

Migraine is the most common brain disorder, affecting approximately 14% of the adult population, but its molecular mechanisms are poorly understood. We report the results of a meta-analysis across 29 genome-wide association studies, including a total of 23 285 migraine cases and 95 425 population-matched controls. We identified 12 loci associated with migraine susceptibility (P < 5 × 10−8). Five loci are new (near AJAP1 on 1p36, near TSPAN2 on 1p13, within FHL5 on 6q16, within c7orf10 on 7p14, and near MMP16 on 8q21). Three of these loci were identified in disease subgroup analyses. Brain tissue eQTL analysis suggests potential functional candidate genes at four loci: APOA1BP, TBC1D7, FUT9, STAT6, and ATP5B.
Recently, significant progress has been made in the identification of common genetic variants associated with migraine susceptibility through genome-wide association (GWA) studies of clinic-based migraine with aura (MA) patients1, migraineurs from the general population2,3, and clinic-based migraine without aura (MO) patients4. To further elucidate the genetic susceptibility of migraine, we performed a meta-analysis of 23 285 individuals with migraine from 29 clinic- and population-based studies (Fig. 1, Supplementary Fig. 1, Supplementary Note). These include 5 175 cases from five clinic-based patient collections, matched to 13 972 population-based controls (Supplementary Table 1), as well as 18 110 cases from 14 population-based studies and 81 453 migraine-free or control individuals from the same studies (Supplementary Table 2). Results from GWA studies of the five clinic-based collections1,4 and four of the population-based collections2,3 have been previously reported (Supplementary Fig. 2).
Figure 1. Description of the studies comprising the International Migraine Genetics Meta-analysis Consortium, and their sample contributions to each analysis.

Each coloured box corresponds to one analysed phenotype and lists the total number of cases and controls, as well as the sample contributions of individual cohorts. Participation in each analysis depended on the availability of the data in question and the recruitment method.
In addition to the primary meta-analysis using all available genotype data, three subgroup analyses were performed in those cohorts where sufficient additional clinical information was available (Supplementary Table 3). The first two subgroups consisted of migraine cases fulfilling the International Headache Society diagnostic criteria5 for either MA or MO. The third subgroup included only the clinic-based samples, under the hypothesis that they represent a group of migraineurs more enriched for severe migraines than cases identified from the general population.
Results from the primary meta-analysis and the three subgroups identify 142 single nucleotide polymorphisms (SNPs), at a total of 12 loci, to be significantly associated with migraine susceptibility (Table 1, Supplementary Fig. 3,4). Eight of those loci contain SNPs that lie within a known transcript. In addition, 1 168 SNPs at 134 loci (Supplementary Table 4) showed suggestive association to migraine, again combining the primary analysis and the three subgroup analyses. The single most significant P value overall was observed for rs11172113 in the primary analysis (P value 2.69 × 10−19; Fig. 2a, Table 1) at the LRP1 locus on 12q13.
Table 1. Results of the association analyses.
| SNP | Chr | Position | Location | Gene | Minor allele | MAF | P-value | OR (95% CI) | q/p-value | I 2 | Group or subgroup with the lowest p-value | Additional significance in |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs2651899 | 1 | 3,073,572 | Genic | PRDM16 * | C | 0.41 | 3.28 × 10−14 | 1.09 (1.07-1.12) | 0.214 | 20% | All samples | |
| rs10915437 | 1 | 4,082,866 | Intergenic | near AJAP1 | G | 0.36 | 2.81 × 10−8 | 0.86 (0.82-0.91) | 0.108 | 47% | Clinics only | |
| rs12134493 | 1 | 115,479,469 | Intergenic | near TSPAN2 | A | 0.46 | 6.71 × 10−14 | 1.14 (1.10-1.18) | 0.480 | 14% | All samples | |
| rs2274316 | 1 | 154,712,866 | Genic | MEF2D * | C | 0.37 | 3.14 × 10−8 | 1.07 (1.04-1.09) | 0.021 | 45% | All samples | |
| rs7577262 | 2 | 234,483,608 | Genic | TRPM8 * | A | 0.10 | 3.27 × 10−13 | 0.87 (0.84-0.90) | 0.070 | 33% | All samples | Clinics only, MO only |
| rs6790925 | 3 | 30,455,089 | Intergenic | near TGFBR2 * | T | 0.38 | 2.16 × 10−8 | 1.15 (1.10-1.21) | 0.780 | 0% | Clinics only | |
| rs9349379 | 6 | 13,011,943 | Genic | PHACTR1 * | G | 0.40 | 2.81 × 10−10 | 0.86 (0.82-0.90) | 0.443 | 0% | MO only | All samples |
| rs13208321 | 6 | 96,967,075 | Genic | FHL5 | A | 0.22 | 2.15 × 10−12 | 1.18 (1.13-1.24) | 0.168 | 0% | All samples | MO only |
| rs4379368 | 7 | 40,432,725 | Genic | c7orf10 | T | 0.12 | 1.46 × 10−9 | 1.11 (1.08-1.15) | 0.441 | 2% | All samples | MO only |
| rs10504861 | 8 | 89,617,048 | Intergenic | near MMP16 | T | 0.16 | 1.32 × 10−8 | 0.86 (0.81-0.90) | 0.755 | 0% | MO only | |
| rs6478241 | 9 | 118,292,450 | Genic | ASTN2 * | A | 0.38 | 1.04 × 10−9 | 1.16 (1.11-1.22) | 0.646 | 0% | Clinics only | All samples |
| rs11172113 | 12 | 55,813,550 | Genic | LRP1 * | C | 0.43 | 2.69 × 10−19 | 0.90 (0.88-0.92) | 0.188 | 22% | All samples | MO only |
When multiple subgroups show significant association, P values and odds ratios (OR) are shown for the analysis with the lowest P value. ORs reported for the minor allele. Chromosomal positions are based on NCBI build 36. In the Location and Gene columns, SNPs located within the gene transcript (‘Genic’) list that gene, while for intergenic SNPs (‘Intergenic’) the gene reported for that locus is listed.
Previously identified migraine locus. MAF – minor allele frequency. q/p value - P value from Cochran’s heterogeneity statistic. I2 – heterogeneity index. MO – migraine without aura.
Figure 2. Manhattan plot of the results of the meta-analysis.
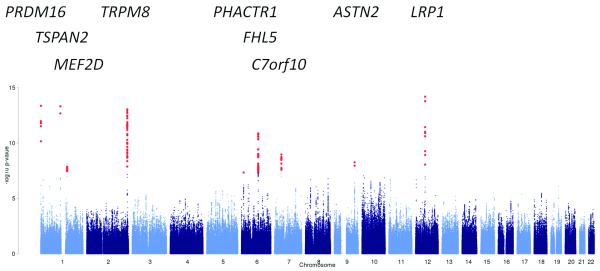
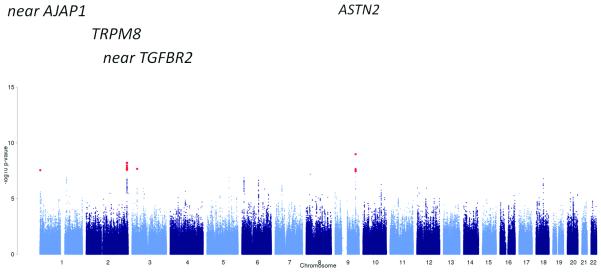
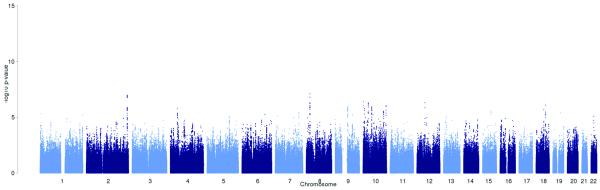
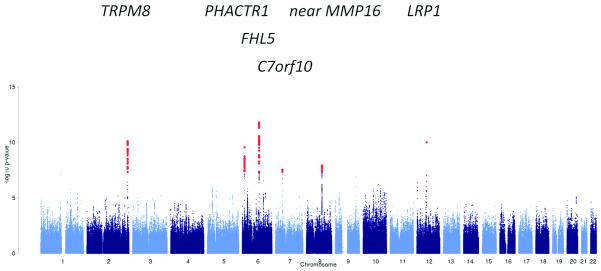
Results of the meta-analysis of all migraine cases, of any migraine subtype or recruiting method, versus all available controls, adjusting for gender. Red dots indicate SNPs with significant (< 5 × 10−8) P values.
A) All migraine (23 285 cases, 95 425 controls)
B) Clinic-based studies only (5 175 cases, 13 972 controls)
C) Migraine with aura (5 118 cases, 74 239 controls)
D) Migraine without aura (7 107 cases, 69 427 controls)
Five of the 12 genome-wide significant loci are new (near AJAP1, near TSPAN2, FHL5, c7orf10, and near MMP16), while seven confirm previously reported migraine loci (PRDM162, MEF2D4, TRPM81,2, near TGFBR24, PHACTR14, ASTN24, and LRP12). All seven previously reported loci seen in this study remained significant (all p < 6.25 × 10−3, correcting for eight previously reported loci) in analyses that exclude samples used in previous reports1,2,4 (Supplementary Fig. 5 and Supplementary Table 5). Among the newly identified loci, two contain SNPs with significant association that are located within known transcripts. On 6q16, FHL5 encodes a transcription factor that regulates cAMP responsive elements CREM and CREB6, which play a role in synaptic plasticity7 and memory formation8. The locus also overlaps KIAA0776, which encodes a hypothetical protein also known as UFL1. On 7p14, mutations in c7orf10 have been found in phenotypically mild or even clinically asymptomatic forms of glutaric aciduria type III9, a rare metabolic abnormality leading to persistent excretion of glutaric acid.
The novel loci on 1p36, 1p13, and 8q21 are located outside known transcripts. On 1p36, rs10915437 is located approximately 500 kb telomeric from AJAP1 and approximately 300kb centromeric from a gene cluster encoding the apoptosis-related proteins DFFB and TP73 as well as centrosomal protein CEP104. AJAP1 is expressed in brain (Supplementary Fig. 6) and has been associated with tumor invasion and regulation of metalloproteinase activity10. On 1p13, rs12134493 is located 87 kb 5′ of TSPAN2, a member of the tetraspanin family, encoding a cell surface protein that mediates signal transduction events involved in the regulation of cell development, activation, growth and motility11. TSPAN2 has further been shown to act as a regulator of metalloproteinase activity11. On 8q21, rs10504861 is located 200 kb telomeric from matrix metalloproteinase MMP16. Members of the metalloproteinase family are widely expressed in human tissues and are involved in the breakdown of extracellular matrix in normal physiological processes. Notably, the protein encoded by MMP16 (MT-MMP2) cleaves LRP112, encoded by a previously reported migraine gene2. In addition, MMP16 has recently been shown to be involved in basal NgR1 (Nogo-66 receptor) shedding in cortical neurons, thereby increasing axonal and synaptic plasticity13.
Four of the twelve loci (near AJAP1, near TGFBR2, PHACTR1, near MMP16), including two of the novel associations, were identified exclusively in the subgroup analyses (Table 1 and Supplementary Table 4). Two of the loci (rs9349379 in PHACTR1 on 6p21, and rs10504861 near MMP16 on 8q21) reached genome-wide significance only in MO, while no SNPs reached genome-wide significance in MA (Fig. 2c, d). The lowest P value in MA was with SNP rs7015657 (p = 7.88 × 10−8), which is located approximately 582 kb 3′ from GFRA2, a member of the glial cell line-derived neurotrophic factor family.
A similar subgroup analysis was performed in only those samples that originate from specialized migraine clinics. Two loci with suggestive association in the primary analysis, rs6790925 (near TGFBR2) and rs6478241 (ASTN2), reached genome-wide significance in the clinic-based subgroup (Table 1 and Supplementary Tables 6,7). All of the 12 genome-wide significant loci associated with migraine had larger estimated effect sizes in the clinic-based subgroup compared to the primary (All) analysis (Supplementary Table 6, 7). A two-tailed binomial test shows the chance of observing larger effects at all 12 loci is significantly different from that expected by chance (P = 4.88 × 10−4). Among all reported loci (P <1 × 10−5), only the clinic-based group showed a number of associated SNPs with higher effect sizes (OR > 1.2) at low frequency (MAF < 0.05; Supplementary Fig. 7). Thus, clinic-based migraine samples may represent a promising subgroup to help prioritise loci in the search for low frequency variants with moderate effects. Overall, among the 146 loci identified, twice as many have causative minor alleles over protective ones (with the ratio increasing towards the lower minor allele frequencies).
To explore the biological context for the identified loci we examined the properties of the most proximal genes to the 12 genome-wide significant top SNPs (Table 1). In expression data from 55 269 samples profiled using the Affymetrix HG-U133 Plus 2.0 microarray (including 1 990 brain and 384 endothelial samples), 11 of the 12 genes nearest to the identified loci (all except FHL5) were at least moderately (>20% of samples of the tissue showing a normalized log2 expression value greater than 6; see Methods) expressed in disease-relevant brain regions (Supplementary Fig. 6). In contrast, only TGFBR2 and MEF2D show moderate or greater expression in the endothelial samples. Possibly reflecting known co-morbidity between migraine and cardiovascular disease14, two of the 12 most proximal genes (TGFBR2 and PHACTR1) have also been associated with cardiovascular traits: TGFBR2 mutations have been reported to cause monogenic Marfan’s syndrome15 and to be involved in abdominal aortic aneurysms16, while PHACTR1 is associated with early onset myocardial infarction17. TSPAN218, MEF2D19, TRPM820, TGFBR221, PHACTR122, MMP1623, ASTN224, and LRP125 have been suggested to have functions in synaptic formation or regulation; PRDM16 has been linked to oxidative stress response, and AJAP1 in maintaining tissue borders (Supplementary Fig. 8).
To identify possible non-proximity based genes underlying these associations, we examined eQTL data among 394 samples of brain tissue from the North American Brain Expression Consortium and the UK Brain Expression Consortium. Among the 12 regions with significant association to migraine, four were found to contain significant eQTLs (see Methods) among the SNP-probe pairs within the tested brain samples (four in the frontal cortex, one in the cerebellum; Table 2). On chromosome 1, rs12136718 is an eQTL for APOA1BP, rather than the gene closest to it, MEF2D. APOA1BP is widely expressed and is potentially linked with cholesterol efflux from cells26. On chromosome 6, rs35128104 is an eQTL for FUT9, encoding α1,3-fucosyl transferase IX. The FUT9 enzyme synthesizes the Lewis X (Lex) carbohydrate structure, which has been implicated in neurite outgrowth in several types of brain neuronal cells27-29. At the chromosome 12 locus, two different eQTLs in brain tissue were found within the peak, for STAT6 (rs4559) and for ATP5B (rs113953523), the former at a very robust P-value (2.16 × 10−22). The STAT genes are known for transducing activation signals to transcription factors in macrophages30, and STAT6 phosphorylation has recently been shown to sense oxidative stress in astrocytes resulting in prostaglandin release31. A second eQTL gene at the same locus, ATP5B, is the β subunit of the mitochondrial ATP synthase, but a potential specific role in neuronal or vascular cells is not known. Finally, on chromosome 6, rs9349379 is an eQTL in cerebellar tissue for TBC1D7, that potentially down regulates the tuberous sclerosis gene, TSC1, through a positive regulation of the mTOR-signalling pathway32. In the central nervous system, TSC1/2 signalling contributes to neural connectivity via its multi-faceted roles33. Based on the available data it is not possible to decide whether a gene identified by the eQTL analysis or the gene closest to the strongest positional association is the most relevant gene contributing to migraine pathogenesis.
Table 2. Significant eQTLs observed in 394 samples of two different brain tissue at loci significantly associated with migraine.
| Frontal cortex | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chr | Position (bp) | Locus | Probe | Gene name | SNPs with sign. eQTL with probe |
SNP with lowest eQTL p-value |
Lowest eQTL p-value |
Highest LD with reported SNP |
SNPs with sign. eQTL with probe |
| 1 | 154,830,352 | MEF2D | ILMN_1729533 | APOA1BP | 1 | rs12136718 | 2.18 × 10−5 | 0.38 | 0 |
| 6 | 13,413,333 | PHACTR1 | ILMN_1661622 | TBC1D7 | 0 | - | - | - | 1 |
| 6 | 96,769,539 | FHL5 | ILMN_1878007 | FUT9 | 2 | rs35128104 | 5.96 × 10−5 | 0.74 | 0 |
| 12 | 55,775,568 | LRP1 | ILMN_1763198 | STAT6 | 40 | rs4559 | 2.16 × 10−22 | 1 | - |
| 12 | 55,318,337 | LRP1 | ILMN_1772132 | ATP5B | 1 | rs113953523 | 1.62 × 10−4 | 0.39 | 0 |
-Chr – chromosome. All basepair positions are given for build 36/hg18. ‘Highest LD…’ column indicates the maximum extent of LD observed between a significant eQTL SNP-probe pair (see Methods), and a SNP passing the reporting threshold (p< × 10−5) for association with migraine. Due to multiple overlapping signals, the HLA region has been excluded from the analysis.
In a hypersensitivity site analysis, associated SNPs in the migraine loci were found to occur significantly more often in DNAse I hypersensitivity sites in a number of tissues (Supplementary Fig. 9). This suggests that the loci associated with migraine are enriched for actively transcribed regions, supporting a regulatory role for the variants. Both neuronal and vascular tissue types carry an enriched set of sites within the detected loci. In addition, querying the RegulomeDB (see URLs) showed that several of the associated SNPs were found to overlap directly with known transcription factor binding motifs (Supplementary Table 8).
The number of significantly associated loci is still modest to form a broad understanding of the disease susceptibility, and any proposed functional hypothesis from the identified loci must thus be taken with caution. Some functional hypotheses could be inferred from the results of this study, as the majority of the identified loci harbour genes that can be linked to neuronal function.
The eQTL analysis further supported that regulatory effects in brain tissue may underlie several of the association signals. However, the lack of replication across brain regions suggests that our ability to use the eQTL data to pinpoint the functionally most significant gene within the locus is limited.
The observed difference between the number of significant loci in the MO and MA groups (6 vs 0, respectively; Fig. 2c, d) despite reasonably similar sample sizes was somewhat unexpected. MA has been shown to have a considerably higher heritability estimate and sibling recurrence risk than MO (3.8 vs 1.9), and has thus been considered to be the more heritable of the two common migraine types34. One possible explanation could be that the genetic susceptibility to MA is mediated more by rare variants with larger effect sizes, although this remains speculative. Another explanation for the difference may be a higher degree of heterogeneity among the MA cases (due to genetically distinct subgroups, for example). No common variants specifically predisposing to aura were identified by this study (the lowest observed P-value was p = 7.88 × 10−8).
In summary, we conducted a large migraine meta-analysis and identified 12 loci associated with migraine susceptibility, including five loci not previously associated with migraine, as well as 134 additional suggestive loci. An eQTL analysis of brain tissue highlighted a further five genes potentially implicated in migraine susceptibility. Two of the 12 loci were observed only in the clinic-based sample group, possibly suggesting more specificity to severe migraine headache, and two only in the MO group. Seven previously reported loci for migraine susceptibility were replicated in independent samples in this study. The difference in the number of identified loci and the strength of association suggest that the genetic background of MA is considerably less influenced by common variants than that of MO, contrary to previous expectations. Finally, while pathway analysis of the 146 loci showed no concentration in any particular pathway or tissue, eight of the 12 identified loci are located in or immediately outside genes with known function in synaptic or neuronal regulation and several of them exert regulatory control on one another.
Methods
Overall study design
For this meta-analysis, we used SNP marker data from 23 285 cases and 95 425 controls of European descent from 29 studies, including five clinic-based studies compared to population-matched control samples with unknown migraine status, as well as 14 entirely population-based cohorts. Four of the population-based cohorts (the B58C, NFBC, Young Finns and FinnTwin; see Supplementary Note for further details) were birth cohorts. The datasets for the meta-analysis included previously genotyped genome-wide association (GWA) study data from migraine-specific studies by the International Headache Genetics Consortium (see URLs) studies and the Women’s Genome Health Study, as well as a number of pre-existing population-based GWA studies (for complete list of references, see Supplementary Table 2). Local research ethics committees approved the individual studies, and informed consent was obtained from all participants when necessary (see the Supplementary Materials for full details of ethics and consent procedures for each study). Additional details on sample recruitment and phenotypes and summary details for each collection are given in the Supplementary Note and in Supplementary Tables 1 and 2. Genome-wide SNP genotyping was performed independently in each cohort with the use of various standard genotyping technologies, and imputed for each study with reference to HapMap release 21 or 22 CEU phased genotypes35.
Study phenotypes
The primary phenotype analysed was migraine of any type, regardless of source. This was followed by a subgroup analysis consisting of 1) analysing only the clinical samples, 2) only samples satisfying criteria for MA, and 3) for MO. Population-based samples were not analysed genome-wide as a subgroup, due to forming 78% of cases and 85% of controls in the main analysis, but associations were calculated for the significant SNPs for comparative purposes. In the clinical cohorts, headache specialist has assigned a migraine diagnosis based on direct or telephone interview or through the use of an extensive questionnaire. For the population studies, migraine status for individuals in a study sample has been determined by a questionnaire (see Supplementary Note).
Statistical analysis of GWA study data
Each study contributed summary statistic data from an association analysis performed using a frequentist additive model based on an expected allelic dosage model for SNP markers, adjusting for gender (using either SNPTEST or ProbABEL [see URLs]). SNPs were filtered on per-study level based on inclusion criteria of MAF>0.1% and imputation quality measures of IA > 0.6 (IMPUTE 236) or r2 > 0.3 (MACH37). Four of the included studies contain novel genotyping (HUNT) or imputation (the Finnish, German and Dutch MA studies and HUNT). In the meta-analysis, combined association data for ~2.3 million imputed and genotyped autosomal SNPs were analysed in a fixed-effects model using GWAMA. At this stage, SNPs with a heterogeneity coefficient I2 exceeding 75% or presence in only four or fewer studies were filtered out. In the meta-analysis, there was little evidence for population stratification at the study level (each genomic inflation factor λ≤ 1.1), though moderate inflation was observed at the meta-analysis level (λ = 1.15; Supplementary Fig. 4). For estimating genome-wide significance, we used the commonly accepted threshold of 5 × 10−8 for primary loci 38, and 1 × 10−5 for secondary loci, in accordance with the reporting threshold for the GWAS catalog39. At secondary loci, to limit spurious associations, at least two SNPs were required to pass the significance threshold within a 50kb window. We also estimated the robustness of this threshold using the false discovery rate method of Benjamini & Hochberg40, showing that the P value threshold corresponding to FDR<0.05 was 2.33 × 10−5. The quantile-quantile plot (Supplementary Fig. 4) of the meta-analysis P values showed a marked excess of association signals well beyond those expected by chance below the suggestive reporting threshold. The significant loci were visualized using the LocusZoom interface41. For the heterogeneity analyses for migraine type, due to shared controls in some of the sets, the available study samples were divided into as equally-sized groups in terms of effective study size as possible, and then the data was analysed using the gender heterogeneity analysis method 42 (–sex option) of GWAMA43, with a dummy variable coding for MA and MO instead of gender. For the heterogeneity analyses for gender, the same method was used to compare P values from males-only and females-only analyses.
Pathway analyses
MAGENTA - The MAGENTA software44 was used to conduct an analysis to evaluate whether P values for association with migraine are enriched to particular biological networks, using pathway lists from GO, PANTHER, INGENUITY, KEGG, REACTOME and BIOCARTA. In the gene set enrichment analysis, P values were estimated via 10,000 permutations of genes evaluated at 75th FDR percentile (due to assumption of high polygenicity), manually corrected to account for FDR across all pathway sets. DAPPLE - Using a refined database of high-confidence protein-protein interactions (InWeb45,46) we used DAPPLE 47 to assess the amount of physical interactions connecting the genes within 50 kb of the 146 reported migraine loci, as well as an analysis of only the 16 proteins from the 12 genome-wide significant loci. Both direct and indirect (through 1st order common interaction partners) were measured and compared to a random expectation over 10,000 permutations, and the resulting network was plotted. GRAIL - The GRAIL web interface 48 was used to explore similarities in published PubMed articles (August 2012 freeze), using data from HapMap release 22 CEU and gene size correction set to on. From the GRAIL results, only genes with significant (P value<0.05) are shown, and the list of similar genes was capped at genes within the top 200 highest ranks.
Overlap with DNAse I hypersensitivity sites
The positions of SNPs from migraine-associated loci were overlapped with DNase 1 ‘hotspot’ regions from the ENCODE project that mark generalized chromatin accessibility mapped for each of 125 diverse cell lines and tissues49. To assess the significance of overlap for the set of SNPs as a whole, 100 background sets of SNPs were chosen from the genome so that each migraine associated SNP was matched in each set by a SNP within the same decile for minor allele frequency, distance to the nearest transcription start site and GC content of the 100 base region surrounding the SNP. The background SNP sets were overlapped with the DNase 1 hotspots, and the enrichment for overlap with the migraine associated SNP set expressed as the Z score relative to the distribution of background SNP set overlaps on a per cell line basis. In addition migraine associated SNPs were analysed for other overlap with ENCODE data including transcription factor motifs using RegulomeDB (http://regulome.stanford.edu)50.
Tissue-based gene expression analysis
For the tissue analysis, a microarray-based analysis of gene expression was performed on a dataset of 55 269 samples in the Gene Expression Omnibus (GEO) database that were measured on the Affymetrix U133 Plus 2.0 Array. Each sample in the raw expression data was first linearly transformed using a modified invariant set normalization method51 on a set of eighty control genes with stable expression on U133 Plus 2.0. The expression data was log2 transformed to stabilize the variance and expression distribution. Finally, the data were quantile-normalized52 to match the expression distribution of each sample. Expression values for genes with multiple probe sets were calculated by taking the median value of all probe sets for that gene. Following normalization, a log2 expression value of 4 is considered baseline and log2 expression values greater than 6 are considered expressed. Sample annotations were curated based on GEO descriptions provided by depositors. To account for variation in the number of samples representing each tissue in the dataset, expression of a gene was plotted to show the fraction of samples of a tissue that exceeds a log2 expression value of 6, with higher fractions indicating more ubiquitous expression in the tissue in question.
eQTL analysis
Based on the meta-analysis results for association with migraine, 146 regions of interest were queried against the expression quantitative trait loci (eQTL) results from the North American Brain Expression and UK Brain Expression Consortium studies (GEO # GSE36192, dbGaP # phs000249). These eQTL results are based on Cerebellum and Frontal Cortex tissue mRNA expression levels from 394 human subjects (see Supplementary methods). Of the 146 associated regions 134 were represented in the eQTL analysis. Within these regions 831 mRNA expression traits and 222 668 cis SNP/expression trait pairs were considered in Cerebellum and 864 mRNA expression traits and 230 660 cis SNP/expression trait pairs were considered in within the Frontal Cortex. 45 SNPs within the migraine associated loci were found to have significant correlation (evaluated at FDR-corrected threshold; 0.0001668 for frontal cortex and 0.0002187 for cerebellum) with the expression of 12 mRNA transcripts in both the Cerebellum and the Frontal Cortex. A more detailed methods description can be found in Supplementary Materials. The extent of linkage disequilibrium between SNPs associated with migraine and the SNPs in the tested eQTL SNP-probe pairs was evaluated using SNAP (see URLs).
Supplementary Material
Acknowledgments
Study-specific acknowledgments appear as a Supplementary Note. We wish to thank Alison Coffey, S. Hunt, R. Gwillian, P. Whittaker, S. Potter and A. Tashakkori-Ghanbarian, for their invaluable help with this study, and to collectively thank everyone who has contributed to the collection, genotyping and analysis of the individual cohorts, as well as all the study participants.
Footnotes
Competing Financial Interests
The authors declare no competing financial interests
URLs.
DAPPLE - www.broadinstitute.org/mpg/dapple
GRAIL - www.broadinstitute.org/mpg/grail
GWAMA - www.well.ox.ac.uk/gwama
IMPUTE2 - mathgen.stats.ox.ac.uk/impute
International Headache Genetics Consortium- www.headachegenetics.org
MAGENTA - www.broadinstitute.org/mpg/magenta
ProbAbel - http://www.genabel.org/packages/ProbABEL
RegulomeDB - http://regulome.stanford.edu
SNAP - http://www.broadinstitute.org/mpg/snap
SNPTEST - https://mathgen.stats.ox.ac.uk/genetics_software/snptest
References
- 1.Anttila V, et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet. 2010;42:869–73. doi: 10.1038/ng.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chasman DI, et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet. 2011;43:695–8. doi: 10.1038/ng.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ligthart L, et al. Meta-analysis of genome-wide association for migraine in six population-based European cohorts. Eur J Hum Genet. 2011;19:901–7. doi: 10.1038/ejhg.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freilinger T, et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet. 2012;44:777–82. doi: 10.1038/ng.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International Headache Society The International Classification of Headache Disorders: 2nd edition. Cephalalgia. 2004;24(Suppl 1):9–160. doi: 10.1111/j.1468-2982.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 6.Fimia GM, De Cesare D, Sassone-Corsi P. A family of LIM-only transcriptional coactivators: tissue-specific expression and selective activation of CREB and CREM. Mol Cell Biol. 2000;20:8613–22. doi: 10.1128/mcb.20.22.8613-8622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dash PK, Hochner B, Kandel ER. Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature. 1990;345:718–21. doi: 10.1038/345718a0. [DOI] [PubMed] [Google Scholar]
- 8.Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–40. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherman EA, et al. Genetic mapping of glutaric aciduria, type 3, to chromosome 7 and identification of mutations in c7orf10. Am J Hum Genet. 2008;83:604–9. doi: 10.1016/j.ajhg.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schreiner A, et al. Junction protein shrew-1 influences cell invasion and interacts with invasion-promoting protein CD147. Mol Biol Cell. 2007;18:1272–81. doi: 10.1091/mbc.E06-07-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lafleur MA, Xu D, Hemler ME. Tetraspanin proteins regulate membrane type-1 matrix metalloproteinase-dependent pericellular proteolysis. Mol Biol Cell. 2009;20:2030–40. doi: 10.1091/mbc.E08-11-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rozanov DV, Hahn-Dantona E, Strickland DK, Strongin AY. The low density lipoprotein receptor-related protein LRP is regulated by membrane type-1 matrix metalloproteinase (MT1-MMP) proteolysis in malignant cells. J Biol Chem. 2004;279:4260–8. doi: 10.1074/jbc.M311569200. [DOI] [PubMed] [Google Scholar]
- 13.Borrie SC, Baeumer BE, Bandtlow CE. The Nogo-66 receptor family in the intact and diseased CNS. Cell Tissue Res. 2012;349:105–17. doi: 10.1007/s00441-012-1332-9. [DOI] [PubMed] [Google Scholar]
- 14.Schurks M, et al. Migraine and cardiovascular disease: systematic review and meta-analysis. Bmj. 2009;339:b3914. doi: 10.1136/bmj.b3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizuguchi T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–60. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biros E, Walker PJ, Nataatmadja M, West M, Golledge J. Downregulation of transforming growth factor, beta receptor 2 and Notch signaling pathway in human abdominal aortic aneurysm. Atherosclerosis. 2012;221:383–6. doi: 10.1016/j.atherosclerosis.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Kathiresan S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–41. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terada N, et al. The tetraspanin protein, CD9, is expressed by progenitor cells committed to oligodendrogenesis and is linked to beta1 integrin, CD81, and Tspan-2. Glia. 2002;40:350–9. doi: 10.1002/glia.10134. [DOI] [PubMed] [Google Scholar]
- 19.Flavell SW, et al. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–38. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsuzuki K, Xing H, Ling J, Gu JG. Menthol-induced Ca2+ release from presynaptic Ca2+ stores potentiates sensory synaptic transmission. J Neurosci. 2004;24:762–71. doi: 10.1523/JNEUROSCI.4658-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diniz LP, et al. Astrocyte-induced synaptogenesis is mediated by transforming growth factor beta signaling through modulation of D-serine levels in cerebral cortex neurons. J Biol Chem. 2012 doi: 10.1074/jbc.M112.380824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen PB, Greenfield AT, Svenningsson P, Haspeslagh DC, Greengard P. Phactrs. 1-4: A family of protein phosphatase 1 and actin regulatory proteins. Proc Natl Acad Sci U S A. 2004;101:7187–92. doi: 10.1073/pnas.0401673101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferraro GB, Morrison CJ, Overall CM, Strittmatter SM, Fournier AE. Membrane-type matrix metalloproteinase-3 regulates neuronal responsiveness to myelin through Nogo-66 receptor 1 cleavage. J Biol Chem. 2011;286:31418–24. doi: 10.1074/jbc.M111.249169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson PM, Fryer RH, Fang Y, Hatten ME. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J Neurosci. 2010;30:8529–40. doi: 10.1523/JNEUROSCI.0032-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.May P, et al. Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol Cell Biol. 2004;24:8872–83. doi: 10.1128/MCB.24.20.8872-8883.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jha KN, et al. Biochemical and structural characterization of apolipoprotein A-I binding protein, a novel phosphoprotein with a potential role in sperm capacitation. Endocrinology. 2008;149:2108–20. doi: 10.1210/en.2007-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gouveia R, et al. Expression of glycogenes in differentiating human NT2N neurons. Downregulation of fucosyltransferase 9 leads to decreased Lewis(x) levels and impaired neurite outgrowth. Biochim Biophys Acta. 2012;1820:2007–2019. doi: 10.1016/j.bbagen.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Lieberoth A, et al. Lewis(x) and alpha2,3-sialyl glycans and their receptors TAG-1, Contactin, and L1 mediate CD24-dependent neurite outgrowth. J Neurosci. 2009;29:6677–90. doi: 10.1523/JNEUROSCI.4361-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishihara S, et al. Alpha1,3-fucosyltransferase IX (Fut9) determines Lewis X expression in brain. Glycobiology. 2003;13:445–55. doi: 10.1093/glycob/cwg048. [DOI] [PubMed] [Google Scholar]
- 30.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–61. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 31.Park SJ, et al. Astrocytes, but not microglia, rapidly sense H(2)O(2)via STAT6 phosphorylation, resulting in cyclooxygenase-2 expression and prostaglandin release. J Immunol. 2012;188:5132–41. doi: 10.4049/jimmunol.1101600. [DOI] [PubMed] [Google Scholar]
- 32.Dibble CC, et al. TBC1D7 Is a Third Subunit of the TSC1-TSC2 Complex Upstream of mTORC1. Mol Cell. 2012;47:535–46. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han JM, Sahin M. TSC1/TSC2 signaling in the CNS. FEBS Lett. 2011;585:973–80. doi: 10.1016/j.febslet.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell MB, Olesen J. Increased familial risk and evidence of genetic factor in migraine. British Medical Journal. 1995;311:541–544. doi: 10.1136/bmj.311.7004.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–8. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 41.Pruim RJ, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Magi R, Lindgren CM, Morris AP. Meta-analysis of sex-specific genome-wide association studies. Genet Epidemiol. 2010;34:846–53. doi: 10.1002/gepi.20540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magi R, Morris AP. GWAMA: software for genome-wide association meta-analysis. BMC Bioinformatics. 2010;11:288. doi: 10.1186/1471-2105-11-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Segre AV, Groop L, Mootha VK, Daly MJ, Altshuler D. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lage K, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol. 2007;25:309–16. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 46.Lage K, et al. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci U S A. 2008;105:20870–5. doi: 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossin EJ, et al. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raychaudhuri S, et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boyle AP, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.