

Preclinical microscopy has greatly enhanced our mechanistic understanding of cancer invasion and metastasis, the contribution of the tumour microenvironment to metastatic progression, and how invasion and the microenvironment jointly support cancer cell survival and resistance. Using organotypic models in vitro, live-cell imaging in three-dimensional (3D) tissue culture has identified how cytoskeletal, adhesion and protease systems drive invasion and metastasis [1]. When altered at the molecular level, these pathways underlie the unexpected diversity of the invasive process [2]. The recent use of intravital microscopy has further suggested that cancer invasion into interstitial stroma in vivo: (1) occurs mostly as collective invasion in which cells remain coupled to neighbouring cancer cells, (2) is guided by and responsive to signals delivered by connective tissue structures and (3) that invasion pathways cross-talk with pathways of cancer cell survival and resistance to anticancer therapy [3].
1. Principles of collective cell invasion
Collective cell migration is defined as the movement of multiple cells that retain cell–cell contacts, coordinate their actin dynamics and intracellular signaling, and thereby form a structural and functional unit for joint translocation [1,4]. In contrast to single-cell migration, moving cell masses remain mechanically coupled by cell–cell adhesion receptors, most notably of the cadherin and integrin families, and form a coordinated cortical structure of the actin cytoskeleton, occasionally referred to as a ‘super-cell’ [4]. Besides cancer invasion and metastasis, collective cell movement contributes to cell migration in morphogenesis and tissue repair [5], suggesting homologous underlying mechanisms.
As in all known types of actomyosin-based cell migration, collective migration is plastic, i.e. it undergoes modification with altered intracellular signaling or an altered environment [2]. Interference with molecules that maintain or regulate collective cell behaviour can lead to single-cell detachment. Depending on the type of single-cell migration obtained after dissociation, two types of conversion are currently known: the epithelial–mesenchymal transition (EMT) and the collective–amoeboid transition (CAT). EMT is a well established molecular process that leads to the down modulation of cell–cell adhesion, whereby the migration machinery remains intact, which induces cell detachment and scattering from multicellular groups [1] (and references therein). Mechanisms that enable single-cell detachment include reduced cadherin expression, loss-of-function mutations in cadherin and catenin [mit Leerzeichen ersetzen] signaling pathways, and deregulated function of proteases degrading cadherins and other cell–cell adhesion molecules [4]. In vivo, EMT corresponds to the loss of differentiated epithelial morphology in usually small regions towards a sarcomatous, stromal and, hence, invasive and likely metastatic phenotype. CAT is the transition from collective invasion to amoeboid single-cell crawling after simultaneous weakening of cell–cell and cell–ECM interactions, such as after EMT-independent down-regulation of cadherins (data not shown) or inhibition of β1 integrins in collectively invading melanoma explants [5] and in tumour xenografts in vivo (data not shown). Detached cells then survive, continue to move via amoeboid shape change (similarly to interstitial migration of amoeboid leukocytes [6]), and eventually cause distant metastasis (S. Alexander, MD Anderson Cancer Center). These findings suggest that collective migration represents an invasion mode of high cellular and molecular order that, after loss of function of particular adhesion pathways, interconverts to single-cell dissemination and metastasis. The understanding of the signals maintained by simultaneous cell–cell and cell–matrix communication during collective invasion and secondary plasticity will be important in defining the cross-talk between strategies of invasion and resistance signaling [3].
2. Intravital multiphoton microscopy of collective cancer invasion in vivo
Multiphoton microscopy (MPM) has become the method of choice for investigating cell structure and function in tissues and organs, including the invasion and progression of cancer lesions [7]. Particularly suited for cancer research is infrared multiphoton microscopy, which enables deep tissue penetration and detects multicellular, collective invasion of melanoma and soft-tissue sarcoma lesions in vivo [8]. Recent evidence from intravital microscopy further suggests that collective invasion is strongly associated with resistance to radiation therapy and chemotherapy.
3. Joint mechanisms of cancer invasion and resistance
Based on the multiple inputs from the tumour microenvironment and their overlapping signaling pathways, invasive tumour-cell migration and resistance are now considered as interconnected cell functions. To gain deeper insight into the steps and niches of concurrent resistance and dissemination, preclinical animal models followed by time- and space-resolved molecular imaging are necessary to detect tumour responses to therapy at a cellular level. The signals required for both single-cell and collective cancer invasion include the activation of integrins, cadherins, small GTPases Rac and Rho, as well as Ras pathways, and the engagement of intracellular signaling networks that include PI3K, mTOR, Src and Map kinases (Fig. 1). Consequently, druggable signaling hubs that may serve to target both tumour invasion and resistance include growth factor and chemokine signaling, integrin engagement, as well as downstream Ras/MAPKs, PI3K and mTOR signaling. Thereby, the residual niches that withstand targeting of conventional therapy can consist of a limited number of cells which, after surviving cycles of therapies, regrow, initiate migration and thereby re-establish an invasive tumour.
Fig. 1.
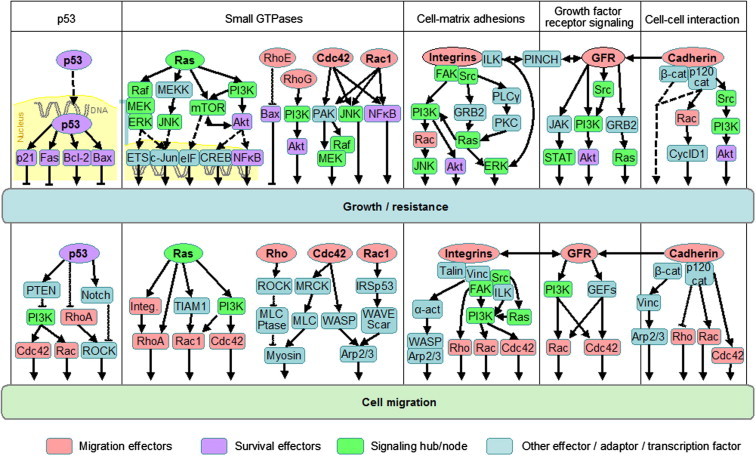
Signaling pathways controlling tumour cell growth, survival and invasion. Example pathways of p53, Ras GTPase, small Rho GTPases, integrins, growth factor receptors and cadherins with a dual role in controlling cell growth (upper row) and survival as well as cell migration and invasion (lower row). Migration effectors are marked in pink, survival effectors in purple, signaling hubs in bright green. Arrows indicate signaling direction. Bound to DNA, transcription factors. Figure taken from Ref. [3]. α-Act., α-actinin; cat, catenin; Cdc42, cell division cycle 42; CREB, cAMP response element-binding; CyclD1, cyclin D1; eIF, eukaryotic initiation factor; ERK, extracellular signal-related kinase; ETS, erythroblast transformation specific (transcription factor); FAK, focal adhesion kinase; GEF, guanine nucleotide exchange factor; GFR, growth factor receptor; GRB2, growth factor receptor-bound protein 2; ILK, integrin-linked kinase; Integ., integrin; JNK, Janus-kinase; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase; MEKK, MEK kinase; mTOR, mammalian target of rapamycin; MLC, myosin light chain; MLCPtase, MLC phosphatase; MRCK, myotonic dystrophy kinase-related Cdc42-binding kinase; NFκB, nuclear factor ‘kappa-light-chain-enhance’ of activated B cells; PAK, p21-activated kinase; PINCH, particularly interesting Cys–His-rich protein; PKC, protein kinase C; PLCγ, phospholipase γ; PI3K, phosphoinosid-3-kinase; PTEN, phosphatase and tensin homologue; ROCK, Rho-activated kinase; STAT, signal tranducer and activator of transcription; TIAM1, T-cell lymphoma invasion and metastasis 1; Vinc, vinculin; WASP, Wiskott–Aldrich syndrome protein; WAVE; WASP family Verprolin-homologous protein. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Outlook
Collective invasion types contribute and provide particular challenges to the progression and therapy of cancer disease. The cell mass likely produces high autocrine concentrations of promigratory factors and matrix proteases. Because many cells move as one functional unit, cells of different clonal origin or different biological abilities may be linked and invade together (‘mixed clone’ behaviour). Furthermore, it can protect inner cells from immunological assault through cytotoxic lymphocytes. As a particular challenge, the joint signaling from tissue structures and cell–cell junctions may activate survival pathways not engaged in quiescent, non-invading tumour regions. Thus, preclinical in vivo microscopy will enable both fascinating insight into the basic mechanisms of cancer biology as well as advance preclinical validation of drugs and the identification of resistance mechanisms.
Conflict of interest statement
None declared.
References
- 1.Friedl P., Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 2.Friedl P., Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol. 2010;188:11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexander S., Friedl P. Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. Trends Mol Med. 2012;18:13–26. doi: 10.1016/j.molmed.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Friedl P., Locker J., Sahai E., Segall J.E. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14:777–783. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- 5.Friedl P., Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 6.Hegerfeldt Y., Tusch M., Brocker E.B., Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell–cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002;62:2125–2130. [PubMed] [Google Scholar]
- 7.Andresen V., Alexander S., Heupel W.-M. Infrared multiphoton microscopy: subcellular-resolved deep tissue imaging. Curr Opin Biotechnol. 2009;20:54–62. doi: 10.1016/j.copbio.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Alexander S., Koehl G.E., Hirschberg M., Geissler E.K., Friedl P. Dynamic imaging of cancer growth and invasion: a modified skin-fold chamber model. Histochem Cell Biol. 2008;130:1147–1154. doi: 10.1007/s00418-008-0529-1. [DOI] [PubMed] [Google Scholar]