeloid dendritic cells. J. Virol. 2005;79:9608–9617. doi: 10.1128/JVI.79.15.9608-9617.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siren J, Imaizumi T, Sarkar D, Pietila T, Noah DL, Lin R, Hiscott J, Krug RM, Fisher PB, Julkunen I, Matikainen S. Retinoic acid inducible gene-I and mda-5 are involved in influenza A virus-induced expression of antiviral cytokines. Microbes Infect. 2006;8:2013–2020. doi: 10.1016/j.micinf.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 24.Jouanguy E, Zhang SY, Chapgier A, Sancho-Shimizu V, Puel A, Picard C, Boisson-Dupuis S, Abel L, Casanova JL. Human primary immunodeficiencies of type I interferons. Biochimie. 2007;89:878–883. doi: 10.1016/j.biochi.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 25.Hartshorn KL, Collamer M, Auerbach M, Myers JB, Pavlotsky N, Tauber AI. Effects of influenza A virus on human neutrophil calcium metabolism. J. Immunol. 1988;141:1295–1301. [PubMed] [Google Scholar]
- 26.Hartshorn KL, White MR, Tecle T, Holmskov U, Crouch EC. Innate defense against influenza A virus: activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J. Immunol. 2006;176:6962–6972. doi: 10.4049/jimmunol.176.11.6962. [DOI] [PubMed] [Google Scholar]
- 27.Rodgers BC, Mims CA. Influenza virus replication in human alveolar macrophages. J. Med. Virol. 1982;9:177–184. doi: 10.1002/jmv.1890090304. [DOI] [PubMed] [Google Scholar]
- 28.Kobasa D, Rodgers ME, Wells K, Kawaoka Y. Neuraminidase hemadsorption activity, conserved in avian influenza A viruses, does not influence viral replication in ducks. J. Virol. 1997;71:6706–6713. doi: 10.1128/jvi.71.9.6706-6713.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006;80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeyaseelan S, Manzer R, Young SK, Yamamoto M, Akira S, Mason RJ, Worthen GS. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am. J. Respir. Cell Mol. Biol. 2005;32:531–539. doi: 10.1165/rcmb.2005-0063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am. J. Physiol. 2004;287:L143–L152. doi: 10.1152/ajplung.00030.2004. [DOI] [PubMed] [Google Scholar]
- 32.Herold S, von Wulffen W, Steinmueller M, Pleschka S, Kuziel WA, Mack M, Srivastava M, Seeger W, Maus UA, Lohmeyer J. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J. Immunol. 2006;177:1817–1824. doi: 10.4049/jimmunol.177.3.1817. [DOI] [PubMed] [Google Scholar]
- 33.Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am. J. Pathol. 2000;156:1951–1959. doi: 10.1016/S0002-9440(10)65068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S. Alveolar macrophages are the primary interferon-α producer in pulmonary infection with RNA viruses. Immunity. 2007;27:240–252. doi: 10.1016/j.immuni.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Erlandsson L, Blumenthal R, Eloranta ML, Engel H, Alm G, Weiss S, Leanderson T. Interferon-β is required for interferon-α production in mouse fibroblasts. Curr. Biol. 1998;8:223–226. doi: 10.1016/s0960-9822(98)70086-7. [DOI] [PubMed] [Google Scholar]
- 36.Pavlovic J, Haller O, Staeheli P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J. Virol. 1992;66:2564–2569. doi: 10.1128/jvi.66.4.2564-2569.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dittmann J, Stertz S, Grimm D, Steel J, Garcia-Sastre A, Haller O, Kochs G. Influenza A virus strains differ in sensitivity to the antiviral action of Mx-GTPase. J. Virol. 2008;82:3624–3631. doi: 10.1128/JVI.01753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benfield CT, Lyall JW, Kochs G, Tiley LS. Asparagine 631 variants of the chicken Mx protein do not inhibit influenza virus replication in primary chicken embryo fibroblasts or in vitro surrogate assays. J. Virol. 2008;82:7533–7539. doi: 10.1128/JVI.00185-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levy DE, Garcia-Sastre A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 2001;12:143–156. doi: 10.1016/s1359-6101(00)00027-7. [DOI] [PubMed] [Google Scholar]
- 40.Holzinger D, Jorns C, Stertz S, Boisson-Dupuis S, Thimme R, Weidmann M, Casanova JL, Haller O, Kochs G. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J. Virol. 2007;81:7776–7785. doi: 10.1128/JVI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fensterl V, White CL, Yamashita M, Sen GC. Novel characteristics of the function and induction of murine p56 family proteins. J. Virol. 2008;82:11045–11053. doi: 10.1128/JVI.01593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, Storey H, Yao L, Liu H, Barahmand-pour F, Sivakumar P, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44:896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- 43.Uze G, Monneron D. IL-28 and IL-29: newcomers to the interferon family. Biochimie. 2007;89:729–734. doi: 10.1016/j.biochi.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 2007;81:7749–7758. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-λ (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meager A, Visvalingam K, Dilger P, Bryan D, Wadhwa M. Biological activity of interleukins-28 and -29: comparison with type I interferons. Cytokine. 2005;31:109–118. doi: 10.1016/j.cyto.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, Klucher K, Staeheli P. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 2008;4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, Kebadze T, Mallia P, Stanciu LA, Parker HL, et al. Role of deficient type III interferon-λ production in asthma exacerbations. Nat. Med. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 49.Jordan WJ, Eskdale J, Boniotto M, Rodia M, Kellner D, Gallagher G. Modulation of the human cytokine response by interferon λ-1 (IFN-λ1/IL-29) Genes Immun. 2007;8:13–20. doi: 10.1038/sj.gene.6364348. [DOI] [PubMed] [Google Scholar]
- 50.Pekarek V, Srinivas S, Eskdale J, Gallagher G. Interferon λ-1 (IFN-λ1/IL-29) induces ELR− CXC chemokine mRNA in human peripheral blood mononuclear cells, in an IFN-γ-independent manner. Genes Immun. 2007;8:177–180. doi: 10.1038/sj.gene.6364372. [DOI] [PubMed] [Google Scholar]
- 51.Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR, Sambhara S, Tumpey TM, Katz JM. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J. Virol. 2007;81:2736–2744. doi: 10.1128/JVI.02336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matikainen S, Siren J, Tissari J, Veckman V, Pirhonen J, Severa M, Sun Q, Lin R, Meri S, Uze G, et al. Tumor necrosis factor α enhances influenza A virus-induced expression of antiviral cytokines by activating RIG-I gene expression. J. Virol. 2006;80:3515–3522. doi: 10.1128/JVI.80.7.3515-3522.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou J, Law HK, Cheung CY, Ng IH, Peiris JS, Lau YL. Functional tumor necrosis factor-related apoptosis-inducing ligand production by avian influenza virus-infected macrophages. J. Infect. Dis. 2006;193:945–953. doi: 10.1086/500954. [DOI] [PMC free article] [PubMed] [Google Scholar]