

Abstract
Rationale
Stimulation of β3-adrenoreceptors (β3-AR) blunts contractility and improves chronic left ventricular function in hypertrophied and failing hearts in a neuronal nitric oxide synthase (nNOS) dependent manner. nNOS can be regulated by post-translational modification of stimulatory phosphorylation residue Ser1412 and inhibitory residue Ser847. However, the role of phosphorylation of these residues in cardiomyocytes and β3-AR protective signaling has yet to be explored.
Objective
We tested the hypothesis that β3-AR regulation of myocyte stress requires changes in nNOS activation mediated by differential nNOS phosphorylation.
Methods and results
Endothelin (ET-1) or norepinephrine induced hypertrophy in rat neonatal ventricular cardiomyocytes (NRVMs) was accompanied by increased β3-AR gene expression. Co-administration of the β3-AR agonist BRL-37433 (BRL) reduced cell size and reactive oxygen species (ROS) generation, while augmenting NOS activity. BRL-dependent augmentation of NOS activity and ROS suppression due to NE were blocked by inhibiting nNOS (L-VNIO). BRL augmented nNOS phosphorylation at Ser1412 and dephosphorylation at Ser847. Cells expressing constitutively dephosphorylated Ser1412A or phosphorylated Ser847D nNOS mutants displayed reduced nNOS activity and a lack of BRL modulation. BRL also failed to depress ROS from NE in cells with nNOS-Ser847D. Inhibiting Akt decreased BRL-induced nNOS-Ser1412 phosphorylation and NOS activation, whereas Gi/o blockade blocked BRL-regulation of both post-translational modifications, preventing enhancement of NOS activity and ROS reduction. BRL resulted in near complete dephosphorylation of Ser847 and a moderate rise in Ser1412 phosphorylation in mouse myocardium exposed to chronic pressure-overload.
Conclusion
β3-AR regulates myocardial NOS activity and ROS via activation of nNOS involving reciprocal changes in phosphorylation at two regulatory sites. These data identify a novel and potent anti-oxidant and anti-hypertrophic pathway due to nNOS post-translational modification that is coupled to β3-AR receptor stimulation.
Keywords: Neuronal nitric oxide synthase, Reactive oxygen species, Hypertrophy, Heart failure, Beta3-adrenergic receptors
1. Introduction
Sustained sympathetic activation of the cardiac myocyte β1/β2 adrenergic-receptor is a potent contributor to myocardial disease. By contrast, β3-receptor stimulation protects the heart from both hypertrophy and failure [1,2]. β3-AR expression is expressed at very low levels in the normal heart, but increases with pathophysiological conditions (e.g. diabetes, hypertrophy, heart failure) unlike β1/β2-AR [3,4]. β3-AR can be stimulated by high levels of catecholamines and is associated with a countering negative inotropic effect mediated by inhibitory G-protein coupled NOS–NO signaling [5,6]. In this manner, β3-AR serves as a physiological “brake” to protect the heart from sympathetic overdrive [1]. β3-AR-mediated responses are maintained despite sustained hyper-sympathetic activity because β3-AR lacks phosphorylation sites for PKA and beta-AR kinase (βARK) that induce receptor desensitization [7–9]. With heart failure, β3-AR signaling can depress contractility and heart rate, but it also provides cardiac protection making it an intriguing therapeutic target.
The negative inotropic effect of β3-AR stimulation involves NOS/NO signaling [2,5,10,11], and while it was initially thought to be due to endothelial NOS (eNOS), new data has shown that neuronal NOS (nNOS) also plays a key role [12–15]. In mice with nNOS genetically deleted or acutely inhibited, β3-AR-dependent cardiac protection, NO generation, and NOS downstream signaling are lacking in failing mouse myocardium [10,11]. Napp et. al reported increased β3-AR-stimulated NO generation that was independent of eNOS in failing human hearts [16], also supporting a role of other NOS isoforms. Myocytes express both eNOS and nNOS, and recent studies have revealed the importance of nNOS to cardiac calcium cycling and contractility [17,18]. Similar to eNOS, nNOS activity can be modified by post-translational modifications (PTMs) at specific residues. Phosphorylation at Ser1412, analogous to Akt-dependent phosphorylation site of eNOS-Ser1177 enhances nNOS activity [19]. By contrast, phosphorylation at Ser847, located on the Ca2+-Calmodulin binding domain, inhibits nNOS [19]. As the β3-AR is known to couple to Gi/o- and Akt-dependent signaling, we hypothesized that these PTMs may mediate the protective impact of β3-AR stimulation in countering myocyte stress.
2. Methods
2.1. Cell culture
Neonatal Rat Ventricular Cardiomyocytes (NRVMs) were isolated from 2 to 4 days old neonatal Sprague–Dawley pups (Taconic Farms, Hudson, NY). Hearts were removed and placed into Krebs buffer containing collagenase and trypsin. Isolated myocytes were plated and incubated in DMEM with 10% FBS at 37 °C. Cells were stimulated with BRL-37344 (Tocris, Bristol, UK). Prior to BRL administration cells were pretreated with endothelin-1(ET-1) or norepinephrine (NE) (Sigma-Aldrich, St. Louis, MO).
2.2. Pressure-overload model
Nine C57BL/6J male mice (9–10 weeks old, Jackson Laboratory, Bar Harbor, ME) were arbitrarily divided into 3 groups. Two-thirds of the mice underwent transverse aortic constriction (TAC) to induce cardiac hypertrophy and heart failure via pressure overload, while the remaining one-third had a sham procedure. Half of the TAC mice were treated with BRL (Tocris Bioscience, Ellisville, MO) at 0.1 mg/kg/day via osmotic mini-pumps (Alzet Inc., Cupetino, CA) which were subcutaneously implanted one day post-TAC, as previously described [10].
2.3. Quantitative PCR
RNA was extracted from cells using Trizol Reagent and cDNA synthesized. PCR reactions were analyzed using Taqman master mix, β3-AR- and β-actin-specific probes, and GeneAmp 7900 sequence detection system (Applied Biosystems, Foster City, CA).
2.4. Radiolabeled NOS activity assay
NOS calcium-dependent activity was determined from cellular lysates by measuring C14 arginine to citrulline conversion (assay kits from Cayman Chemical, Ann Arbor, MI) as previously described [2].
2.5. Lucigenin-enhanced chemiluminescence
Myocardial superoxide was assayed by lucigenin-enhanced chemiluminescence. Cells were lysed and equilibrated in 1× Cell Lysis Buffer (Millipore, Billerica, MA). After sonification, the lysates were added to a low concentration of lucigenin-solution (5 μM) with 150 μM NADPH to alleviate artifacts [20]. Baseline and maximum lucigenin-enhanced chemiluminescent signal were detected by a liquid scintillation counter (LS6000IC, Beckman Instruments, Fullerton, CA), with data reported as counts per minute per milligrams of protein after background subtraction (cpm/mg) [21].
2.6. Western blotting
Cell and myocardial lysates were subjected to SDS-PAGE and proteins were electrophoretically transferred to a nitrocellulose membrane as previously described. The membranes were then exposed to primary antibodies overnight at 4 °C. Antibody against phospho-nNOS-Ser847 was purchased from Abcam (1:1000, Cambridge, MA) and anti-phosphonNOS-Ser1412 was a gift from K.J. Hurt (1:5000, UC-Denver). Anti-Myc (1:5000), anti-nNOS (1:1000), anti-eNOS (1:1000), and anti-phospho-eNOS-Ser1177 and −114 (1:1000) were purchased from Cell Signaling (Boston, MA). Immunoreactive proteins were then incubated with the peroxidase-linked secondary antibody (GE Healthcare, Piscataway, NJ) for 1 h at room temperature, afterwards visualized by Thermo Scientific Pierce Enhanced Chemiluminescence Western Blotting Substrate kit (Rockford, IL).
2.7. cGMP
After treatment, cardiac myocytes were lysed with 0.1 M HCL and centrifuged to pellet cellular debris. Supernatant was saved and assayed using the acetylated format of the cGMP complete ELISA Kit (Enzo Life Sciences, Farmingdale, NY).
2.8. Co-IP
Myocardial lysates were precipitated with affinity-purified rabbit anti-recombinant nNOS antibody. Antibody–protein complexes were collected by Immobilized Protein A/G (Pierce) and eluted by boiling in Laemmli sample buffer containing β-mercaptoethanol. The samples were subjected to SDS-PAGE as described in the Western blotting section.
2.9. Data analysis
The data presented is representative of at least three separate experiments yielding similar results. Intensity of the immunoblot bands were measured by UN-SCAN-IT gel Automated Digitizing System Version 5.1. Counts per minute (cpm) detected from NOS activity and assays were measured by the LS6000IC liquid scintillation counter. Data was analyzed with Prism 5.0 statistical program software using one- or two-way analysis of variance (ANOVA) followed by Tukey or Bonferroni post-test analysis where appropriate. Significance was shown at *p < 0.05.
3. Results
3.1. Cardiac myocyte hypertrophy augments β3-AR expression
Prior studies have shown that sustained stimulation of β-AR receptors results in augmentation of the β3-AR expression [4,22]. We tested if similar upregulation was coupled to alternative GCPR-stimuli — specifically the Gq-coupled agonists ET-1 and NE. NRVMs were exposed to either agonist for 48–72 h, which resulted in a 40–60% increase in myocyte size. Co-incubation with β3-AR agonist BRL-37344 (75 nM) reduced this significantly in both cases (Figs. 1A, B). With either Gq stimulant, β3-AR mRNA expression rose (123.0 ± 1.7% ET-1, 131.4 ± 1.4% NE, both P < 0.05). For simplicity, the subsequent experiments were conducted using NE.
Fig. 1.

Norepinephrine and ET-1 induced hypertrophy and β3-AR expression in isolated NRVMs. Cells were treated with or without ET-1 (100 nM) for 48 h, and NE (100 mM) for 72 h. A–B.) Cell surface area was determined by phase contrast microscopy. C.) β3-AR mRNA expression was measured by qPCR, and normalized to baseline expression. *p < 0.05 vs. basal; #p < 0.05 vs. respective agonist control in immunostaining experiment; and #p < 0.05 vs. ET-1 in qPCR experiment.
3.2. BRL restores NOS activity and attenuates superoxide generation via a nNOS dependent mechanism
NE pre-treatment for 72 h induced marked suppression of NOS activity in NRVMs. This was reversed by subsequent addition of BRL over a 2-hour period, still in the presence of NE (Fig. 2A, left). The peak increase was observed after 45 min, essentially restoring NOS activity to normal basal levels (Fig. 2A, right). BRL had no significant effect on NOS activity in non-hypertrophied (non-stimulated) controls (Fig. 2A). The fall in NOS activity with NE-pretreatment was accompanied by a reciprocal rise in superoxide formation (Fig. 2B). BRL had a marked and rapid effect on O2− production, reducing it by >70% within 10 min (Fig. 2B, left, right shows summary data after 45 min). To test the importance of nNOS to the total NOS activation, cells pre-treated with NE were subsequently exposed to BRL in the presence or absence of the nNOS-specific inhibitor, LVNIO. LVNIO treated cells showed a marked reduction in BRL-enhanced NOS activity (Fig. 2C) and reduction in . In addition, BRL increased cGMP levels via an nNOS dependent mechanism (Supplemental figure). This supports an important role of nNOS in β3-mediated myocyte signaling.
Fig. 2.

BRL restores NOS activity and suppresses hypertrophy-induced superoxide generation via a nNOS-dependent manner in hypertrophied cardiomyocytes. Cells were stimulated for 45 min with BRL in the presence or absence of 72 h NE pretreatment, or LVNIO, nNOS inhibitor. Hypertrophied cells were stimulated at indicated time points to measure A.) NOS activity, B.) superoxide generation in the absence or C–D.) presence of LVNIO. NOS activity was measured by 14C arginine to citrulline conversion and superoxide was measured by Lucigenin Assay. Data is presented as mean ± SEM for 3–6 experiments. *p < 0.001 vs. hypertrophied control; #p < 0.05 vs. LVNIO treated.
3.3. BRL mediates post-translational modification of nNOS and eNOS
The time-course of BRL-induced modification of both NOS activity and modulation was most compatible with post-translational changes of existing protein. To test this, we examined nNOS phosphorylation at Ser1412 and Ser847, sites of activation and deactivation, respectively [19]. BRL induced a 1-fold increase in Ser1412 phosphorylation and virtually complete dephosphorylation at Ser847 (Fig. 3A), both consistent with enhanced nNOS activity and reduced production after 45 min. eNOS phosphorylation was also modified by BRL. The negative regulatory site at Ser114 became phosphorylated over 45 min, while the stimulatory residue, Ser1177, remain unchanged (Fig. 3B). These are consistent with reduced eNOS activation, opposite to that observed for nNOS.
Fig. 3.

BRL alters phosphorylation of nNOS and eNOS in hypertrophied cardiomyocytes. Cells were stimulated for 45 min with BRL in the presence or absence of 72 h NE pretreatment. Cells were stimulated at indicated time points to measure A.) nNOS-Ser1412 and -Ser847, and B.) eNOS-Ser1177 and -Ser114 phosphorylations. Phosphorylation was measured by Immunoblot analysis with phosphospecific antibodies. Data is presented as mean ± SEM for 3–6 experiments. *p < 0.05 vs. basal.
3.4. nNOS phosphorylation plays a key role in β3 signaling in vivo
To further test if BRL modulation of nNOS residue phosphorylation was relevant in vivo, mice were subjected to transverse aortic constriction (TAC) [10] for 21 days with or without concomitant treatment with BRL (0.1 mg/kg/day). We previously demonstrated that BRL rescues the myocardium from TAC-induced heart failure and NO reduction via an nNOS dependent manner [10]. In this study, TAC induced a 40% increase in Ser847 phosphorylation over sham control, and this was reversed by BRL, mimicking the in vitro findings (Fig. 4A). There was also a trend for increased Ser1412 phosphorylation with TAC, and a significant rise, with BRL treatment as compared with sham control (Fig. 4B). Thus, BRL-induced nNOS PTMs are similarly observed in a relevant in vivo model.
Fig. 4.
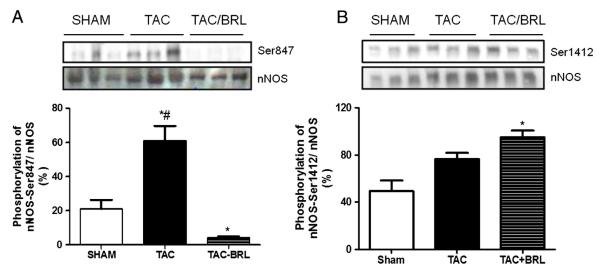
BRL administration alters nNOS phosphorylation in mice myocardium after pressure overload. C57BL/6J mice underwent 3-weeks of transverse aortic constriction (TAC) in the presence or absence of BRL administration (0.1 mg/kg/day). Left ventricular myocardium was homogenized and probe against phospho-specific antibodies for A.) nNOS-Ser847 and B) Ser1412. Phosphorylation was measured by western blot analysis. Data is presented as mean ± SEM for three experiments. *p < 0.05 vs. SHAM; #p < 0.05 vs. TAC-BRL.
3.5. BRL-induced NOS activity and O2 levels are altered by nNOS phosphomimetic mutants
To further investigate the relation between β3-regulated nNOS phosphorylation and both NOS activation and O2 suppression, myocytes were infected with Sindbis viruses encoding for a c-myc-tagged site-mutated nNOS with either Ser1412A, Ser847A, or Ser1412D, Ser847D (Fig. 5A). This provided either phosphomimetic (aspartate, D) or silenced (alanine, A) mutants [14]. Virus expressing green-fluorescent protein (GFP) served as negative controls. GFP viral constructs displayed an increased NOS activity at baseline when compared to prior experiments with non-viral infected cells. Nevertheless, BRL was consistent in increasing NOS activity by at least 55% from baseline in infected cells. BRL-dependent NOS activity was attenuated in cells expressing Ser847D or Ser1412A (enhancing the negative regulating site, or blocking the activation site, respectively). This indicates that β3-AR NOS activation is dependent upon PTM. The alternative Ser847A mutant had no effect on BRL-induced NOS activity as might be predicted, while Ser1412 mutant enhance NOS activity at baseline (Fig. 5B). The Ser847D mutant also prevented a BRL-mediated decline in (Fig. 5C). Unexpectedly, however, this was not observed with the Ser1412A mutant. This suggests that β3-AR regulation of is dissociable from its activation of nNOS, and is linked to dephosphorylation of Ser847 and not Ser1412 phosphorylation.
Fig. 5.

nNOS phosphomimetic mutants alter BRL-induced NOS activity and superoxide suppression in hypertrophied cardiomyocytes. Hypertrophied cells were infected with Myc-tagged Sindbis viruses encoding an aspartate (D) or alanine (A) site-mutation for Ser847 or Ser1412 for 24 h prior to 45 min BRL treatment. A.) Infection was determined using Immunoblot analysis with antibodies against Myc protein. B.) NOS activity was measured by 14C arginine to citrulline conversion and C.) superoxide was measured by Lucigenin Assay. GFP Sindbis virus was used to define basal levels of transformation. Data is presented as mean ± SEM for 3–6 experiments. *p < 0.05 vs. respective viral control. #p < 0.05 vs. GFP baseline.
3.6. BRL-induced nNOS-phosphorylation of Ser1412 is Akt-dependent
Previous studies have shown that nNOS phosphorylation of Ser1412 is Akt dependent [19,23]. Thus, we tested if the selective Akt-inhibitor (Akt-i) could also prevent BRL modulation of nNOS activity. Following BRL stimulation, hypertrophied myocytes treated with Akt-i showed a significant decrease in Ser1412 phosphorylation (Fig. 6A) but no change in Ser847 phosphorylation (Fig. 6B) as compared to controls, accompanied by a 50% reduction in BRL-stimulated NOS activity (Fig. 6C). However, the antioxidant response to BRL was unaltered by Akt-i (Fig. 6D). In our in vivo studies we witnessed an increase in nNOS-Akt interaction, as assessed by co-immunoprecipitation, in TAC and TAC + BRL animals (Fig. 6E). This correlated with the increase in Ser1412 phosphorylation seen in Fig. 4B. Interestingly, we observed no change in Akt phosphorylation in isolated cells or heart extracts (data not shown).
Fig. 6.

Akt inhibition blocks β3-AR-induced NOS activity and Ser1412 phosphorylation in hypertrophied cardiomyocytes. Cells were stimulated for 45 min with BRL in the presence or absence of Akt-i, Akt inhibitor, after NE pretreatment. Phosphorylation, NOS activity and superoxide were measured by A–B.) immunoblotting, C.) 14C arginine to citrulline conversion, and D.) lucigenin assay, respectively. E.) Left ventricular myocardium was homogenized and immunoprecipitated with anti-nNOS then probed with anti-Akt. Data is presented as mean ± SEM for three experiments. p < 0.05 vs. basal; #p < 0.05 vs. Akt-i + BRL group.
3.7. Gi/o inhibition alters β3-regulated nNOS phosphorylation, NOS activity, and superoxide generation
β3-AR has been shown to signal eNOS via Gi/o in the ventricular myocardium [24], however a role in nNOS stimulation has not been previously reported. Myocytes treated with pertussis toxin to block Gαi/o signaling displayed a marked decline in BRL-induced S1412 phosphorylation, and significant increases in S847 phosphorylation (Figs. 7A–B). PTX treatment also resulted in blunted BRL-stimulated NOS activity and its antioxidant effects (Figs. 7C–D).
Fig. 7.

Gi/o inhibition blocks β3-induced Ser1412 phosphorylation, NOS activity, and superoxide suppression while partially restoring Ser847. Cells were pretreated with NE for 72 h and stimulated for 45 min with BRL in the presence or absence of Pertussis Toxin, Gi/o inhibitor. Phosphorylation was detected with A.) Ser1412 and B) nNOS-Ser847 phospho-specific antibodies. C.) NOS activity and D.) superoxide generation were measured by 14C arginine to citrulline conversion and Lucigenin assay, respectively. Data is presented as mean ± SEM for three experiments. *p < 0.05 vs. basal; #p < 0.05 vs. BRL alone.
4. Discussion
While basal β3-AR expression and response to stimulation in the heart is low to undetectable [4,10], growing evidence supports its role in cardiac pathophysiological disorders where levels rise and signaling becomes significant [4,13,25,26]. Furthermore, recent data has shown that genetic deletion of the pathway or its stimulation by selective agonists can respectively worsen or improve the pathological response to cardiac stress remodeling [2,10]. This modulation in the diseased heart involves an interaction between the β3-AR and nNOS [10,11]. In the current study, we provide novel evidence that the nNOS modulation occurs rapidly, and involves phosphorylation changes at two residues, Ser1412 and Ser847, one enhancing and the other depressing nNOS activity (Fig. 8). Corresponding antioxidant effects from β3-AR are also observed and shown linked to the altered Ser847 phosphorylation in nNOS. In addition to revealing a mechanistic link between β3-AR myocyte modulation and nNOS activity, these results are the first evidence of a role for nNOS phosphorylation changes and myocyte-NO and ROS generation in hypertrophied cells.
Fig. 8.
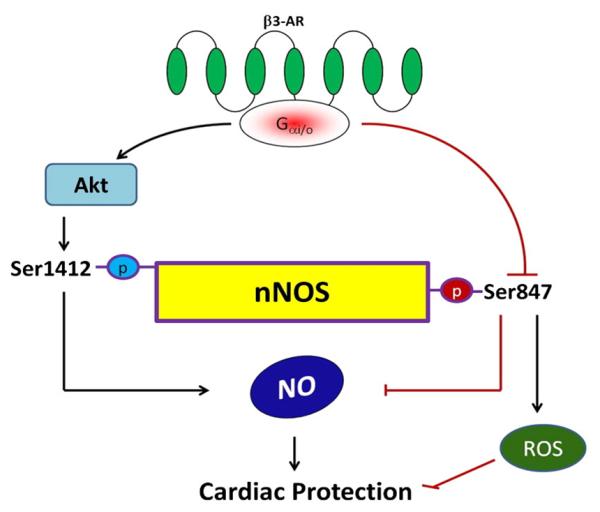
Proposed signal transduction cascade by which β3-AR activation leads to NOS signaling and ROS scavenging in cardiac myocytes.
4.1. β3-Agonism restores NOS activity and decreases ROS generation via myocardial nNOS rather than eNOS
Activation of the β3-AR pathway in the ventricular myocardium is accompanied by decreased contractility in humans and rodents due to β3-AR's ability to generate NO via NOS signaling downstream of Gi/o activation [10,11,16,24]. Negative inotropy from the β3-AR agonist, BRL, is inhibited by both NO and non-selective NOS inhibitors and can be reversed by an excess of the substrate for NO production, L-arginine [24]. While this initially leads to the conclusion that the primary target of β3-AR-induced NO generation was eNOS [2,24,27,28], recent studies have suggested differently. In human failing left ventricular myocardium, Napp et al. observed that BRL was capable of inducing NO-dependent negative inotropy even if myocardial eNOS was inactivated, suggesting that another NOS isoform or paracrine signaling from endothelial eNOS was involved [16]. In failing mouse hearts, we found that β3-agonism led to nNOS-dependent protection of left ventricular function, while inducing eNOS deactivation via Ser114 phosphorylation [10] Furthermore, Aragon et al. demonstrated that the dual β1-blocker and β3-agonist, nebivolol, failed to reduce myocardial ischemic-reperfusion infarct size in nNOS−/− animals as compared with WT [11]. This refocused attention on β3-AR interaction with nNOS.
In the current study, we established a hypertrophy-pre-stimulation cell model, capable of activating β3-AR expression and myocyte hypertrophy, depressing NOS activity while concomitantly enhancing super-oxide generation. The model facilitated the analysis of β3-AR and nNOS signaling without interference by vascular cells. The finding that BRL stimulation reversed these cellular responses, which was prevented by co-treatment with the nNOS inhibitor LVNIO, strongly supports this isoform to be a determining factor of pathophysiologies in myocytes. These data further show that nNOS is acting not only as an important β3-AR modulated source of NOS activity, but also as a regulator of ROS induced by NE-stimulation. The source of ROS is not necessarily NOS itself, however. This was suggested by our studies in the S1412A mutants that inhibited the BRL-induced augmentation of NOS activity, yet still exhibited an antioxidant effect from β3-AR stimulation. Other studies have also suggested that myocardial nNOS can interact specifically with xanthine oxidase reductase (XOR) derived [15,29,30], thought to be primarily due to co-localization at the sarcoplasmic reticular membrane. More recently, Idigo et al. showed that XOR inhibition restores β3 negative inotropy as well as eNOS coupling in nNOS−/− myocytes [15]. Whether and how nNOS may suppress other sources of ROS, such as from NADPH oxidases or monoamine oxidases, remains to be determined.
In hypertrophied myocytes, an increase in β3-AR mRNA was observed, which is consistent with literature that demonstrates a functional significance of β3-AR under pathophysiological conditions [3,31]. Though myocyte β3-AR levels are modest, the relative effect may be magnified due to the amplification of downstream signaling pathways in addition to concomitant down-regulation of β1-AR, which has been demonstrated in the setting of hypertrophy [3,31]. Under normal conditions cardiac β1 to β3 ratio is 8:1 as compared with 2:1 during cardiomyopathies [31].
4.2. Phosphorylation of nNOS provides differential regulation of downstream β3-signaling
Post-translational modification plays an important role in the functionality of NOS isoforms [10,19,32]. nNOS undergoes post-translational modification via phosphorylation [33]. We found β3-agonism induced nNOS phosphorylation at Ser1412, a positive regulatory site, and dephosphorylation at site Ser847 – a negative modulator. While some insight into nNOS – PTMs has been reported from neuroscience, nothing has been previously reported in cardiac myocytes or hearts. In neurons, phosphorylation of Ser1412 is required for nNOS-dependent NO production induced by N-methyl-d-aspartate (NMDA) receptors, estradiol, and adrenomedullin [33,34]. Further, inhibition of AngII in spontaneous hypertensive rats increases nNOS-Ser1412 phosphorylation in neurons of the nucleus tractus solitarii (NTS) [35]. In myocytes, nNOS is thought to be constitutively activated and regulating SR-calcium cycling [36], though the level of basal S1412 phosphorylation is low, so this PTM would seem more important to stress-induced activation.
The current study used both gain and loss of phospho-function mutations to examine their respective roles. Besides finding correlations between the time-course of enhanced NOS activity and these PTMs following exposure to BRL, we show their individual contribution to both enhanced NOS activity and ROS modulation. Analogous to the eNOS positive regulatory site, nNOS-Ser1412 has been observed to be primarily PI3K/Akt dependent [19,33], although Yen et al. suggested a possible role for PKA as well [34]. The Akt-inhibitor studies, however showed that this is likely the primary targeting kinase for this site PTM in myocytes, at least as coupled to β3-AR activation. Here again, the data showed a selective influence of this phosphorylation site on NOS activity, but not on modulation, supporting alternative changes in nNOS for the latter.
Dephosphorylation at Ser847 also increases nNOS activity and associated NO generation [19,34]. This residue is located in the Ca2+/Calmodulin binding domain of the nNOS enzyme, and phosphorylated Ser847 blocks Ca2+/Calmodulin binding which is necessary for nNOS activation [37]. In murine cerebellum and hippocampus, spinal cord injury and forebrain ischemia impair nNOS/NOS signaling pathways through Ser847 phosphorylation via Ca2+/Calmodulin Kinase II modulation [38,39]. Our analysis has clarified that in heart, modification of this site is coupled to both NO generation and the capacity of nNOS activity to suppress ROS signaling. Further, we show that this is uncoupled from Akt, but is linked to a Gi/o-activation cascade. This could occur by Gi/o-coupled phosphatase activation or suppression of the required kinase. Potential candidates are PP1/PP2A, and PP2B (calcineurin) which enhance hippocampal NMDA-mediated nNOS-Ser847 phosphorylation in an ischemia model [34]. However, others have found a decline in phosphorylation following PP1/PP2A and PP2B inhibition in rat cortical neurons [35], so the precise targeting is unclear.
5. Conclusion
We conclude that myocardial β3-mediated NOS signaling is dependent upon alterations in phosphorylation of nNOS at sites Ser1412 and Ser847 in cardiomyocytes under pathophysiological conditions. In addition, we conclude that β3-mediated nNOS-dependent ROS scavenging is solely dependent upon dephosphorylation of negative regulatory site Ser847. To our knowledge this is the first study to demonstrate a role for nNOS phosphorylation as a key mediator in cardiac signaling, in addition to β3-AR protective signaling cascade. These results contribute significantly to our understanding of the negative inotropic properties of myocardial β3-AR at the cellular level during cardiac sympathetic overstimulation, and will ultimately aid in drug discoveries that target the molecular mechanisms associated with cardiac hypertrophy and heart failure.
Supplementary Material
Acknowledgments
We would like to thank Dr. K. Joseph Hurt of the University of Colorado-Denver for generously providing the phospho-specific nNOS-Ser1412 antibody.
Footnotes
Funding sources Funding: National Institutes of Health [5T32HL007227 to V.L.W]; American Heart Association [to V.L.W]; STEP-UP-NIDDK [to F.S.]; American Heart Association Beginning Grant-In-Aid [to L.A.B.]; American Diabetes Association [to L.A.B.]; and the China Scholarship Council [to X. N].
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.yjmcc.2013.04.025.
Disclosures There are no relationships to disclose.
References
- [1].Arch JR, Ainsworth AT. Thermogenic and antiobesity activity of a novel beta-adrenoceptor agonist (BRL 26830A) in mice and rats. Am J Clin Nutr. 1983;38:549–58. doi: 10.1093/ajcn/38.4.549. [DOI] [PubMed] [Google Scholar]
- [2].Moens AL, Leyton-Mange JS, Niu X, Yang R, Cingolani O, Arkenbout EK, et al. Adverse ventricular remodeling and exacerbated NOS uncoupling from pressure-overload in mice lacking the beta3-adrenoreceptor. J Mol Cell Cardiol. 2009;47:576–85. doi: 10.1016/j.yjmcc.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Moniotte S, Kobzik L, Feron O, Trochu JN, Gauthier C, Balligand JL. Upregulation of beta(3)-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001;103:1649–55. doi: 10.1161/01.cir.103.12.1649. [DOI] [PubMed] [Google Scholar]
- [4].Germack R, Dickenson JM. Induction of beta3-adrenergic receptor functional expression following chronic stimulation with noradrenaline in neonatal rat cardiomyocytes. J Pharmacol Exp Ther. 2006;316:392–406. doi: 10.1124/jpet.105.090597. [DOI] [PubMed] [Google Scholar]
- [5].Angelone T, Filice E, Quintieri AM, Imbrogno S, Recchia A, Pulera E, et al. Beta3-adrenoceptors modulate left ventricular relaxation in the rat heart via the NO-cGMP-PKG pathway. Acta Physiol (Oxf) 2008;193:229–39. doi: 10.1111/j.1748-1716.2008.01838.x. [DOI] [PubMed] [Google Scholar]
- [6].Moens AL, Yang R, Watts VL, Barouch LA. Beta 3-adrenoreceptor regulation of nitric oxide in the cardiovascular system. Journal of Molecular Cellular Cardiology. 2010;48:1088–95. doi: 10.1016/j.yjmcc.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liggett SB, Freedman NJ, Schwinn DA, Lefkowitz RJ. Structural basis for receptor subtype-specific regulation revealed by a chimeric beta 3/beta 2-adrenergic receptor. Proc Natl Acad Sci U S A. 1993;90:3665–9. doi: 10.1073/pnas.90.8.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Soeder KJ, Snedden SK, Cao W, Della Rocca GJ, Daniel KW, Luttrell LM, et al. The beta3-adrenergic receptor activates mitogen-activated protein kinase in adipocytes through a Gi-dependent mechanism. J Biol Chem. 1999;274:12017–22. doi: 10.1074/jbc.274.17.12017. [DOI] [PubMed] [Google Scholar]
- [9].Nantel F, Bonin H, Emorine LJ, Zilberfarb V, Strosberg AD, Bouvier M, et al. The human beta 3-adrenergic receptor is resistant to short term agonist-promoted desensitization. Mol Pharmacol. 1993;43:548–55. [PubMed] [Google Scholar]
- [10].Nui X, Watts VL, Cingolani OH, Sivakumaran V, Leyton-Mange JS, Ellis CL, et al. Cardioprotective effect of beta-3 adrenergic receptor agonism: role of neuronal nitric oxide synthase. J Am Coll Cardiol. 2012;59(22):1979–87. doi: 10.1016/j.jacc.2011.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Aragon JP, Condit ME, Bhushan S, Predmore BL, Patel SS, Grinsfelder DB, et al. Beta3-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J Am Coll Cardiol. 2011;58:2683–91. doi: 10.1016/j.jacc.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Amour J, Loyer X, Le Guen M, Mabrouk N, David JS, Camors E, et al. Altered contractile response due to increased beta3-adrenoceptor stimulation in diabetic cardiomyopathy: the role of nitric oxide synthase 1-derived nitric oxide. Anesthesiology. 2007;107:452–60. doi: 10.1097/01.anes.0000278909.40408.24. [DOI] [PubMed] [Google Scholar]
- [13].Birenbaum A, Tesse A, Loyer X, Michelet P, Andriantsitohaina R, Heymes C, et al. Involvement of beta 3-adrenoceptor in altered beta-adrenergic response in senescent heart: role of nitric oxide synthase 1-derived nitric oxide. Anesthesiology. 2008;109:1045–53. doi: 10.1097/ALN.0b013e31818d7e5a. [DOI] [PubMed] [Google Scholar]
- [14].Dawson D, Lygate CA, Zhang MH, Hulbert K, Neubauer S, Casadei B. nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation. 2005;112:3729–37. doi: 10.1161/CIRCULATIONAHA.105.539437. [DOI] [PubMed] [Google Scholar]
- [15].Idigo WO, Reilly S, Zhang MH, Zhang YH, Jayaram R, Carnicer R, et al. Regulation of endothelial nitric-oxide synthase (NOS) S-glutathionylation by neuronal NOS: evidence of a functional interaction between myocardial constitutive NOS isoforms. J Biol Chem. 2012;287:43665–73. doi: 10.1074/jbc.M112.412031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Napp A, Brixius K, Pott C, Ziskoven C, Boelck B, Mehlhorn U, et al. Effects of the beta3-adrenergic agonist BRL 37344 on endothelial nitric oxide synthase phosphorylation and force of contraction in human failing myocardium. J Card Fail. 2009;15:57–67. doi: 10.1016/j.cardfail.2008.08.006. [DOI] [PubMed] [Google Scholar]
- [17].Vandsburger MH, French BA, Kramer CM, Zhong X, Epstein FH. Displacement-encoded and manganese-enhanced cardiac MRI reveal that nNOS, not eNOS, plays a dominant role in modulating contraction and calcium influx in the mammalian heart. Am J Physiol Heart Circ Physiol. 2012;302:H412–9. doi: 10.1152/ajpheart.00705.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang YH, Casadei B. Sub-cellular targeting of constitutive NOS in health and disease. J Mol Cell Cardiol. 2012;52:341–50. doi: 10.1016/j.yjmcc.2011.09.006. [DOI] [PubMed] [Google Scholar]
- [19].Rameau GA, Tukey DS, Garcin-Hosfield ED, Titcombe RF, Misra C, Khatri L, et al. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J Neurosci. 2007 doi: 10.1523/JNEUROSCI.4799-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Afanas'ev IB. Lucigenin chemiluminescence assay for superoxide detection. Circ Res. 2001;89:E46. [PubMed] [Google Scholar]
- [21].Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2006;26:2439–44. doi: 10.1161/01.ATV.0000243924.00970.cb. [DOI] [PubMed] [Google Scholar]
- [22].Moens AL, Yang R, Watts VL, Barouch LA. Beta 3-adrenoreceptor regulation of nitric oxide in the cardiovascular system. J Mol Cell Cardiol. 2010;48:1088–95. doi: 10.1016/j.yjmcc.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ferreira JM, Burnett AL, Rameau GA. Activity-dependent regulation of surface glucose transporter-3. J Neurosci. 2011;31:1991–9. doi: 10.1523/JNEUROSCI.1850-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gauthier C, Leblais V, Kobzik L, Trochu JN, Khandoudi N, Bril A, et al. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest. 1998;102:1377–84. doi: 10.1172/JCI2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mallem MY, Toumaniantz G, Serpillon S, Gautier F, Gogny M, Desfontis JC, et al. Impairment of the low-affinity state beta1-adrenoceptor-induced relaxation in spontaneously hypertensive rats. Br J Pharmacol. 2004;143:599–605. doi: 10.1038/sj.bjp.0705990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kong YH, Zhang Y, Li N, Zhang L, Gao YH, Xue HJ, et al. Association between beta3-adrenergic receptor and oxidative stress in chronic heart failure rats. Zhonghua Xin Xue Guan Bing Za Zhi. 2010;38:435–9. [PubMed] [Google Scholar]
- [27].Brixius K, Bloch W, Pott C, Napp A, Krahwinkel A, Ziskoven C, et al. Mechanisms of beta 3-adrenoceptor-induced eNOS activation in right atrial and left ventricular human myocardium. Br J Pharmacol. 2004;143:1014–22. doi: 10.1038/sj.bjp.0705983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Brixius K, Bloch W, Ziskoven C, Bolck B, Napp A, Pott C, et al. Beta3-adrenergic eNOS stimulation in left ventricular murine myocardium. Can J Physiol Pharmacol. 2006;84:1051–60. doi: 10.1139/y06-033. [DOI] [PubMed] [Google Scholar]
- [29].Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101:15944–8. doi: 10.1073/pnas.0404136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kinugawa S, Huang H, Wang Z, Kaminski PM, Wolin MS, Hintze TH. A defect of neuronal nitric oxide synthase increases xanthine oxidase-derived superoxide anion and attenuates the control of myocardial oxygen consumption by nitric oxide derived from endothelial nitric oxide synthase. Circ Res. 2005;96:355–62. doi: 10.1161/01.RES.0000155331.09458.A7. [DOI] [PubMed] [Google Scholar]
- [31].Dincer UD, Bidasee KR, Guner S, Tay A, Ozcelikay AT, Altan VM. The effect of diabetes on expression of beta1-, beta2-, and beta3-adrenoreceptors in rat hearts. Diabetes. 2001;50:455–61. doi: 10.2337/diabetes.50.2.455. [DOI] [PubMed] [Google Scholar]
- [32].Watts VL, Motley ED. Role of protease-activated receptor-1 in endothelial nitric oxide synthase-Thr495 phosphorylation. Exp Biol Med. 2009;234:132–9. doi: 10.3181/0807-RM-233. [DOI] [PubMed] [Google Scholar]
- [33].Yen DH, Chen LC, Shen YC, Chiu YC, Ho IC, Lou YJ, et al. Protein kinase A-dependent neuronal nitric oxide synthase activation mediates the enhancement of baroreflex response by adrenomedullin in the nucleus tractus solitarii of rats. J Biomed Sci. 2011;18:32. doi: 10.1186/1423-0127-18-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhou C, Li C, Yu HM, Zhang F, Han D, Zhang GY. Neuroprotection of gamma-aminobutyric acid receptor agonists via enhancing neuronal nitric oxide synthase (Ser847) phosphorylation through increased neuronal nitric oxide synthase and PSD95 interaction and inhibited protein phosphatase activity in cerebral ischemia. J Neurosci Res. 2008;86:2973–83. doi: 10.1002/jnr.21728. [DOI] [PubMed] [Google Scholar]
- [35].Rameau GA, Chiu LY, Ziff EB. NMDA receptor regulation of nNOS phosphorylation and induction of neuron death. Neurobiol Aging. 2003;24:1123–33. doi: 10.1016/j.neurobiolaging.2003.07.002. [DOI] [PubMed] [Google Scholar]
- [36].Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–9. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- [37].Rameau GA, Chiu LY, Ziff EB. Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-d-aspartate receptor. J Biol Chem. 2004;279:14307–14. doi: 10.1074/jbc.M311103200. [DOI] [PubMed] [Google Scholar]
- [38].Osuka K, Watanabe Y, Usuda N, Nakazawa A, Fukunaga K, Miyamoto E, et al. Phosphorylation of neuronal nitric oxide synthase at Ser847 by CaM-KII in the hippocampus of rat brain after transient forebrain ischemia. J Cereb Blood Flow Metab. 2002;22:1098–106. doi: 10.1097/00004647-200209000-00007. [DOI] [PubMed] [Google Scholar]
- [39].Osuka K, Watanabe Y, Usuda N, Atsuzawa K, Aoshima C, Yamauchi K, et al. Phosphorylation of neuronal nitric oxide synthase at Ser847 in the nucleus intermediolateralis after spinal cord injury in mice. Neuroscience. 2007;145:241–7. doi: 10.1016/j.neuroscience.2006.10.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.