

Abstract
Suppressors of cytokine signaling (SOCS) proteins are negative-feedback regulators of JAK/STAT pathway and SOCS3 contributes to host immunity by regulating the intensity/duration of cytokine signals and inflammatory responses. Mice with Socs3 deletion in myeloid cells exhibit enhanced STAT3-signaling, expansion of Th1 and Th17 cells and developed severe experimental autoimmune encephalomyelitis (EAE). Interestingly, development of the unique IL-17/IFN-γ-double producing (Th17/IFN-γ and Tc17/IFN-γ) subsets that exhibit strong cytotoxic activities and associated with pathogenesis of several autoimmune diseases, has recently been shown to depend on epigenetic suppression of SOCS3 expression, further suggesting involvement of SOCS3 in autoimmunity and tumor immunity. In this study, we generated mice with Socs3 deletion in CD4 T cell compartment (CD4-SOCS3KO) to determine in vivo effects of the loss of Socs3 in the T cell-mediated autoimmune disease, experimental autoimmune uveitis (EAU). In contrast to the exacerbation of EAE in myeloid-specific SOCS3-deleted mice, CD4-SOCS3KO mice were protected from acute and chronic uveitis. Protection from EAU correlated with enhanced expression of CTLA4 and expansion of IL-10 producing Tregs with augmented suppressive activities. We further show that SOCS3 interacts with CTLA4 and negatively regulates CTLA4 levels in T cells, providing mechanistic explanation for the expansion of Tregs in CD4-SOCS3 during EAU. Contrary to in vitro epigenetic studies, Th17/IFN-γ and Tc17/IFN-γ populations were markedly reduced in CD4-SOCS3KO, suggesting that SOCS3 promotes expansion of Th17/IFN-γ subset associated with development of severe uveitis. Thus, SOCS3 is a potential therapeutic target in uveitis and other auto-inflammatory diseases.
Introduction
The JAK/STAT pathway is an evolutionarily conserved signal transduction mechanism that regulates a myriad of physiological processes in mammals (1). The importance of regulating the initiation, duration and intensity of STAT signals is underscored by the diverse array of pathologic conditions that arise from disruption or aberrant activation of STATs (2). JAK/STAT pathways are therefore under stringent regulation by a number of cytoplasmic proteins including PIAS (protein inhibitors of activated STAT), SHP-1 (SH2-containing phosphatase 1), SHIP-2 and suppressors of cytokine signaling (SOCS) family of proteins. In context of immune regulation or immune modulation therapy, much interest has focused on SOCS proteins, particularly SOCS1 and SOCS3 (3, 4). SOCS proteins are rapidly induced in response to cytokines (IFN-γ, IL-2, IL-4, IL-6, IL-10, IL-12, IL-21, IL-23, IL-27) or growth factors (CNTF, LIF, FGF, IGF-1, insulin) and their inhibitory effects derive from direct interaction with cytokine/growth-factor receptors or signaling proteins, leading to proteosomal degradation of the receptor complex and termination of the signal (5). Because of the relatively short half-life of SOCS proteins, their negative regulatory effects are generally transient. However, unabated stimulation of STAT signaling pathway by chronic inflammation can induce constitutive activation of SOCS expression (6). In some tissues this may result in persistent silencing of critical cellular pathways and pre-disposition to development of organ-specific diseases (7). SOCS proteins have now been implicated in diverse diseases including, autoimmune diseases, diabetes, and cancer (6–9).
SOCS3 regulates the activation and differentiation of naïve CD4 T cells, preferentially promoting Th2 and inhibiting Th1 differentiation via the inhibition of IL-12-mediated STAT4 activation (10, 11). It is constitutively expressed in naïve CD4+ T cells and its expression is inversely correlated with the level of IL-2 (11, 12). SOCS3 mediates the IL-27-induced suppression of CD28-mediated IL-2 production (13) and it blocks IL-2 production in response to TCR activation by suppressing calcineurin-dependent dephosphorylation and activation of NFATp (14). Unlike T-helper cells, Tregs are deficient in SOCS3 protein expression and in vitro over-expression of SOCS3 in Treg decreased their proliferation and expression of Foxp3, suggesting the SOCS3/IL-2 axis plays critical role in controlling physiological levels of Tregs. In terms of the potential involvement of SOCS3 in autoimmune diseases, it has recently been shown that mice with Socs3 deletion in myeloid cells develop severe EAE, suggesting that STAT3/SOCS3 axis regulates neuroinflammation (15, 16). On the other hand, expression of SOCS3 in human arthritic chondrocytes contributes to cartilage damage during arthritis (17, 18). Interestingly, epigenetic suppression of SOCS3 expression in T cells promotes the expansion of a unique Tc17/IFN-γ-double producing CD8+ T cells implicated in several autoimmune diseases (19–22). These observations thus underscore the complexity of SOCS3 functions in the immune system and mechanisms that regulate autoimmune pathology.
Intraocular inflammation or uveitis is a major cause of severe visual handicap and includes sight-threatening diseases such as Behcet disease, birdshot retinochoroidopathy, Vogt-Koyanagi-Harada, sympathetic ophthalmia and ocular sarcoidosis (23). Although the cause of most chronic ocular inflammatory diseases of non-infectious etiology is largely unknown, studies in the mouse indicate that it is predominantly a T cell mediated disease. Experimental autoimmune uveoretinitis (EAU) shares essential immunopathologic features with human uveitis and is the animal model of human uveitis (24). The disease is characterized by massive infiltration of inflammatory cells into the retina, destruction of retinal architecture and Th1 and Th17 cells were thought to be the etiologic agents (25, 26). Expression of SOCS3 is induced in the retina during uveitis with its highest level at the peak of the disease and coinciding with infiltration of the retina by CD4+ T cells (27). However, it is not clear whether expression of SOCS3 by T cells contribute disease pathology or if its function is to ameliorate uveitis. In this report, we show that mice that lack SOCS3 in CD4+ T cells were protected from chronic uveitis by inducing an immune suppressive state characterized by enhanced expression of CTLA4 and expansion of IL-10 producing Tregs with augmented suppressive activities.
Methods
Mice
We derived mice with conditional deletion of Socs3 in CD4+ T cells (CD4-SOCS3KO) by breeding Socs3fl/fl (provided by Dr. Akihiko Yoshimura, Kyushu University, Japan) with CD4-Cre (Taconic, Hudson, NY) mice. The CD4-SOCS3KO mouse strain was established by brother/sister inbreeding for more than 10 generations. The CD4-SOCS3−/+ hemizygous mouse strain was derived after the second-generation filial (F2) breeding. Littermate Socs3fl/fl mice, in C57BL/6 background, were used as wild type (WT) controls. Mice were maintained and used in accordance with NEI/NIH Animal Care and Use Committee guidelines.
Induction of experimental autoimmune uveoretinitis (EAU) by active immunization
Mice were immunized with 150 µg interphotoreceptor retinoid-binding protein (IRBP) and 300µg of human IRBP peptide (1–20) in 0.2 ml emulsion 1:1 v/v with Complete Freund’s adjuvant (CFA) containing Mycobacterium tuberculosis strain H37Ra (2.5 mg/ml). Mice also received Bordetella pertussis toxin (0.2 µg/mouse) concurrent with immunization and clinical disease was established by fundoscopy as described previously (28). Fundoscopy and/or histology were used to monitor progression of pathology in the eyes at 0, 7, 14, 21, 60 or 90 days post-immunization. For histology, eyes were harvested, fixed in 4% glutaraldehyde for 30 minutes, and transferred to 10% buffered formalin. After adequate fixation, specimens were dehydrated through graded alcohols and embedded in methacrylate. Serial vertical sections through the papillary-optic nerve plane were cut and stained with hematoxylin and eosin (H&E) as described (28). Clinical disease was established and scored by fundoscopy as described (28) (29). Briefly, following intraperitoneal injection of ketamine (1.4 mg/mouse) and xylazine (0.12 mg/mouse), pupils were dilated by topical administration of 1% tropicamide ophthalmic solution (Alcon Inc, Fort Worth, Texas). To avoid a subjective bias, evaluation of the fundus photographs was conducted without knowledge of the mouse identity by a masked observer. At least six images (2 posterior central retinal view, 4 peripheral retinal views) were taken from each eye by positioning the endoscope and viewing from superior, inferior, lateral and medial fields and each individual lesion was identified, mapped and recorded. The clinical grading system for retinal inflammation was as previously established (30).
Isolation, propagation and characterization of CD4+ T-cells
Naïve CD4+ T cells (> 97%) were isolated from mouse peripheral blood (PBMC), lymph nodes (LN) or spleen as described early (10). CD4+ T-cells were activated in plate-bound anti-CD3 Ab (3µg/ml) (BD BioScience) and anti-CD28 Ab (3 µg/ml) in complete medium (Th0). For propagation under Th17 condition the medium contained TGF-β (2 ng/ml), IL-6 (10 ng/ml), IL-1β (10 ng/ml), anti-IFN-γ Ab (10 µg/ml) and anti-IL-4 Ab (10 µg/ml). Where indicated CD4+ T cells were analyzed without stimulation while most cultures were stimulated for 4 days. For intracellular cytokine detection, freshly isolated or cultured CD4+ T-cells were re-stimulated for 5 h with PMA (20ng/ml)/ionomycin (1µM) in the presence of Golgi-stop at the recommended concentrations (BD Pharmingen, San Diego, CA). The cells were permeabilized, and stained with the requisite Abs using the BD Biosciences Cytofix/Cytoperm kit according to the manufacturer’s instructions. FACS analysis using anti-CD3, CD4, IFN-γ, IL-17, Foxp3, mAbs and corresponding isotype control Abs (PharMingen) was performed on Becton Dickinson FACSCalibur or LSRII (BD Biosciences, San Jose, CA) as previously described (31).
Cytokine Analysis
WT or CD4-SOCS3KO T cells were activated for 4 days with anti-CD3/CD28 Abs and multiplex ELISA of supernatants for cytokine secretion was performed using a commercial ELISA kit (R&D Systems, Minneapolis, MN).
Lymphocyte proliferation assay
Naïve T cells or LN cells from IRBP-immunized mice were cultured for 2–3 d in quintuplet cultures containing anti-CD3/CD28 Abs or IRBP. For the T cell suppression study, naïve T cells from WT or CD4-SOCS3KO were cultured with LN cells from WT mice with EAU for 3 days in medium containing IRBP. After 36 h, some cultures were pulsed with 3H-thymidine (0.5 µCi/10 µl/well) for 12 additional hours and analyzed. Data are mean CPM ± S.E. of responses of five replicate cultures.
Quantitative and Semi-quantitative RT PCR analyses
Total RNA was extracted using the TriZol reagent according to the procedures recommended by the manufacturer (Life Technologies, Gaithersburg, MD). All RNA samples were digested with RNase-free DNase 1 (Life Technologies), purified by phenol/chloroform extractions. RNA integrity was verified by analysis of 18S and 28S ribosomal RNA expression on RNA gels. RNA (10 µg), SuperScript III Reverse Transcriptase (Life Technologies, Gaithersburg, MD), and oligo (dT)12–16 were used for first-strand synthesis as previously described (32). First-strand synthesis containing each mRNA sample but no reverse transcriptase was performed to control for possible DNA contamination; failure to obtain RT-PCR products with any of the PCR amplimers confirmed the absence of DNA templates. All cDNA preparations used were suitable substrates for PCR amplification on the basis of efficient amplification of a β-actin sequence. Real-time PCR was performed on an ABI 7500 (Applied Biosystems) and PCR parameters were as recommended for the TaqMan Universal PCR kit (Applied Biosystems). Primers and probes were purchased from (Applied Biosystems).
Western blotting analysis
Preparation of whole cell lysates and performance of Western blot analysis were as described (10). Cell extracts (20–40 µg/lane) were fractionated on 4–12% gradient SDS-PAGE, and antibodies used were: pSTAT3 (Cell Signaling Technology); CTLA-4, CD28, SOCS3 and β-Actin (Santa Cruz Biotechnology, Santa Cruz, CA). Pre-immune serum was used in parallel as controls and signals were detected with HRP-conjugated secondary F(ab')2 Ab (Zymed Laboratories) using the ECL-PLUS system (Amersham, Arlington Heights, IL).
SOCS3-GST pull-down assays
pGEX6p1 vector and GST-gene fusion reagents were purchased from GE Healthcare Bioscience. Briefly, mSOCS3 cDNA was cloned into pGEX6p1 vector via BamH1 and Sal I cloning sites. Plasmid sequence was verified by DNA sequencing. The empty vector and GST-SOCS3 plasmid were transformed into Escherichia coli BL21 strain. The GST fusion protein was induced by IPTG (Isopropyl β-D-1-thiogalactopyranoside) and analyzed by SDS-PAGE, coomassie blue staining and Western blotting. GST or GST-SOCS3 fusion protein purified by Glutathione Sepharose 4B affinity was used for protein pull-down assays. Protein extracts (500 µg) isolated from activated CD4+ T cells were incubated with the purified GST-SOCS3 protein for 1 h at 4°C, washed twice and the precipitates were analyzed by immunoblotting with CTLA-4 Abs.
Statistical analysis
Statistical analyses were performed by independent two-tailed Student’s t-test. Data presented as mean + SD. Asterisks denote p value (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Results
Generation and functional charecterization of CD4-SOCS3 mice
We generated mice with targeted deletion of SOCS3 in the CD4 compartment (CD4-SOCS3KO) to investigate the function of SOCS3 in T cells during an autoimmune disease. PCR analysis of tail DNA of mice from the cross between CD4-Cre and SOCS3fl/fl mouse strains on C57BL/6 background confirmed the generation of CD4-Cre/SOCS3fl/fl double positive mice (Fig. 1A). Expression of the Cre recombinase under the CD4 promoter element (CD4-Cre strain) is initiated at the CD4/CD8 double positive stage of the developing T cell, resulting in targeted expression the Cre protein in both CD4+ and CD8+ T cells (33, 34). We therefore expected that the expression of Cre at the double positive stage would mediate SOCS3-specific deletion in both CD4+ and CD8+ T cells of the CD4-SOCS3KO mouse strain. To confirm that SOCS3 was indeed deleted in CD4+ and CD8+ T cells of the CD4-SOCS3KO T cells, we isolated CD4+ and CD8+ T cells from WT and CD4-SOCS3KO mice, purified the cells by sorting. We prepared cDNA from RNA isolated from the sorted CD4+ or CD8+ T cells and RT-PCR and qRT-PCR analyses revealed that SOCS3 was deleted from CD4+ and also CD8+ T cells (Fig. 1B). Analysis of whole cell extracts prepared from the sorted CD4+ T cells by the Western blot assay, confirmed specific deletion of the SOCS3 protein from the CD4 compartment (Fig, 1C). Western blot analysis further show that SOCS3 is constitutively expressed in naive CD4+ T cells as previously reported (10) and we detected constitutive expression of pSTAT3 in CD4-SOCS3KO T cells but not in WT or hemizigous (CD4-SOCS3−/+) CD4+ T cells (Fig. 1D). To verify that functional SOCS3 function is lost in CD4-SOCS3KO T cells we examined whether the CD4-SOCS3KO T cells had lost capacity to terminate STAT signals. Naïve CD4+ T cells from WT or CD4-SOCS3KO mice were stimulated with anti-CD3/CD28 Abs for 3 days. The cells were then washed, starved in serum free medium for 3 hours on ice and stimulated in medium containing IL-6 as indicated. The protein extracts were analyzed by Western blotting (Fig. 1E) and showed that the pSTAT3 signal was inhibited after 120 min in WT but not CD4-SOCS3KO T cells, confirming complete loss SOCS3 function in the CD4-SOCS3KO mice.
Figure 1.

Generstion and characterization of CD4-SOCS3KO mice. (A) SOCS3fl/fl mice were cross-bred with CD4-Cre mice to generate mice with deletion of SOCS3 in the CD4+ T cells (CD4-SOCS3KO) and the SOCS3fl/fl/Cre alleles were identified by PCR analysis of mouse tail genomic DNA. (B–D) RNA or protein was isolated from naïve CD4+ T cells (B, C, D) or CD8+ T cells (B) and analyzed by RT-PCR and qPCR (B) or Western blotting (C, D). (E) Naïve CD4+ T cells were activated by anti-CD3/CD28 for three days, then starved for two hours in 1% BSA before stimulation with IL-6 (20ng/ml) at indicated time points. Protein extracts were analyzed by Western blotting (E). Results are representative of at least three independent experiments.
SOCS3 deficiency promoted the expansion of IL-10-expressing regulatory T cells
Previous studies suggested that SOCS3 is an essential negative regulator of IL-23 signaling and because IL-23 mediates STAT3 phosphorylation and Th17 generation, it was suggested that SOCS3 might therefore constrain the generation of Th17 differentiation (35). We therefore next investigated whether loss of SOCS3 in T cells would promote the expansion of Th17 cells. Naïve CD4+ T cells were isolated and stimulated under non-polarizing condition or to mature into Th17 by using the appropriate cytokine and abs cocktail. The cytokine expression profile of the cultured cells was assessed by intracellular staining assay while cytokine secretion was detected using ELISA. Contrary to prediction, we show here that the CD4+ T cells lacking Socs3 generated comparable frequency of IL-17-expressing Th17 cells as WT T cells (Fig 2A), suggesting that SOCS3 is not a critical regulator of Th17 polarization. By contrast, SOCS3 was found to be a major regulator of IL-10-expressing cells (Fig. 2A). These results were confirmed by our ELISA data showing high levels of secretion of IL-10 by CD4-SOCS3KO T cells while IL-17 secretion was comparable between the WT and SOCS3KO T cells (Fig.2B). The high level of IL-10-expressing T cells appeared to have produced an overall anti-proliferative condition in the CD4-SOCS3KO cultures characterized by relatively low percentage of activated IL-2- and IFN-γ-expressing T cells (Fig. 2A). These results thus suggest that SOCS3 may function to promote inflammatory activities of T cells.
Figure 2.

SOCS3-deficient CD4+ T cells promote the expansion of IL-10-producing cells. Naïve CD4+ T cells isolated from WT and CD4-SOCS3KO mice were stimulated with anti-CD3/CD28 for four days under Th0 or Th17 polarizing condition and then analyzed for cytokine expression by the intracellular cytokine staining assay (A) or for cytokine secretion by ELISA (B). Numbers in quadrants (A) indicate percent of T cells expressing IL-2, IL-10, IL-17 or IFN-γ. Results are representative of at least three independent experiments.
Socs3 deletion in T cells confers protection from experimental autoimmune uveitis (EAU)
To investigate the impact of deleting SOCS3 in T cells on development and severity of acute and chronic uveitis, we induced EAU in WT (Socs3fl/fl) and CD4-SOCS3KO mice by active immunization with IRBP in CFA and monitored the course of the disease for 90 days by funduscopy. During EAU in mice, the first 28 days constitutes the acute phase of EAU and disease activity is most prominent between days 12 to 22 (36). Fundus images taken at days 14 and 21 show that both strains developed acute uveitis. However, EAU in the immunized WT mice was characterized by the infiltration of massive numbers of inflammatory cells into the retina resulting in destruction of retinal cells, development of retinal folding, serous retinal detachment, vasculitis, retinitis, choroiditis, vitritis and these pathological features are hallmarks of severe acute uveitis. Although very few cells are detectable in the retina after day-28 post-immunization, the chronic phase ensue and it is associated with significant ocular pathology characterized by severe papillitis, vasculitis with cuffing, retinal degenerative changes and choroidal-neovascularization (36–38). Thus, the clinical scores shown in Figure 3B is mono-phasic and reflects continued progression of ocular pathology that culminates in vision loss (36). In contrast to EAU in the WT mice, the disease in the CD4-SOCS3KO mice is very mild as indicated by the disease score (Fig. 3B) and histology (Fig. 3C, 3D). Contemporaneous analysis of fundus images did not reveal overt pathology at day-60 or day-90, suggesting that the CD4-SOCS3KO mice were protected from developing chronic EAU.
Figure 3.

Loss of SOCS3 in CD4+ T cells confers protection from uveitis. EAU was induced in WT or CD4-SOCS3KO mice by active immunization with IRBP in CFA. Progression and severity of EAU was monitored for 90 days by funduscopy or histology. (A) Fundus images were taken using an otoendoscopic imaging system, and the development of papillitis (black arrows), retinal vasculitis (blue arrows) and inflammatory infiltrates (white arrows) are indicated. (B) Clinical score was based on changes at the optic nerve disc, retinal vessels, and surrounding tissues as described in the methods section. (C) Eyes were enucleated on day-21 post-immunization, fixed in paraffin and stained with H&E. Infiltrated inflammatory cells in the vitreous (V) are denoted by black arrows; blue asterisks indicate retinal folds. OpN (optic nerve), GCL (ganglion cell layer), INL (inner nuclear layer), ONL (outer nuclear layer), RPE (retinal pigment epithelial layer), CH (choroid). (D) Disease scored based on histology.
The resistance of CD4-SOCS3KO mice to EAU correlates with expansion of regulatory T cells
In order to investigate the mechanistic basis for the resistance of CD4-SOCS3KO mice to EAU, we analyzed the immune-phenotype and cytokines expression profile of T cells in the peripheral blood of IRBP/CFA immunized WT or SOCS3KO mice (day-21 post-immunization), as well as, T cells from unimmunized C57BL/6 mice. Analysis of the expression of activation markers revealed that the frequency of activated CD4+ T cells is much reduced in CD4-SOCS3KO blood compared to the WT (Fig. 4A), indicating that SOCS3 may promote an anti-inflammatory environment during EAU. Both Th1 and Th17 cells have been implicated in the etiology of human and mouse uveitis (25, 26) and SOCS3 is thought to be an essential negative regulator of IL-23 signaling and Th17 differentiation (35). Contrary to the expectation that Th17 cells would be aberrantly expanded in the CD4-SOCS3KO mice during EAU, we observed a slight decrease in the percent of IL-17-expressing Th17 cells as well as a decrease in IFN-γ-expressing Th1 cells (Fig. 4B). On the other hand, we observed higher frequency of IL-10-expressing cells in the blood of IRBP-immunized CD4-SOCS3KO compared to WT mice by FACS (Fig. 4C) and ELISDA (D). The latter result may partially explain the lower numbers of activated cells and Th1 or Th17 in CD4-SOCS3KO mice. Of significant interest is the >3.5-folds reduction of IL-17/IFN-γ-double producing Th17 cells (Fig. 4B) associated with several inflammatory diseases, including EAU (19–22). Because SOCS3 is also deleted in CD8+ T cells, we also examined whether development of the unique Tc17/IFN-γ CD8+ population is also compromised in CD4-SOCS3KO mice. Similar to CD4+ T cells, we observed a 4.4-folds reduction in Tc17/IFN-γ CD8+ population (Fig. 4E) and in contrast to CD4+ T cells, the CD4-SOCS3KO mice have higher percent Tc17 cells (Fig. 4F).
Figure 4.

SOCS3 promotes an anti-inflammatory response in vivo. (A) Freshly isolated PBMCs of un-immunized mice or IRBP-immunized mice were analyzed for expression of T cell cell-surface proteins by FACS. (B–D) The CD4+ T cells were also analyzed by the intracellular cytokine staining assays (B, C) or ELISA (D) for the expression IL-7, IFN-γ and/or IL-10. (E, F) CD8+ T cells isolated from IRBP-immunized mice were analyzed by the intracellular cytokine staining assays for the expression IL-7, IFN-γ and/or IL-10. The cells were gated on CD3/CD4 or CD3/CD8 and numbers in quadrants indicate percent of CD4+ T cells expressing IL-10, IL-17 and/or IFN-γ. Statistical analysis of the percentage of cytokine-expressing T cells was based on analysis of 6 mice per group. Experiments were repeated at least three times.
Socs3 inhibits the suppressive activity of Tregs
Regulatory T cells (Treg cells) expressing the transcription factor Foxp3 and induced T regulatory type 1 (Tr1) cells that lack Foxp3 expression play crucial roles in maintaining immune homeostasis and they both induce immunosuppressive functions that regulate effector T cell function through the production of IL-10 (39). We isolated naïve CD4+ T cells from the WT or CD4-SOCS3KO mice, purified the cells using anti-CD4 magnetic beads, stimulated them for 3 days with anti-CD3/CD28 in the presence of TGF-β1 and examined whether SOCS3 regulates the expansion of Foxp3+ Tregs. The analysis of Foxp3+CD25+ Tregs by intracellular flow cytometry showed a tremendous expansion of this regulatory cell population in the CD4-SOCS3KO T cell cultures (Fig. 5A), suggesting that SOCS3 inhibits their expansion. We next examined whether the IL-10 producing CD4-SOCS3KO T cells possess T cell suppressive activities. We induced EAU in WT mice and on day-21 post-immunization we havested cells from the LN of the mice with EAU. The freshly isolated draining LN cells were then co-cultured with naïve CD4+ T cells from either WT or CD4-SOCS3KO mice (ratios of naïve CD4+ T cells: uveitogenic LN cells are indicated). The proliferation of the uveitogenic T cells in response to re-stimulation with IRBP was assessed by the [3H]-thymidine incorporation assay. In the first series of experiments performed at the 1:1 ratio of naïve CD4+ T cells: uveitogenic LN cells, SOCS3KO CD4 T cells did not appreciably alter the baseline proliferation and the observe differences between the cultures containing WT or SOCS3KO can be attributed to background stimulation by WT cells vs no stimulation by SOCS3KO (Fig. 5B; left-most panel). However, the next series of experiments performed at the 1:4 ratio (naïve CD4+ T cells: uveitogenic T cells), we observed significant suppression of proliferation in cultures containing CD4-SOCS3KO T cells while the WT T cells did not inhibit proliferation of the IRBP-stimulated uveitogenic LN cells (Figure 5B; middle panel). On the other hand, the inhibitory effect of the CD4-SOCS3KO T cells was lost in experiments performed at the 1:16 ratio (naïve CD4+ T cells: uveitogenic T cells) and this can be attributed to the relatively low frequency of inhibitory CD4-SOCS3KO T cells in the cultures (Fig. 5B (right-most panel). Taken together, these results suggest that the suppressive activities of the CD4-SOCS3KO T cells might have contributed to the suppression of EAU in the CD4-SOCS3KO mice.
Figure 5.
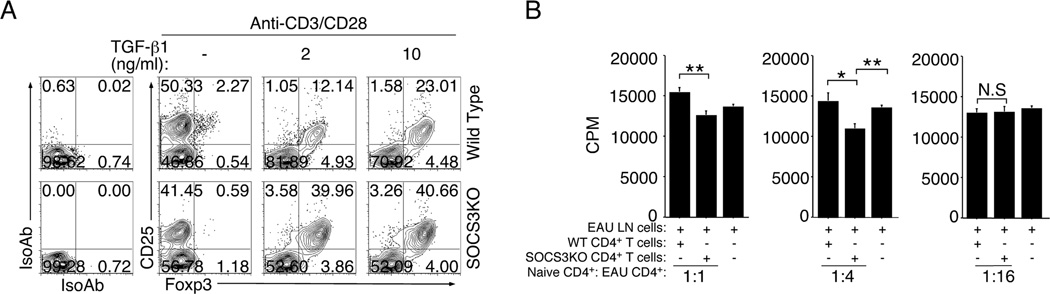
Loss of SOCS3 in CD4+ T cells promotes the expansion of regulatory T cells and their suppressive activities. (A) Naïve CD4+ T cells were isolated and purified from lymph nodes and spleen of WT or CD4-SOCS3KO mice and stimulated with anti-CD3/CD28 for three days in the presence or absence of TGF-β1. The cells were analyzed by FACS and the intracellular cytokine staining assays. (B) Naïve T cells from WT or CD4-SOCS3KO were cultured with LN cells from WT mice with EAU for 3 days in medium containing IRBP. Ratios of naïve CD4+ T cells: uveitogenic T cells are indicated. T cell proliferation was assessed by [3H]-thymidine incorporation assay. Results represent of at least three independent experiments.
SOCS3 interacts with CTLA-4 and regulates its steady-state level in CD4+ T cells
It was previously reported that SOCS3 prevents excessive CD28-mediated IL-2 production by interacting with the CD28 co-stimulatory receptor and targeting it for proteosomal degradation (11). This report also indicated that the association between CD28 and SOCS3 required the interaction of SOCS3 with the tyrosine phosphorylation of a motif in the cytoplasmic region of CD28 (11). CD28 and CTLA-4 share the highly conserved tyrosine-containing motifs (e.g. MYPPPY) (40). We therefore speculated that SOCS3 might also interact with CTLA-4 and regulate its expression. We isolated naïve CD4+ T cells from WT or CD4-SOCS3KO mice, stimulated the cells for 3 days with anti-CD3/CD28 Abs and examined whether the loss of SOCS3 correlates with high levels of CTLA-4. As predicted, Western blot analysis of extracts from the T cells showed increase of CTLA-4 protein in the naïve or activated CD4-SOCS3KO compared to WT T cells (Fig. 6A). Consistent with previous reports, we also found increase of CD28 in the CD4-SOCS3KO T cells, suggesting that SOCS3 is a negative regulator CTLA-4 and CD28. To investigate whether SOCS3 regulates the level of CTLA-4 in vivo, we analyzed the level of CTLA4-expressing CD4+ T cells in the blood of IRBP/CFA immunized WT or SOCS3KO mice (day-21 post-immunization). The frequency of CTLA4-expressing T cells was doubled in CD4-SOCS3KO compared to WT mice (Fig. 6B), further supporting the observed augmented suppressive activity of Tregs generated from CD4-SOCS3KO mice (Fig. 5). Although the reduced EAU severity in CD4-SOCS3KO mice is associated with increased CTLA-4 expression, more studies are required to firmly establish a causal relationship between SOCS3 deletion, CTLA-4 increase, and suppression of uveitis. To directly examine whether SOCS3 interacts with CTLA-4, we generated a GST-SOCS3 fusion protein in a bacterial expression system. We characterized and purified the recombinant protein by affinity chromatography following induction with IPTG (Fig. 6C, 6D). The GST-SOCS3 protein was identified and verified by Western blotting (Fig. 6E). Pull-down protein assays demonstrated that SOCS3 binds to CTLA-4 in CD4 T cells (Fig. 6F).
Figure 6.

SOCS3 interacts with CTLA-4 and down-regulates its expression in CD4+ T cells. (A) Naïve CD4+ T cells were stimulated by anti-CD3/CD28 for three days and analyzed by Western blotting. (B) Freshly isolated CD4+ T cells isolated from IRBP-immunized mice were analyzed by the intracellular cytokine staining assays. The cells were gated on CD4 and numbers in quadrants indicate percent of CD4+ T cells expressing CTLA-4 and/or IFN-γ. (C–F) A GST-SOCS3 fusion protein was produced by cloning the full-length mouse SOCS3 cDNA into the pGEX6p1 expression vector and analyzed by SDS-PAGE, western blotting and/or immunoprecipitation. (C) Crude extracts (20µg) from transformed E. coli BL21 bacterial GST-SOCS3 recombinants bacteria before and after induction with IPTG were fractionated SDS-PAGE and stained with Coomassie blue dye. (D, E) Affinity purified GST-SOCS3 protein was analyzed by SDS-PAGE/Coomassie blue staining (D) or Western blotting using anti-SOCS3 Abs (E). (F) Western blot analysis following pull-down of CTLA-4 from activated CD4+ T cell extracts by GST-SOCS3. Results are representative of at least three independent experiments.
Discussion
SOCS3 is constitutively expressed in naïve T cells and following TCR activation, the SOCS3 protein is rapidly down regulated with concomitant up-regulation of IL-2 levels, leading to the suggestion that SOCS3 levels are inversely correlated with the amount of IL-2 secretion and proliferative responses of differentiating T-helper cells (11, 12). In this study, we have shown that T cells that lack SOCS3 produce less IL-2 (Fig. 2A) and exhibit a less activated phenotype (Fig. 4A) and have provided mechanistic insights into how the antagonism between SOCS3 and IL-2 contribute to the regulation of T cell activation/proliferation.
Socs3 deletion is embryonic lethal. Thus, mouse strains with tissue-specific Socs3 deletion have been generated by Cre-Lox technology as an approach to understand in vivo functions of Socs3. For example, the CreMMTV/Socs3fl/fl strain generated by mating Socs3fl/fl with mice expressing Cre under the control of the mouse mammary tumor virus (MMTV) promoter (CreMMTV), exhibited loss of >99% of Socs3 mRNA in thymocytes, lymphocytes, and activated CD4+ T cells (35). Consistent with our findings in the CD4-SOCS3KO mice, the CreMMTV/Socs3fl/fl mice had normal numbers of lymph node CD4 and CD8 cells. In vitro analysis of CD4+ T cells from the CreMMTV/Socs3fl/fl mice revealed that SOCS3 is an essential negative regulator of IL-23 signaling, inhibition of which constrained the generation of Th17 differentiation (35). However, in vivo function of Socs3 deletion was not examined. In this study, we have used the CD4-SOCS3KO mice to examine the role of SOCS3 in an organ-specific autoimmune disease.
Analysis of the CD4-SOCS3KO mice revealed that SOCS3 is essential for the regulation of physiological levels of two essential T cell co-signaling receptors, CD28 (Co-stimulatory receptor) and CTLA-4 (co-inhibitory receptor), in naïve and activated CD4+ T cells. CD28 is constitutively expressed on naive CD4+ and CD8+ T cells and following ligation by B7-1 and B7-2 on antigen-presenting cells, it provides essential co-stimulatory signals for IL-2 production, T cell proliferation and survival. However, CD28 and CTLA-4 share the highly conserved MYPPPY motif through which they competitively bind to B7-1 and B7-2 (40). Thus following T cell activation, CTLA-4 expression is up-regulated, resulting in the suppression of CD28-mediated T cell responses by outcompeting CD28 for binding to B7-1 and B7-2. Although the level of CTLA-4 is relatively low in naïve T cells, we observed substantial increase of CTLA-4 in naïve T cells of the CD4-SOCS3KO mice (Fig. 6A). We also show that the activated T cells that lack SOCS3 maintained their relatively high levels of CD28 compared to WT (Fig. 6A). These results suggest that SOCS3 may serve a gatekeeper function in T cells. On the one hand, constitutive expression of SOCS3 in naïve cells would ensure maintenance of low levels of CTLA-4, allowing the cell to be poised to receive co-stimulatory CD28 signals for IL-2 production and growth. The increase in IL-2 production that follows T cell activation induces a transient diminution of SOCS3 level as previously reported (12) and as shown in this study, decrease in SOCS3 promotes the elevation of CTLA-4 level (Fig. 6A, 6B). It has been suggested that following TCR-mediated increase in CTLA-4 expression, CTLA-4 accumulates in the same region of the immunological synapse as CD28, physically excluding CD28 from the cSMAC and disrupting positive co-stimulatory signaling (41). Here, we provide empirical evidence that SOCS3 indeed physically interacts with CTLA-4 and might therefore regulate the steady state levels of CTLA-4. Interestingly, CD4-SOCS3KO T cells continue to express high levels of CD28 after TCR activation (Fig. 6A), consistent with reports that SOCS3 regulates CD28 levels by physically interacting with and targeting CD28 for proteosome-mediated degradation (11). It is therefore intriguing that the dynamic regulation of steady-state levels of CD28 and CTLA-4 by SOCS3 might contribute to mechanisms that initiate T cell activation and those that eventually restore the activated T cell to its resting state.
Because of the mutual antagonism between IL-2 and SOCS3 and the fact that different T cell subsets require different amounts of IL-2 for differentiation, growth or effector functions, we expected the loss of SOCS3 to confer selective advantages to some T cell subsets. Thus, the CD4-SOCS3KO T cells produced >3-folds less IL-2-expressing CD4+ T cells following TCR activation (Fig. 2A; top left panels) and this favored the expansion of IL-10-producing T regulatory cells (Fig. 4C, 5B) at the expense of IL-17-producing Th17 and IFN-γ-producing Th1 cells (Fig. 4B) during EAU. In this context it is notable that SOCS1 and SOCS3 play essential roles in T cell development, function and lineage commitment and various CD4+ T cell subsets differ in the repertoire and amount of SOCS proteins they express (10). For example all T helper cells express SOCS1 and SOCS3 proteins during their development and constitutive expression of SOCS1 or SOCS3 promotes commitment and stability of Th1 and Th2 lineage, respectively (10). In contrast, Tregs do not express SOCS3 protein and in vitro over-expression of SOCS3 in Treg cells decreased their proliferation, Foxp3 expression and suppressive function (42). It is also of note that targeted degradation of SOCS1 is required to maintain competitive fitness of Tregs, underscoring the importance of stringent regulation of the levels of SOCS1 and SOCS3 proteins in Tregs (43, 44).
EAU is a CNS autoimmune disease that shares essential features with EAE and these diseases serve as models of human uveitis and multiple sclerosis (24, 45). Studies utilizing these model diseases have provided valuable insights into pathogenic processes mediated by Th1 and Th17 cells in immune privileged organs like the eye and brain (24, 45). The role of Th1 and Th17 cells in both diseases is still controversial. However, there is emerging consensus that production of both IL-17 and IFN-γ is required for pathology observed in these CNS autoimmune diseases (21, 26, 46). In EAU the earliest T cells detected in the blood or retina after disease induction are Th17 cells (21, 25). The highest level is detected at peak of disease and rapidly declines upon disease resolution. On the other hand, Th1 cells are relatively low at early stages of EAU but steadily increase and become abundant at resolution stage of the acute disease, leading to the notion that Th17 cells initiate uveitis while Th1 cells may be protective (21, 25). Of particular interest are reports showing that the Th17 subset is plastic and rapidly converts to Th1-like cells, accounting for the appearance of IL-17/IFN-γ-double producing Th17 population in several diseases (19–22). The IL-17/IFN-γ-double producing Th17 cells accumulate in the blood and retina at peak of EAU but disappear at resolution of the disease (21). Because both IFN-γ and IL-17 contribute to EAU pathology we believe that targeting this population would ameliorate uveitis. In this study we have shown that loss of SOCS3 in CD4+ or CD8+ T cells leads to dramatic reduction of the IL-17/IFN-γ-double producing Th17 population and correlates with resistance to EAU (Fig. 4B, 4D). It is also remarkable that during EAU, the main impact of the loss of SOCS3 is on the level of IL-17-expressing
Uveitis is a potentially sight-threatening idiopathic disease often characterized by repeated cycles of remission and recurrent intraocular inflammation. This makes prolonged use of the standard therapy, steroids, problematic because of their serious adverse effects including glaucoma and cataract. This concern has been the impetus for seeking alternative therapeutic approaches. CTLA-4 has been shown to limit the expansion of encephalitogenic T cells in EAE and mediates remission in relapsing EAE models (47–49). In this study, we have shown that targeting SOCS3 may be useful in modulating CTLA-4 levels and regulating Tregs and IL-17/IFN-γ-double producing Th17 population, suggesting that further understanding the role of STAT3/SOCS3 axis during intraocular inflammation may lead to development of rational therapeutics for uveitis.
Acknowledgments
Authors thank Dr. Marie Mameza (NEI, NIH) for technical assistance. We also thank Dr. Ivy Dambuza (NEI, NIH) for critical reading of the manuscript.
References
- 1.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 2.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36:515–528. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- 4.Hilton DJ. Negative regulators of cytokine signal transduction. Cell Mol Life Sci. 1999;55:1568–1577. doi: 10.1007/s000180050396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander WS. Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol. 2002;2:410–416. doi: 10.1038/nri818. [DOI] [PubMed] [Google Scholar]
- 6.Evans MK, Yu CR, Lohani A, Mahdi RM, Liu X, Trzeciak AR, Egwuagu CE. Expression of SOCS1 and SOCS3 genes is differentially regulated in breast cancer cells in response to proinflammatory cytokine and growth factor signals. Oncogene. 2007;26:1941–1948. doi: 10.1038/sj.onc.1209993. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Mameza MG, Lee YS, Eseonu CI, Yu CR, Kang Derwent JJ, Egwuagu CE. Suppressors of cytokine-signaling proteins induce insulin resistance in the retina and promote survival of retinal cells. Diabetes. 2008;57:1651–1658. doi: 10.2337/db07-1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seki Y, Inoue H, Nagata N, Hayashi K, Fukuyama S, Matsumoto K, Komine O, Hamano S, Himeno K, Inagaki-Ohara K, Cacalano N, O'Garra A, Oshida T, Saito H, Johnston JA, Yoshimura A, Kubo M. SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat Med. 2003;9:1047–1054. doi: 10.1038/nm896. [DOI] [PubMed] [Google Scholar]
- 9.Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab. 2006;17:365–371. doi: 10.1016/j.tem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Egwuagu CE, Yu CR, Zhang M, Mahdi RM, Kim SJ, Gery I. Suppressors of cytokine signaling proteins are differentially expressed in Th1 and Th2 cells: implications for Th cell lineage commitment and maintenance. J Immunol. 2002;168:3181–3187. doi: 10.4049/jimmunol.168.7.3181. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto A, Seki Y, Watanabe R, Hayashi K, Johnston JA, Harada Y, Abe R, Yoshimura A, Kubo M. A role of suppressor of cytokine signaling 3 (SOCS3/CIS3/SSI3) in CD28-mediated interleukin 2 production. J Exp Med. 2003;197:425–436. doi: 10.1084/jem.20020939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu CR, Mahdi RM, Ebong S, Vistica BP, Gery I, Egwuagu CE. Suppressor of cytokine signaling 3 regulates proliferation and activation of T-helper cells. J Biol Chem. 2003;278:29752–29759. doi: 10.1074/jbc.M300489200. [DOI] [PubMed] [Google Scholar]
- 13.Owaki T, Asakawa M, Kamiya S, Takeda K, Fukai F, Mizuguchi J, Yoshimoto T. IL-27 suppresses CD28-mediated [correction of medicated] IL-2 production through suppressor of cytokine signaling 3. Journal of immunology (Baltimore, Md. 2006;176:2773–2780. doi: 10.4049/jimmunol.176.5.2773. [DOI] [PubMed] [Google Scholar]
- 14.Banerjee A, Banks AS, Nawijn MC, Chen XP, Rothman PB. Cutting edge: Suppressor of cytokine signaling 3 inhibits activation of NFATp. J Immunol. 2002;168:4277–4281. doi: 10.4049/jimmunol.168.9.4277. [DOI] [PubMed] [Google Scholar]
- 15.Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, Reynolds SL, Yanagisawa LL, Fox TH, 3rd, Park K, Harrington LE, Raman C, Benveniste EN. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A. 2012;109:5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, Saris CJ, O'Shea JJ, Hennighausen L, Ernst M, Hunter CA. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 17.Smeets RL, Veenbergen S, Arntz OJ, Bennink MB, Joosten LA, van den Berg WB, van de Loo FA. A novel role for suppressor of cytokine signaling 3 in cartilage destruction via induction of chondrocyte desensitization toward insulin-like growth factor. Arthritis Rheum. 2006;54:1518–1528. doi: 10.1002/art.21752. [DOI] [PubMed] [Google Scholar]
- 18.van de Loo FA, Veenbergen S, van den Brand B, Bennink MB, Blaney-Davidson E, Arntz OJ, van Beuningen HM, van der Kraan PM, van den Berg WB. Enhanced suppressor of cytokine signaling 3 in arthritic cartilage dysregulates human chondrocyte function. Arthritis Rheum. 2012;64:3313–3323. doi: 10.1002/art.34529. [DOI] [PubMed] [Google Scholar]
- 19.Satoh T, Tajima M, Wakita D, Kitamura H, Nishimura T. The development of IL-17/IFN-gamma-double producing CTLs from Tc17 cells is driven by epigenetic suppression of Socs3 gene promoter. Eur J Immunol. 2012;42:2329–2342. doi: 10.1002/eji.201142240. [DOI] [PubMed] [Google Scholar]
- 20.Kuang DM, Peng C, Zhao Q, Wu Y, Zhu LY, Wang J, Yin XY, Li L, Zheng L. Tumor-activated monocytes promote expansion of IL-17-producing CD8+ T cells in hepatocellular carcinoma patients. J Immunol. 2011;185:1544–1549. doi: 10.4049/jimmunol.0904094. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Lee YS, Yu CR, Egwuagu CE. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol. 2008;180:6070–6076. doi: 10.4049/jimmunol.180.9.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tajima M, Wakita D, Satoh T, Kitamura H, Nishimura T. IL-17/IFN-gamma double producing CD8+ T (Tc17/IFN-gamma) cells: a novel cytotoxic T-cell subset converted from Tc17 cells by IL-12. Int Immunol. 2011;23:751–759. doi: 10.1093/intimm/dxr086. [DOI] [PubMed] [Google Scholar]
- 23.Nussenblatt RB. The natural history of uveitis. Int Ophthalmol. 1990;14:303–308. doi: 10.1007/BF00163549. [DOI] [PubMed] [Google Scholar]
- 24.Caspi RR. A look at autoimmunity and inflammation in the eye. J Clin Invest. 2010;120:3073–3083. doi: 10.1172/JCI42440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. T(H)17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nature medicine. 2007;13:711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- 26.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan CC, Caspi RR. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205(4):799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takase H, Yu CR, Liu X, Fujimoto C, Gery I, Egwuagu CE. Induction of suppressors of cytokine signaling (SOCS) in the retina during experimental autoimmune uveitis (EAU): potential neuroprotective role of SOCS proteins. Journal of neuroimmunology. 2005;168:118–127. doi: 10.1016/j.jneuroim.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 28.Oh HM, Yu CR, Lee Y, Chan CC, Maminishkis A, Egwuagu CE. Autoreactive Memory CD4+ T Lymphocytes That Mediate Chronic Uveitis Reside in the Bone Marrow through STAT3-Dependent Mechanisms. J Immunol. 2011;187(6):3338–3346. doi: 10.4049/jimmunol.1004019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu CR, Mahdi RR, Oh HM, Amadi-Obi A, Levy-Clarke G, Burton J, Eseonu A, Lee Y, Chan CC, Egwuagu CE. Suppressor Of Cytokine Signaling-1 Inhibits Lymphocyte Recruitment into the Retina and Protects SOCS1 Transgenic Rats and Mice from Ocular Inflammation. Invest Ophthalmol Vis Sci. 2011;52(9):6978–6986. doi: 10.1167/iovs.11-7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu H, Koch P, Chen M, Lau A, Reid DM, Forrester JV. A clinical grading system for retinal inflammation in the chronic model of experimental autoimmune uveoretinitis using digital fundus images. Exp Eye Res. 2008;87:319–326. doi: 10.1016/j.exer.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 31.Yu CR, Oh HM, Golestaneh N, Amadi-Obi A, Lee YS, Eseonu A, Mahdi RM, Egwuagu CE. Persistence of IL-2 expressing Th17 cells in healthy humans and experimental autoimmune uveitis. Eur J Immunol. 2011;41:3495–3505. doi: 10.1002/eji.201141654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee YS, Amadi-Obi A, Yu CR, Egwuagu CE. Retinal cells suppress intraocular inflammation (uveitis) through production of interleukin-27 and interleukin-10. Immunology. 2011;132:492–502. doi: 10.1111/j.1365-2567.2010.03379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellmeier W, Sawada S, Littman DR. The regulation of CD4 and CD8 coreceptor gene expression during T cell development. Annu Rev Immunol. 1999;17:523–554. doi: 10.1146/annurev.immunol.17.1.523. [DOI] [PubMed] [Google Scholar]
- 34.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'Shea JJ. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh HM, Yu CR, Lee Y, Chan CC, Maminishkis A, Egwuagu CE. Autoreactive memory CD4+ T lymphocytes that mediate chronic uveitis reside in the bone marrow through STAT3-dependent mechanisms. J Immunol. 2011;187:3338–3346. doi: 10.4049/jimmunol.1004019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Copland DA, Wertheim MS, Armitage WJ, Nicholson LB, Raveney BJ, Dick AD. The clinical time-course of experimental autoimmune uveoretinitis using topical endoscopic fundal imaging with histologic and cellular infiltrate correlation. Invest Ophthalmol Vis Sci. 2008;49:5458–5465. doi: 10.1167/iovs.08-2348. [DOI] [PubMed] [Google Scholar]
- 38.Chu CJ, Herrmann P, Carvalho LS, Liyanage SE, Bainbridge JW, Ali RR, Dick AD, Luhmann UF. Assessment and in vivo scoring of murine experimental autoimmune uveoretinitis using optical coherence tomography. PLoS One. 2013;8:e63002. doi: 10.1371/journal.pone.0063002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- 40.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, Yagita H, Tokunaga M, Saito T. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity. 2010;33:326–339. doi: 10.1016/j.immuni.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 42.Pillemer BB, Xu H, Oriss TB, Qi Z, Ray A. Deficient SOCS3 expression in CD4+CD25+FoxP3+ regulatory T cells and SOCS3-mediated suppression of Treg function. Eur J Immunol. 2007;37:2082–2089. doi: 10.1002/eji.200737193. [DOI] [PubMed] [Google Scholar]
- 43.Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–929. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, Loeb GB, Lee H, Yoshimura A, Rajewsky K, Rudensky AY. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat Rev Immunol. 2009;9:440–447. doi: 10.1038/nri2548. [DOI] [PubMed] [Google Scholar]
- 46.Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248:205–215. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hurwitz AA, Sullivan TJ, Sobel RA, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) limits the expansion of encephalitogenic T cells in experimental autoimmune encephalomyelitis (EAE)-resistant BALB/c mice. Proc Natl Acad Sci U S A. 2002;99:3013–3017. doi: 10.1073/pnas.042684699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuhns MS, Epshteyn V, Sobel RA, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates the size, reactivity, and function of a primed pool of CD4+ T cells. Proc Natl Acad Sci U S A. 2000;97:12711–12716. doi: 10.1073/pnas.220423597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karandikar NJ, Eagar TN, Vanderlugt CL, Bluestone JA, Miller SD. CTLA-4 downregulates epitope spreading and mediates remission in relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 2000;109:173–180. doi: 10.1016/s0165-5728(00)00322-2. [DOI] [PubMed] [Google Scholar]