

Abstract
New catalysts for non-directed hydrocarbon functionalization have great potential in organic synthesis. We hypothesized that incorporating a Mn-terpyridine cofactor into a protein scaffold would lead to artificial metalloenzymes (ArMs) in which the selectivity of the Mn cofactor could be controlled by the protein scaffold. We designed and synthesized a maleimide-substituted Mn-terpyridine cofactor and demonstrated that this cofactor could be incorporated into two different scaffold proteins to generate the desired ArMs. The structure and reactivity of one of these ArMs was explored, and the broad oxygenation capability of the Mn-terpyridine catalyst was maintained, providing a robust platform for optimization of ArMs for selective hydrocarbon functionalization.
Keywords: Artificial metalloenzyme, Manganese, C-H functionalization, Epoxidation, Biocatalysis
1. Introduction
New catalysts for non-directed hydrocarbon functionalization have great potential for applications in organic synthesis.1,2 By avoiding the need for directing groups, these species expand the range of substrates on which they can act and eliminate synthetic steps and byproducts associated with directing group installation and removal.3 A number of catalysts, including dirhodium tetracarboxylate complexes4 and several different iron5 and manganese6,7 complexes, are particularly notable in this regard and are widely used for insertion of carbene,8 nitrene,8 and oxo9 fragments into C-H bonds and olefins. While reactivity trends for such catalysts have been outlined, controlling their selectivity,9 particularly on complex substrates,1,2 remains difficult.
Crabtree and Brudvig have demonstrated that the selectivity of Kemp’s-triacid-based Mn-terpyridine complexes for oxygenation of certain carboxylic acid-substituted substrates can be controlled by catalyst-substrate hydrogen bonding.10–13 The reported examples illustrate the potential for supramolecular interactions to control the selectivity of Mn-terpyridine catalysts but require the use of a carboxylic acid directing group. We hypothesized that incorporating Mn-terpyridine cofactors into protein scaffolds would lead to artificial metalloenzymes (ArMs)14 in which the selectivity of the Mn cofactor could be controlled by substrate binding to the protein. Mn-terpyridine complexes catalyze a number of oxygenation reactions,15 ethereal α-C-H oxidation,15 and hydrocarbon desaturation,16 suggesting that the proposed ArMs could possess broad reaction scope. The utility of ArMs for selective catalysis has been demonstrated for a number of different reactions.17 Hayashi recently demonstrated that myoglobin reconstituted with a Mn-porphycene cofactor catalyzes benzylic oxygenation,18 but we are unaware of any reports of similar reactions using ArMs generated via covalent scaffold modification14. While biocatalysts (e.g. cytochromes P450) for this reaction exist, altering their substrate scope and site selectivity can be challenging.19,20 The potential ease with which a Mn-terpyridine cofactor could be incorporated into different proteins21 to modulate selectivity led us to develop a robust platform to explore the feasibility of this approach.
2. Result and discussion
Maleimide-substituted cofactor 3 was synthesized22,23 from the known amine-substituted terpyridine, 124,25 (Scheme 1).
Scheme 1.
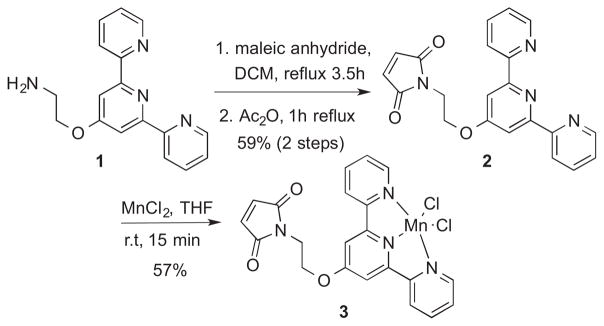
Synthesis of cofactor 3
Cysteine mutants of tHisF26 and aponitrobindin (Nb)27 were then prepared as scaffolds for initial bioconjugation studies. Both of these proteins have been used as ArM scaffolds by us28 and by others29,30 and therefore serve as ideal substrates for initial studies on cofactor bioconjugation using 3. Reacting tHisF-C48 with 3 provided modest conversion (ca. 40%) to tHisF-C48-3 within 2h (Table 1, entry 1), but longer reaction times appeared to give multiple addition products (ESI-MS, data not shown), a common problem with maleimide bioconjugation reactions31. On the other hand, we found that mutating a leucine residue (L50) located directly above C48 in the β-barrel of tHisF26 led to a marked increase in bioconjugation rate and selectivity, and over 80% conversion to tHisF-C48A50-3 was achieved within 2h (entry 2). These improvements presumably resulted from decreased steric hindrance proximal to the cysteine nucleophile.
Table 1.
Mass spectrometry and conversion data for ArMs
| Entry | Scaffold (MW)a | Cofactor (MW)b | MWArMa | MWobsc | Conv (%)c |
|---|---|---|---|---|---|
| 1 | tHisF-C48 (28754) | 3 (372) | 29126 | 29128 | 40 |
| 2 | tHisF-C48A50 (28712) | 3 (372) | 29084 | 29082 | 85 |
| 3 | Nb-C96 (19490) | 3 (372) | 19862 | 19862 | 90 |
| 4 | Nb-C125 (19547) | 3 (372) | 19919 | 19919 | 95 |
Protein MW calculated by using tools at http://www.scripps.edu/~cdputnam/protcalc.html.
The MnCl2 fragment not detected.
Observed MW and approximate conversion from ESI-MS after 2h reaction.
Nitrobindin is a heme protein with a β-barrel structure and a large hydrophobic pocket for heme binding. Cysteine mutations at suitable locations within the β-barrel can be used to position cofactors within the heme binding pocket of the apo enzyme (Nb).30 Bioconjugation of two cysteine mutants of Nb, C96 within the β-barrel and C125 on the protein surface, were examined, and both of these provided greater than 90% conversion to the corresponding Nb-C-3 bioconjugates (entries 3 and 4). These results, in addition to those noted above using tHisF, highlight two key advantages of covalent bioconjugation for ArM formation:21 the nature of both the scaffold itself and the local environment of the bioconjugation site can be readily manipulated to facilitate cofactor installation14.
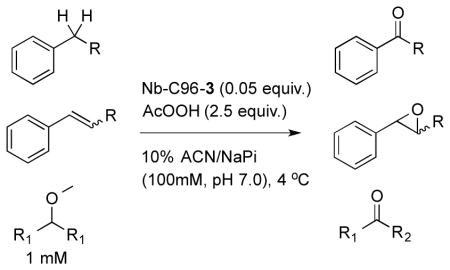
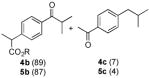
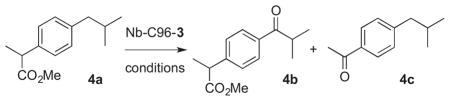
Following characterization of Nb-C96-3, the reactivity of this ArM under a variety of conditions was explored to examine the impact of confining the Mn center within the heme binding pocket. Benzylic oxygenation of ibuprofen methyl ester (4a) was initially investigated based on previous reports from Crabtree and Brudvig.13 As previously noted, these researchers reported that ibuprofen (5a) oxygenation can be catalyzed by Kemp’s-triacid-based Mn-terpyridine complexes to provide a mixture of 5b and 5c, the latter resulting from decarboxylation following oxygenation (Table 3). The selectivity of this reaction could be controlled using hydrogen bonding between carboxylic acid groups on ibuprofen and the catalyst, with the presence of the carboxylic acid on the catalyst improving the 5b:5c ratio from 77:23 to 98.5:1.5.
Table 3.
Substrate scope for ArM catalyzed oxidation
| ||
|---|---|---|
| Entry | Substrate | Products (% conv.) |
| 1a |
 4a (R=Me) 5a (R=H) |
|
| 2b |
 6a |
|
| 3b |
 7a |
|
| 4a |
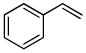 8a |
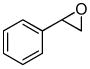 8b (65) |
| 5c |
9a |
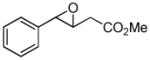 9b (80) |
| 6a |
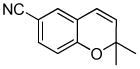 10a |
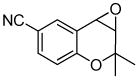 10b (93) |
| 7a |
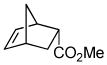 11a |
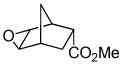 11b (99) |
| 8a |
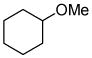 12a |
 12b (59) |
Conversion determined using GC integration values for starting materials and products.
Conversion determined using 1H-NMR signals for starting materials and products.
Conversion determined using HPLC integration values for starting materials and products.
We found that similar benzylic oxygenation and oxygenation/decarboxylation of 4a was catalyzed by Nb-C96-3 (Table 2). Oxone, peracids, and a number of additional oxidants are known to promote Mn-terpyridine catalyzed oxygenation, and both oxone and peroxyacetic acid proved to be compatible with Nb-C96-3. Crucial to this reactivity, however, was the use of phosphate buffer (Table 2, entries 1–3). A modest excess of peroxyacetic acid was sufficient to achieve maximum conversion, and this helped to minimize pH changes over the course of the reaction (entries 3–5). While the Nb scaffold can tolerate 20% v/v acetonitrile, greater than this amount led to protein precipitation and dramatically reduced conversion. Maximum conversion was observed using 10% v/v acetonitrile, and omitting acetonitrile entirely led to reduced conversion, presumably due to poor substrate solubility (entries 5–8). Conducting the reactions at room temperature or decreasing ArM loading to 2.5 mol% both led to significant decreases in conversion (entries 9–10). All reactions in Table 2 provided a similar 4b:4c ratio of 88:12. Nb-C125-3 provided this same ratio with slightly higher conversion, suggesting that improved substrate access to 3 attached to the protein surface (in Nb-C125-3) offset any potential scaffold acceleration32 afforded by the heme binding pocket (in Nb-C96-3).
Table 2.
Optimization of ArM catalyzed oxidationa
| |||
|---|---|---|---|
| Entry | Solvent | Oxidant | Convb. (%) |
| 1 | Tris (25mM, 7.0), 20%ACN | Oxone, 10eq | 0 |
| 2 | NaPi (100mM, 7.0), 20%ACN | Oxone, 10eq | 77 |
| 3 | NaPi (100mM, 7.0), 20%ACN | AcOOH, 10eq | 85 |
| 4 | NaPi (100mM, 7.0), 20%ACN | AcOOH, 2.5eq | 83 |
| 5 | NaPi (100mM, 7.0), 20%ACN | AcOOH, 2eq | 76 |
| 6 | NaPi (100mM, 7.0), 30%ACN | AcOOH, 2.5eq | 12e |
| 7 | NaPi (100mM, 7.0), 10% ACN | AcOOH, 2.5eq | 91 |
| 8 | NaPi (100mM, 7.0) | AcOOH, 2.5eq | 24 |
| 9c | NaPi (100mM, 7.0), 10% ACN | AcOOH, 2.5eq | 45e |
| 10d | NaPi (100mM, 7.0), 10% ACN | AcOOH, 2.5eq | 58 |
Reaction conditions: 1 mM substrate, 5% ArM, 4 °C, overnight in dark.
Determined by GC analysis of DCM extract.
Reaction conducted at room temperature
2.5% ArM loading used.
Protein precipitation was observed.
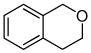
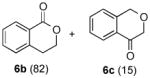
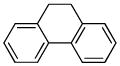
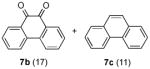
The optimized conditions identified in Table 2, entry 7 were then used to explore the substrate scope of Nb-C96-3 catalyzed oxygenation.15 Both ibuprofen methyl ester and ibuprofen provided high conversions (Table 3, Entry 1), and benzylic oxygenation (4/5b) and decarboxylation (4/5c) products were formed in similar ratios. Isochroman (6a) underwent benzylic oxygenation with a preference for the more activated 1-position (entry 2), while 9,10-dihydrophenanthrene (7a) was oxidized to a mixture of diketone 7b and phenanthrene (entry 3). Epoxidation of different olefins was also examined. Styrene and the β-substituted styrylacetic acid methyl ester were both epoxidized in good yield (entries 4 and 5). Cyclic cis-internal olefins were particularly reactive substrates, and nearly quantitative epoxidation of both chromene 10a and norbornene-2-carboxylate methyl ester 11a was observed (entries 6 and 7). Finally, ethereal α-C-H oxidation of cyclohexyl methyl ether 12a, which proceeds via C-H hydroxylation followed by hemiacetal decomposition, was also observed in moderate yield (Entry 8).
The substrate scope outlined in Table 3 shows that the broad reactivity of Mn-terpyridine complexes toward hydrocarbon oxidation15 is not compromised in the context of a protein scaffold. In general, a slight decrease in reaction rate was observed using Nb-C96-3 relative to 3 (see supporting information), but otherwise similar reactivity was observed. In entries 1–3, product mixtures were obtained, and while no difference between the selectivities of 3 and Nb-C96-3 was observed, these substrates will serve as valuable probes to explore the impact of scaffold mutations on oxygenation selectivity in future studies.33,34 A preliminary investigation of epoxidation enantioselectivity revealed that an e.r. of 53.5:46.5 was observed in the reaction of 10a (racemic product was obtained using 3; see supporting information), while the remaining olefins provided racemic product. While obviously low, this result provides a starting point for engineering scaffold mutants with improved selectivity.33,34
3. Conclusions
Maleimide-substituted manganese terpyridine cofactor 3 was prepared and reacted with cysteine mutants of tHisF and Nb scaffold proteins to generate the desired ArMs in good to excellent yield based on HR ESI-MS. Spectroscopic characterization (CD and UV) of Nb-C96-3 provided additional details on the structure of this bioconjugate, confirming both the nature of the scaffold fold and the presence of the MnCl2 fragment. Nb-C96-3 catalyzed hydrocarbon oxygenation in the presence of either Oxone or AcOOH, and optimized reaction conditions enabled benzylic oxygenation, olefin epoxidation, and heteroatom demethylation15. The substrates examined, in conjunction with the robust ArM reactivity observed, will enable exploration of the impact of scaffold structure on the regio-, enantio-, and chemo-selectivity of 3. Toward this end, we are currently examining a range of cofactor linkage sites within the Nb scaffold as well as mutagenesis of residues proximal to the Mn center in Nb-C96-3. The generality of the covalent linkage method employed21 will also enable facile exploration of additional scaffold proteins to identify systems with unique hydrocarbon oxygenation selectivity.
4. Experimental Section
All reagents were obtained from commercial suppliers and used without further purification. Acetonitrile (ACN), tetrahydrofuran (THF) and dichloromethane (DCM) were obtained from a PureSolv MD solvent purification system by Innovative Technology (solvent deoxygenated by N2 sparge and dried over alumina). “Extra Dry” grade methanol purchased from Acros was used. Deuterated solvents were obtained from Cambridge Isotope Labs. Known compound 1 was prepared as previously reported.
4.1. Cloning, Expression and Purification of Nitrobindin Protein
A codon optimized gene for Nitrobindin protein (Nb, PDB ID 3EMM) was obtained from GenScript (Piscataway, NJ) and cloned into pET22b using NdeI and XhoI restriction sites. A gene encoding the cyclase subunit of the imidazole glycerol phosphate synthase enzyme complex from Thermotoga maritima (tHisF, PDB ID 1THF) was amplified from pET11c-tHisF20 by PCR using gene specific primers containing NdeI (forward) and XhoI (reverse) restriction sites. Both the genes were cloned such that they would expresses with a C-terminal hexa-histidine tag for Ni-NTA affinity chromatography. Overlap extension PCR was used to introduce point mutations, and all constructs were confirmed by sequencing. Mutations encoding a Cysteine (TGC) at desired locations (C96 and C125 for Nb and C48 for tHisF) were introduced for ArM formation. For the tHisF double mutant (C48A50), leucine was replaced by alanine using overlap extension PCR with the gene encoding tHisF-C48 as a template.
For expression, mutants were transformed into E. coli BL21 (DE3) cells. The cells were recovered in LB for 1 h at 37 °C and then plated on LB/agar plates containing 100μg/mL ampicillin. Single colonies were picked and used to inoculate 2YT media containing 100μg/mL ampicillin. The resulting cultures were incubated at 37 °C while shaking at 250 RPM until the OD600 reached approximately 1.0, at which point IPTG was added to final concentrations of 1mM. After incubating for 16 h post induction, cells were harvested by centrifugation and lysed in phosphate-buffered saline (pH 7.5) by sonication. The crude lysate was clarified by centrifugation at 16000g at 4 °C for 30 min. The clarified lysate was then purified by Ni-NTA affinity chromatography at 4 °C. Pure protein was eluted using 50 mM NaH2PO4/300 mM NaCl/250 mM imidazole (pH 8.0) and the purified Nb was concentrated and stored in Tris buffer (10mM pH 7.5) using 10 kDa cutoff spin filters (Millipore).
4.2. Cofactor (3) synthesis
4.2.1. N-[2-([2,2′:6′,2″-terpyridin]-4′-yloxy) ethylmaleimide (2)
To a solution of 1 (292 mg, 1 mmol) in CH2Cl2 (10 mL) was added maleic anhydride (490 mg, 5 mmol). The mixture was heated at reflux for 3.5 h and subsequently cooled to room temperature. A white precipitate formed, was collected by filtration, and was washed with cold CH2Cl2. The solid was then added to a suspension of NaOAc (1 g) in Ac2O (10 mL), and the resulting mixture was heated at reflux for 1 h. The mixture was cooled to room temperature, poured into ice-water (40 mL), and stirred for 2 h. The mixture was extracted with CH2Cl2 (3×100 mL), and the combined organic extracts were washed sequentially with saturated NaHCO3 (3×50mL), H2O (1×50 mL), and brine (1×50 mL), and dried over MgSO4. Removing the solvent using rotary evaporation provided the desired product as a white solid; yield: 219 mg (0.589 mmol, 59%). 1H NMR (500 MHz, CDCl3): δ 8.72 (d, J = 4.6 Hz, 2H), 8.64 (d, J = 7.9 Hz, 2H), 8.02 (s, 2H), 7.88 (td, J = 7.7, 1.7 Hz, 2H), 7.37 (dd, J = 6.9, 5.3 Hz, 2H), 6.79 (s, 2H), 4.45 (t, J = 5.6 Hz, 2H), 4.08 (t, J = 5.6 Hz, 2H). 13C NMR (500 MHz, CDCl3): δ 170.5, 166.6, 157.3, 156.0, 149.2, 137.0, 134.4, 124.0, 121.5, 107.4, 64.9, 37.1. HRMS-ESI (m/z): for C21H16N4O3, calculated: 373.1295, found: 373.1290.
4.2.2. Manganese (II) terpyridine complex (3)
Manganese chloride tetrahydrate (212 mg, 1.07 mmol) was dissolved in 10 mL dry THF using sonication. This solution was added to a stirring solution of ligand 2 (50 mg, 0.134 mmol) dissolved in 1.07 mL dry THF. A fine yellow precipitate formed immediately upon addition. The yellow solid was collected on a fine frit and washed with copious THF to obtain 38 mg of product (0.076 mmol, 57% yield). IR nmax cm−1 (KBr pellet): 3486 (br), 3074 (w), 2062 (w), 1712 (s), 1598 (s), 1474 (m), 1435 (m), 1404 (m), 1384 (s), 1355 (m), 1255 (w), 1220 (s), 1161 (m), 1098 (m), 1063 (m), 1041 (w), 1014 (s), 982 (w), 947 (w), 837 (s), 799 (s), 729 (w), 694 (s), 668 (w), 660 (w), 639 (w), 621 (m). UV (1:1 ACN/water) λmax, nm: 310, 316. ESI-MS (m/z): 462.0298.
4.3. Bioconjugation and purification
50 μL of a solution of cofactor 3 (1.99 mg/mL in 1:1 water/acetonitrile, 2.0 equiv) was added to a solution of protein scaffold (950 μL, 100 μM protein in Tris buffer, 25 mM pH 7.5 for Nb and 50mM pH 7.2 for tHisF) in a microcentrifuge tube and shaken in the dark at 4 °C for 1–2 h. The final concentrations were: 100 μM protein, 200 μM cofactor 3, 2.5 vol% acetonitrile. Nb-C96-3 was purified by four cycles of centrifugal filtration using 10 kDa cutoff spin filters (Millipore) with NaPi buffer (100 mM, pH 7.0 with 10% ACN). The removal of excess cofactor was confirmed by HPLC analysis, and the conversion was estimated by ESI analysis.
4.4. General procedure for biocatalysis
A solution of Nb-C96-3 (500 μL, 100 μM), 500 μL NaPi buffer (100 mM, pH 7.0 with 10% ACN), and 12.5 μL substrate (80 mM stock in ACN) were added to 1.5 mL microcentrifuge tube, and incubated at 4 °C for 15 min. 5 μL of peracetic acid aqueous solution (1M) was added to the mixture. The resulting solution shaken at 4 °C overnight. The final concentrations were: 1 mM substrate, 2.5 mM AcOOH, and 50 hybrid. The reaction was quenched by adding 350 μL DCM. pH was adjusted to 2 for ibuprofen case. The closed vials were agitated using a vortexer for 30s and centrifuged at 15000 rpm for 3 min. The organic layer was isolated. 10 μL sample was injected on HPLC.
Supplementary Material
Figure 1.

A) HR ESI-MS spectra of Nb-C96 and Nb-C96-3 showing the efficiency of Nb bioconjugation. B) UV spectra of Nb-C96, Nb-C96-3, 2, and 3 showing the presence of MnCl2 in Nb-C96-3. C) CD spectra of Nb-C96 and Nb-C96-3 showing proper Nb folding following bioconjugation. D) Overlay of a DFT-optimized structure of 3 (red) covalently linked (Pymol) to a crystal structure of Nb (PDB ID 3EMM27) and the native heme (gray).
Acknowledgments
This work was supported by The David and Lucile Packard Foundation and the NSF under the CCI Center for Selective C–H Functionalization (CHE-1205646). K.E.-G. is a trainee funded by an NIH Chemistry and Biology Interface Training Grant (T32 GM008720). MS data were acquired using instruments purchased using an NSF instrumentation grant (CHE-1048528). We thank Dr. Elena Solomaha at the UC Biophysics Core Facility for assistance with CD spectra.
Footnotes
Supplementary materials, including ESI-MS, HPLC, and NMR characterization of synthetic intermediates and more experimental details associated with this article, can be found online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Davies HML, Morton D. Chem Soc Rev. 2011;40:1857. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]
- 2.Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem Rev. 2010;110:704. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- 3.White MC. Science. 2012;335:807. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 4.Kuhl N, Hopkinson MN, Wencel-Delord J, Glorius F. Angew Chem Int Ed. 2012;51:10236. doi: 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]
- 5.Costas M, Chen K, Que L., Jr Coord Chem Rev. 2000;200:517. [Google Scholar]
- 6.Talsi EP, Bryliakov KP. Coord Chem Rev. 2012;256:1418. [Google Scholar]
- 7.Tanase S, Bouwman E. Advances in Inorganic Chemistry. Vol. 58. Elsevier; 2006. Advances in Inorganic Chemistry; pp. 29–75. [Google Scholar]
- 8.Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- 10.Das S, Brudvig G, Crabtree R. J Am Chem Soc. 2008;130:1628. doi: 10.1021/ja076039m. [DOI] [PubMed] [Google Scholar]
- 11.Hull J, Sauer E, Incarvito C, Faller J, Brudvig G, Crabtree R. Inorg Chem. 2008;48:488. doi: 10.1021/ic8013464. [DOI] [PubMed] [Google Scholar]
- 12.Balcells D, Moles P, Blakemore JD, Raynaud C, Brudvig GW, Crabtree RH, Eisenstein O. Dalton Trans. 2009:5989. doi: 10.1039/b905317d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das S, Incarvito CD, Crabtree RH, Brudvig GW. Science. 2006;312:1941. doi: 10.1126/science.1127899. [DOI] [PubMed] [Google Scholar]
- 14.Lewis JC. ACS Catal. 2013;3:2954. [Google Scholar]
- 15.Kamijo S, Amaoka Y, Inoue M. Synthesis. 2010;2010:2475. [Google Scholar]
- 16.Hull JF, Balcells D, Sauer ELO, Raynaud C, Brudvig GW, Crabtree RH, Eisenstein O. J Am Chem Soc. 2010;132:7605. doi: 10.1021/ja908744w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinreiber J, Ward TR. Top Organomet Chem. 2009;25:93. [Google Scholar]
- 18.Oohora K, Kihira Y, Mizohata E, Inoue T, Hayashi T. J Am Chem Soc. 2013;135:17282. doi: 10.1021/ja409404k. [DOI] [PubMed] [Google Scholar]
- 19.Lewis JC, Arnold FH. CHIMIA. 2009;63:309. [Google Scholar]
- 20.Fasan R. ACS Catal. 2012;2:647. [Google Scholar]
- 21.Qi D, Tann C, Haring D, Distefano M. Chem Rev. 2001;101:3081. doi: 10.1021/cr000059o. [DOI] [PubMed] [Google Scholar]
- 22.Reetz MT, Rentzsch M, Pletsch A, Taglieber A, Hollmann F, Mondiere RJG, Dickmann N, Höcker B, Cerrone S, Haeger MC, Sterner R. Chem Bio Chem. 2008;9:552. doi: 10.1002/cbic.200700413. [DOI] [PubMed] [Google Scholar]
- 23.Chen H, Tagore R, Das S, Incarvito C, Faller J, Crabtree R, Brudvig G. Inorg Chem. 2005;44:7661. doi: 10.1021/ic0509940. [DOI] [PubMed] [Google Scholar]
- 24.Maayan G, Yoo B, Kirshenbaum K. Tetrahedron Lett. 2008;49:335. doi: 10.1016/j.tetlet.2007.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Constable EC, Ward MD. J Chem Soc Dalton Trans. 1990:1405. [Google Scholar]
- 26.Beismann-Driemeyer S, Sterner R. J Biol Chem. 2001;276:20387. doi: 10.1074/jbc.M102012200. [DOI] [PubMed] [Google Scholar]
- 27.Bianchetti CM, Blouin GC, Bitto E, Olson JS, Phillips GN., Jr Proteins. 2009;78:917. doi: 10.1002/prot.22617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang H, Srivastava P, Zhang C, Lewis JC. Chem Bio Chem. 2013;15:223. doi: 10.1002/cbic.201300661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reetz MT, Rentzsch M, Pletsch A, Taglieber A, Hollmann F, Mondiere RJG, Dickmann N, Hoecker B, Cerrone S, Haeger MC, Sterner R. Chem Bio Chem. 2008;9:552. doi: 10.1002/cbic.200700413. [DOI] [PubMed] [Google Scholar]
- 30.Onoda A, Fukumoto K, Arlt M, Bocola M, Schwaneberg U, Hayashi T. Chem Commun. 2012;48:9756. doi: 10.1039/c2cc35165j. [DOI] [PubMed] [Google Scholar]
- 31.Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego, CA, 92101, USA: 1996. [Google Scholar]
- 32.Collot J, Humbert N, Skander M, Klein G, Ward TR. J Organomet Chem. 2004;689:4868. [Google Scholar]
- 33.Reetz MT, Rentzsch M, Pletsch A, Maywald M, Maiwald P, Peyralans JJP, Maichele A, Fu Y, Jiao N, Hollmann F, Mondiere R, Taglieber A. Tetrahedron. 2007;63:6404. [Google Scholar]
- 34.Creus M, Ward TR. Org Biomol Chem. 2007;5:1835. doi: 10.1039/b702068f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.