

Abstract
Human amylin-derived oligomers and aggregates are believed to play an important role in the pathogenesis of type II diabetes mellitus (T2DM). In addition to amylin-evoked cell attrition, T2DM is often accompanied by elevated serum copper levels. Although previous studies have shown that human amylin, in the course of its aggregation, produces hydrogen peroxide (H2O2) in solution, and that this process is exacerbated in the presence of copper(II) ions (Cu2+), very little is known about the mechanism of interaction between Cu2+ and amylin in pancreatic β-cells, including its pathological significance. Hence, in this study we investigated the mechanism by which Cu2+ and human amylin catalyze formation of reactive oxygen species (ROS) in cells and in vitro, and examined the modulatory effect of Cu2+ on amylin aggregation and toxicity in pancreatic rat insulinoma (RIN-m5F) β-cells. Our results indicate that Cu2+ interacts with human and rat amylin to form metalo-peptide complexes with low aggregative and oxidative properties. Human and non-amyloidogenic rat amylin produced minute (nM) amounts of H2O2, the accumulation of which was slightly enhanced in the presence of Cu2+. In a marked contrast to human and rat amylin, and in the presence of the reducing agents glutathione and ascorbate, Cu2+ produced μM concentrations of H2O2 surpassing the amylin effect by several fold. The current study shows that human and rat amylin not only produce but also quench H2O2, and that human but not rat amylin significantly decreases the amount of H2O2 in solution produced by Cu2+ and glutathione. Similarly, human amylin was found to also decrease hydroxyl radical formation elicited by Cu2+ and glutathione. Furthermore, Cu2+ mitigated the toxic effect of human amylin by inhibiting activation of pro-apoptotic caspase-3 and stress-kinase signaling pathways in rat pancreatic insulinoma cells in part by stabilizing human amylin in its native conformational state. This sacrificial quenching of metal-catalyzed ROS by human amylin and copper’s anti-aggregative and anti-apoptotic properties suggest a novel and protective role for the copper–amylin complex.
Introduction
Diabetes is a complex heterologous disease that affects over 8% of the US human population. Adult onset or type II diabetes mellitus (T2DM) is the most common form of diabetes accounting for more than 90% of all diabetic cases. It is primarily characterized by insulin resistance, a loss of pancreatic β-cell mass and secretory function along with the formation of islet amyloid.1 Islet amyloid polypeptide (IAPP) or amylin is a 37-amino acid peptide hormone that is co-expressed and co-secreted with insulin from pancreatic islet β-cells in both diabetic and non-affected individuals. Two of the well-characterized physiological functions of amylin include delaying gastric emptying, thereby suppressing appetite, and inhibiting the appearance of glucose in the blood, hence acting as a synergistic partner to insulin. It has also been suggested that amylin has a direct paracrine effect on β-cells by inhibiting insulin secretion.1 Under normal physiological conditions, human amylin exists as a soluble monomer. However, in T2DM, human amylin undergoes a conformational change from normally soluble monomers to oligomers and finally to insoluble fibrils that are deposited in pancreatic islets.2 That islet amyloid evokes degeneration of β-cells has been a well-documented phenomenon and led to the amyloid hypothesis: that extracellular amyloid fibril deposits are toxic and induce β-cell death.1–4 Although it has been assumed that the association of extracellular amylin fibrils with cell death implied causality, a growing body of evidence has recently emerged supporting the toxic oligomer hypothesis: that small membrane-permeant human amylin oligomers, rather than large amylin fibrils, are the toxic form of amylin that cause beta-cell death.1,5 Current studies performed in primates strongly support the concept that developing islet amyloidosis and β-cell apoptosis are two key determinants of islet dysfunction.6,7 There is a consensus in the field that activation of stress-activated kinases and induction of free radicals by human amylin oligomers and aggregates represent major pathways in the programmed cell death or apoptosis of pancreatic islet β-cells.8–11 However, amylin-evoked membrane destabilization and cation channel formation in cell membranes are also considered to play an important role in amylin toxicity in the β-cells.12,13
In addition to the aforementioned hallmarks of T2DM, elevated serum Cu2+ levels are also commonly associated with the disease.14–16 While Cu2+ plays an essential role in human physiology, it has been linked to the progression of several diseases, including Alzheimer’s and Wilson’s disease.17,18 Cu2+ has been found to interact with a host of proteins and enzymes, and is required for normal cellular functioning.19 However, uncomplexed Cu2+ is potentially hazardous, particularly if it is exposed to reducing environment such as the cytosol. Under reducing cell-free conditions, Cu2+ is readily converted to the potent and reactive Cu+ ion that has the ability to produce tissue damaging ROS such as hydrogen peroxide (H2O2). Many plaque-forming proteins including the brain β-amyloid and prions are found to interact and affect the copper redox cycle leading to diminished or enhanced H2O2 production in vitro (in cell-free environment).19 Electrochemical studies revealed that amyloid proteins such as α-synuclein and β-amyloid form a redox active complex with Cu2+ that can be oxidized with molecular oxygen. The redox potentials for α-synuclein (E0 = 0.018 V vs. Ag/AgCl) and β-amyloid (E0 = 0.295 V vs. NHE) Cu2+ complexes are sufficient enough to allow reduction of Cu2+ by reducing agents such as ascorbate, leading to H2O2 production.20–22 Given the relatively low affinities of these amyloid peptides towards Cu2+ (Kd ~ 1–10 × 10−6 M),19,23 as compared to other Cu2+-binding proteins such as human serum albumin (Kd ~ 10−17 M),24 it is still uncertain if amyloids complexed with Cu2+ may alter redox homeostasis in cells. For example, studies show that β-amyloid and other brain amyloids promote oxidative stress in some cells by catalyzing redox cycling of Cu2+.19,25–27 However, this view is challenged by findings suggesting that binding and sequestration of Cu2+ by β-amyloid actually protect nerve cells from Cu2+-induced oxidative stress.19,28–31 In T2DM, while it has clearly been found that serum concentrations of Cu2+ are elevated,14,15 copper’s potential role in the pathogenesis of the disease has only recently been investigated and still remains to be clarified. Furthermore, to our knowledge, there has yet to be an investigation of a possible links between human amylin and Cu2+ interactions in the progression of T2DM.
The very few studies on the significance of Cu2+ and amylin interactions have focused on the formation of H2O2 by human amylin in vitro, the production of which is modestly elevated in the presence of Cu2+.32,33 H2O2 can elicit oxidative stress, an imbalance between the generation of ROS and cells’ antioxidant defense capacities provided by glutathione and other reducing molecules. Oxidative stress is associated with a number of diseases, including cancer, heart diseases, Alzheimer’s disease, and T2DM. At high concentrations, ROS can cause significant damage to cell structures, nucleic acids, lipids, and proteins, leading to apoptosis.34 As noted above, Cu2+-dependent generation of H2O2 was found to directly contribute to the toxicity of the β-amyloid peptide in primary neural cell cultures.35 There are strikingly similar pathological parallels between Alzheimer’s disease and T2DM such as oligomerization/aggregation of β-amyloid and human amylin that induce oxidative stress in neurons and β-cells, and the elevated tissue copper levels found in the brain and pancreas of the subjects harboring amyloid plaques.1,2,14,15,25–27,36 Thus, we hypothesized that Cu2+ and human amylin act in a synergistic manner to produce ROS and induce oxidative stress in β-cells.
In order to test our hypothesis, we addressed two important biophysical and pathological questions concerning the aggregation of human amylin and the generation of ROS, particularly H2O2: (1) Does the fibrilization process itself cause H2O2 formation and accumulation in vitro? (2) Is there a causal relationship between the Cu2+ human amylin-catalyzed generation of ROS and the extent of oxidative stress and apoptosis in pancreatic cells evoked by these two diabetic factors? Unexpectedly, and in contrast to its pro-oxidative activity in vitro,32 we report here that Cu2+ protects pancreatic cells from amylin’s toxicity in part through reduction of amylin aggregation, by increasing the activation energy needed (Ea) for amylin aggregation, and by inhibiting amylin-induced stress-kinase activation and signaling. Interestingly, the interaction of Cu2+ with amyloidogenic human but not with rat amylin attenuates Cu2+/glutathione-catalyzed H2O2 production. We also found that, at its pathophysiologically-relevant concentrations, Cu2+ does not promote human amylin-evoked oxidative stress in pancreatic cells. In line with these findings, the current study demonstrates no causality between human amylin-evoked H2O2 production and peptide’s fibrilization suggesting that amylin misfolding and aggregation is not a cause of rat and human amylin-generated H2O2 in solution.
Materials and methods
RINm5F cell culture
The insulin-secreting RINm5F pancreatic beta insulinoma cell line, which was derived from a rat islet cell tumor, was obtained from ATCC (Manassas, VA) and cultured in RPMI-1640 medium (ATCC, Manassas, VA) and supplemented with 10% fetal bovine serum and 1% pen-strep. Cells were maintained at 37 °C in a humidified incubator containing 5% CO2 in air and passaged bi-weekly.
Preparation of amylin
Lyophilized C-terminal amidated synthetic human (hA) and rat amylin (rA) (American Peptide, Sunnyvale, CA) was used for preparation of amylin stock solutions using hexafluoro-2-isopropanol (HFIP) as a solvent. The measured amounts of hA and rA were solubilized in HFIP overnight to completely dissolve the amylin. This approach efficiently removes any preformed human amylin aggregates.37,38 Prior to an experiment, the HFIP solvent was evaporated with a gentle stream of nitrogen, and the peptide was then reconstituted in reaction buffer to yield final monomer concentration of 20 μM. Except for binding studies, 20 μM human and rat amylin were used in all experiments for consistency.
MTT reduction assay
The reduction of (3-(4,5-dimethylthiazol-2-yl))-2,5-diphenyl-tetrazolium bromide (MTT) was used to assess mitochondrial metabolic activity and cell viability.39 Following the treatments, an MTT stock of 5 mg mL−1 was added to each culture being assayed equal to one tenth of the original culture volume for 3 hours followed by addition of two volumes of DMSO containing 0.04 M HCl. Colorimetric measurements of viable cell number were made at 570 nm against a background measurement of 660 nm with a Spectra Max M5 spectrofluorometer.
Reactive oxygen species assay
Intracellular reactive oxygen species (ROS) were detected using ROS-sensitive probe CM-H2DCFDA from Invitrogen.40 Cells were treated with various reagents in 96 well plates at 37 °C in RPMI-1640 phenol red-free medium. Cells were washed with 1× PBS and loaded with CM-H2DCFDA dye (10 μM in PBS) for 30 minutes in dark at 37 °C. Cells were then washed again and allowed to recover for 30 minutes. Fluorescence intensity was measured using Spectra Max M5 spectrofluorometer with excitation and emission filter set at 485 nm and 535 nm, respectively.
Copper reduction assay
A colorimetric bicinchoninic (BCA) protein detection kit (Pierce) was used to detect the reduction of Cu2+ to Cu1+ by human and rat amylin in an alkaline medium (modified biuret reaction) and in neutral pH. The assay measures reduction of Cu2+ to Cu1+ by protein or a peptide in an alkaline medium with the highly sensitive and selective colorimetric detection of the cuprous cation (Cu1+) by bicinchoninic acid.41 Thus, human or rat amylin (20 μM) was incubated with the BCA reagent at pH 7 or 11 for 60 min, and changes in absorbance were measured at 562 nm using the Spectramax M5 spectrofluorometer, Molecular Devices (Sunnyvale, CA).
Terephthalic acid assay for hydroxyl radical detection
Formation of hydroxyl ion radicals by various combinations of copper, human/rat amylin and/or reducing agents was detected using the hydroxyl radical probe, terephthalic acid (TPA) from Sigma.42 The reducing agents were mixed with 5 mM TPA in 0.2 M phosphate buffer (pH 7) and the reaction mixtures were incubated at room temperature for 60 minutes. Total fluorescent intensity was measured at an excitation of 326 nm and emission of 432 nm using the Spectramax M5 spectrofluorometer, Molecular Devices (Sunnyvale, CA). The fluorescence intensities of TPA are proportional to the amount of hydroxyl radicals generated in the system.42
Amplex UltraRed assay for H2O2 detection
H2O2 production by human and rat amylin, in the presence or absence of reducing agents and copper, was investigated using the Amplex UltraRed hydrogen peroxide detection assay from Invitrogen. A working solution of Amplex UltraRed/HRP was made fresh using final concentrations of 100 μM Amplex UltraRed reagent and 0.2 U mL−1 HRP in 0.01 M PBS buffer (pH 7). Standard curves were obtained using H2O2 concentrations ranging from 100 nM to 20 μM. Following treatment, an equal reaction volume of Amplex UltraRed/HRP was mixed with each sample and incubated for 20 minutes at room temperature. Total fluorescent intensity was measured at an excitation of 530 nm and emission of 590 nm in the Spectra Maxm5 spectrofluorometer, Molecular Devices (Sunnyvale, CA). Buffer (blank) was subtracted from treatments, and the fluorescence intensities obtained were converted to the peroxide concentrations using the standard curve.
Circular dichroism (CD) spectroscopy
CD spectra were recorded on a Jasco J-815 Spectropolarimeter (JASCO Co., Japan) at a temperature of 25 ± 0.2 °C maintained by a Peltier thermostat. Far-UV CD measurements were performed between 190 and 260 nm in a rectangular quartz cuvette of 2 mm pathlength, using a scan speed of 50 nm min−1, bandwidth of 1.0 nm and resolution of 0.2 nm. The initial concentration of hA and rA in the samples was 20 μM in PBS pH 7.4, either without Cu2+ (control) or with equimolar amount of Cu2+. Each spectrum represents an average of five accumulated scans. The relevant baseline was subtracted by running PBS alone or PBS containing Cu2+ as a blank. The ellipticity of the CD spectra was expressed in millidegrees (mdeg). The time-course spectral measurements were performed at 2 min (initial), and 1, 2, 4, 24 and 48 hours maintaining the same experimental parameters.
Western blot analysis
The effects of Cu2+ and human amylin on the activity of stress-kinases and apoptotic proteins were analyzed by western blot, as described previously.39 Following treatments, cells were solubilized with RIPA buffer, and 10 μg of cell extract was loaded onto 10% SDS-polyacrylamide gels. The protein bands were then transferred electrophoretically to PVDF membranes, and non-specific IgG binding sites were blocked by incubation with 5% nonfat dry milk. The membrane was probed with primary antibody, followed by horseradish peroxidase-conjugated secondary antibody. Cleaved caspase-3 (Asp 175), phospho-specific JNK (Tyr 183/Tyr185), JNK, actin and phosphor-specific c-Jun (Thr 180/Thr 182) antibodies were used. Signal was developed using the enhanced chemiluminescence detection procedure according to the manufacturer’s recommendations (Amersham, Pharmacia Biotech).
Caspase-3/7/annexin V apoptotic assay
The extent of apoptosis in pancreatic cells was investigated by a double staining protocol, using the confocal microscope as described previously.39 This approach allows simultaneous detection of caspase-3 proteolytic activity and phosphatidylserine (PS) externalization in apoptotic cells, both of which are hallmarks of apoptosis. The caspase-3 substrate, which is initially non-fluorescent, crosses the cell membrane to enter the cytoplasm, where it is cleaved by caspase-3 to release a high-affinity DNA dye, which migrates to the cell nucleus and stains it with green fluorescence. Phosphatidylserine (PS) externalization during apoptosis is detected by annexinV-Texas Red, a phospholipid binding protein with a high affinity for PS. Nuclei of all cells in the well are stained with a nuclear dye DRAQ5 (Cell Signaling Tech., Danvers, MA) (blue pseudocolor). The number of apoptotic cells in the wells are scored, averaged, and expressed as % caspase-3/annexinV-positive cells per treatment, each treatment performed in triplicate.
Amylin fibril formation assay
The thioflavin-T (Th-T) assay was used to determine the formation of amylin aggregation as explained previously.37 Freshly prepared stock solutions of the human and rat amylin in the organic solvent HFIP were used to ensure the absence of proto-fibrils in incubation medium. Prior to experiments, HFIP was evaporated with a stream of nitrogen, and the final amylin concentrations were prepared in PBS containing 3 μM Th-T. Real-time changes (1 min interval) in Th-T fluorescence intensities, expressed as arbitrary units (au), were recorded using Spectramax M5 spectrofluorometer, Molecular Devices (Sunnyvale, CA) with the excitation wavelength set at 280 nm and the emission recorded at 303 nm. An increase in emission maximum at 482 nm was obtained for human but not rat amylin, thus confirming that increase in Th-T fluorescence is due to peptide aggregation. In order to avoid any possible secondary effect of Th-T on amylin aggregation, we also verified amylin fibrilization using the end-point approach: following incubation, aliquots from each sample were withdrawn and then mixed with Th-T reagent to detect the aggregated species. Data were collected, plotted and compared using the GraphPad Prism 5 graphic program. All experiments were performed at room temperature. We followed our published approach to determine the kinetics of amylin aggregation in solution.37,38 Briefly, the resultant time courses of amylin aggregation, in the presence or absence of Cu2+, were fitted to first-order kinetic curves of the form:
| (1) |
where Imax is the maximum fluorescence intensity and It is fluorescence intensity at time t. Data were plotted and the rate and order of reaction determined using linear regression analysis.
Determination of dissociation constants
In order to determine the binding affinities between Cu2+ and human or rat amylin peptides, we adopted a published fluorescence-based approach used for determination of apparent dissociation constant (Kd) for β-amyloid–Cu2+ complex as a measure of complex binding strength.23 The assay is based on the inverse relationship between tyrosine (Tyr) fluorescence and the amount of Cu2+ bound to the peptide. Following the binding of Cu2+ to β-amyloid there is a decrease in Tyr fluorescence, quenching of which is used to construct a binding curve.23 As both human and amylin amylin isoforms possess Tyr, we investigated their interactions with Cu2+ by measuring changes in Tyr fluorescence using the Spectramax M5 spectro-fluorometer, Molecular Devices (Sunnyvale, CA) set at ex. 280 nm and em. 303 nm. A lower 10 μM concentration of human amylin in binding studies was used to avoid any potential secondary effect of amylin aggregation on Cu2+ binding to the peptide. At this concentration, human amylin retains its soluble, random-coil conformation.37,38 Following dissolution, 10 μM human or rat amylin were incubated for 1 h with increasing concentrations of CuCl2 (0–100 μM) and changes in the Tyr fluorescence were recorded. Data were plotted and analyzed using the GraphPad Prism software program. Nonlinear regression analysis and least squares fitting method were used to plot the binding curves. The mean Kd and standard errors of mean (SEM) were calculated using 3 random points from the fits and the following standard eqn (2):
| (2) |
where [A] and [B] are free (unreacted) Cu2+ and amylin concentrations respectively, and [AB] is concentration of the complex in the solution. The concentration of amylin–Cu2+ complex was determined using eqn (3):
| (3) |
where Fc is the fluorescence intensity of the peptide measured at particular CuCl2 concentration [Bc], Fmax is fluorescence value obtained in the absence of Cu2+, Fmin is the value at which no further quenching occurs (in which peptide is saturated with CuCl2), ΔFc is the difference between Fmax and Fc, and [AT] is total (10 μM) concentration of the peptide used in the binding studies. Free (unreacted) amylin [A] and Cu2+ [B] concentrations were calculated using eqn (4) and (5):
| (4) |
| (5) |
Statistical analysis
The GraphPad Prism 5 Program was used for plotting of graphs and statistical analysis. Results are expressed as mean ± standard error of mean (SEM) of three or more independent experiments. The unpaired Student’s t test or one-way ANOVA followed by Newman–Keul post hoc test were used for pairwise comparisons among groups when appropriate with significance established at P < 0.05.
Results
Amylin produces and quenches H2O2 in vitro
It has been recently reported that human amylin in the course of its aggregation produces small (nM) concentrations of H2O2.32 Although a link between amylin aggregation and amylin-induced H2O2 production has been postulated by a previous study, the causality between the two processes remains enigmatic. Here, we examined the effect of amylin’s conformational changes and aggregation on H2O2 production, and vice versa. Our results are consistent with a previous study showing that human amylin indeed catalyzed formation and prolonged accumulation of H2O2 in solution (Fig. 1A). Interestingly, we were able to detect comparable levels (0.1–0.6 μM) of H2O2 in solution using amylin concentrations that are 5–10 fold lower than those previously used (100–200 μM).32,33 We also observed distinct kinetics for amylin aggregation and amylin-induced H2O2 production (Fig. S1, ESI‡). While amylin induced linear increase of H2O2 in solution over time (Fig. S1A, ESI‡), amylin aggregated with lag phase followed by accelerating fibrilization phase, yielding a sigmoidal curve typical for nucleation-dependent aggregation processes (Fig. S1B, ESI‡). Kinetic traces (Fig. S1, ESI‡) show that H2O2 formation precedes the onset of amylin aggregation by ~ 2 h, indicating that human amylin aggregation and amylin-evoked H2O2 production are two separate processes and possibly independent of each other. Also, non-amyloidogenic rat amylin was equal to human amylin in catalyzing H2O2 production and accumulation in solution (Fig. 1A). Addition of catalase quenched H2O2 production by human amylin without having any significant effect on the extent of its aggregation (Fig. 1A and B). Thus, amylin-catalyzed H2O2 formation did not influence its aggregation. Consistent with this finding, exogenously supplemented H2O2 failed to significantly modulate the fibrilization process (Fig. 2A). As expected, non-amyloidogenic rat amylin, bearing three proline residues that break β-sheets, did not aggregate in solution (Fig. 2A). In contrast, both peptides significantly reduced externally added H2O2 (Fig. 2B), analogous to the ROS quenching effect of some amyloid proteins. For example, oxidation of methionine residues in β-amyloid–Cu2+ complex to sulfoxide and sulfone groups by H2O2 and OH• radical has been demonstrated.43 The quenching effect of rat and human amylin was saturable (Fig. 2C) and somewhat larger (0–1.5 μM, Fig. 2C) as compared to the amount of H2O2 produced by these two peptides (0.1–0.6 μM, Fig. 1A).32 Therefore, our results suggest that the small amounts of H2O2 detected in solution as a result of two opposing reactions: H2O2 production and H2O2 quenching by the two amyloid peptides, and that reaction equilibrium shifts toward quenching upon excess of externally added H2O2. Taken together, these results indicate that human amylin aggregation and amylin-evoked H2O2 production are two separate and unrelated processes, and that an intrinsic ability of amylin peptides to produce small amounts of H2O2 is paralleled by their ability to quench ROS. This redox buffering activity of human amylin was also observed in the presence of Cu2+ and the cells’ main reducing agent glutathione, as demonstrated below.
Fig. 1.

Aggregation of human amylin is not affected by catalytically produced H2O2. 20 μM of human amylin (hA) and rat amylin (rA) were incubated in phosphate buffer, in the absence or in presence of catalase, for 24–72 h and the extent of H2O2 accumulation (A) and amylin-aggregation (B) quantified using Amplex-Red H2O2 and Th-T-aggregation fluorescent assays, respectively. (A) Stimulatory effect of human and rat amylin on accumulation of peroxide in solution is shown. (B) Catalase had no significant effect on the aggregation of human amylin (P > 0.05, n = 6). Significance established at ***P < 0.001, hA vs. hA + catalase, n = 6, ANOVA followed by Newman–Keul post hoc test.
Fig. 2.

Both amylin isoforms produce and quench H2O2 in vitro. Human and rat amylin were incubated for 24 h in the absence or in the presence of increasing concentrations of H2O2 (1–20 μM). The extent of amylin-aggregation (A) and H2O2 accumulation in solution (B) were quantified using Th-T and Amplex-Red fluorescent assays, respectively. (A) Addition of H2O2 to solutions containing human amylin had no significant effect (P > 0.5, n = 6) on its aggregation. (B and C) Human and rat amylin quenched exogenously added H2O2 in a dose-dependent and saturable manner. Significance established at *P <0.05 and **P <0.01, control vs. hA or rA, n = 6, ANOVA followed by Newman–Keul post hoc test.
Cu2+ inversely modulates amylin-evoked H2O2 production and amylin aggregation in solution
If amylin aggregation catalyzes H2O2 formation then changes in the rate and/or extent of amylin fibrilization, should have a corresponding effect on H2O2 generation. Hence, we tested the modulatory effect of Cu2+ on amylin aggregation and production using the standard Th-T aggregation (Fig. 3A and B, Fig. S2, ESI‡) and Amplex-UltraRed H2O2 detection assays (Fig. S4, ESI‡), respectively. Human amylin aggregated at a slow rate following a first order kinetic law with k = 1.4 × 10−2 min−1, exhibiting a typical sigmoidal curve (Fig. 3A and B). The presence of Cu2+ slowed down the rate of amylin aggregation by ~ 3-fold (hA + Cu2+, k = 5 × 10−3 min−1, Fig. 3B), prolonged the lag phase by ~ 90 min, and decreased the extent of amylin aggregation by ~ 45% relative to control (Fig. 3A). Hence, our data is in agreement with recent reports showing an inhibitory effect of Cu2+ on amylin aggregation.44 To avoid a possible secondary effect of Th-T on amylin aggregation, we also analyzed amylin aggregation using end-point Th-T assay (see Method section). Supplementation of equimolar Cu2+ blocked amylin aggregation (Fig. S2, ESI‡), confirming the results obtained using standard Th-T approach (Fig. 3A and B). Applying the Arrhenius equation and taking into account rate constants for amylin aggregation above, we calculated that Cu2+ increases the activation energy (Ea) for the aggregation process by ΔEa = 2.55 kJ mol −1 at 25 °C. Interestingly, despite marked differences in their aggregation rates, both curves exhibit first-order kinetics (R2 > 0.9; Fig. 3B). Binding studies reveal that human amylin (Kd = 2.2 ± 0.4 × 10−6 M, Fig. 3C) and rat amylin (Kd = 11.2 ± 1.1 × 10−6 M, Fig. 3D) form complexes with Cu2+ in solution, with stoichiometry of 1:1 (mol/mol). Interestingly, human amylin shows a higher affinity for binding to Cu2+ as compared to rat amylin. Thus, the obtained Kd values explain the high (μM) concentrations of Cu2+ required to inhibit aggregation of human amylin (Fig. 3A and B; Fig. S2, ESI‡).44
Fig. 3.

Cu2+ forms a low-affinity complex with human amylin and inhibits its aggregation. (A) 20 μM human amylin and rat amylin were incubated in phosphate buffer, in the absence or presence of 20 μM CuCl2 and the extent of amylin-aggregation analyzed by Th-T fluorescent assay. (B) Kinetics of human amylin aggregation in the presence or absence of Cu2+ is shown. (C and D) Determination of amylin–Cu2+ binding constants using the quenching of the tyrosine fluorescence by CuCl2 at pH = 7.4. 10 μM of human (C) and rat amylin (D) solutions were allowed to interact with increasing Cu2+ concentrations, and their apparent Kd determined by measuring the decrease in Tyr fluorescence at 303 nm.
We previously demonstrated that acquisition of the β-sheet secondary conformation is a rate-limiting step in the aggregation of human amylin.37 It is likely that Cu2+ inhibits amylin aggregation by increasing the activation energy of the amylin phase transition from random coil to β-sheets, an idea that was further tested by aid of CD spectroscopy (Fig. 4). The far-UV CD time course measurements reveal marked differences between amylin aggregation in the absence and presence of Cu2+, and between human and rat amylin. The initial far-UV CD spectrum of hA measured immediately after hA dilution in PBS, shows a strong negative Cotton Effect (CE) at 202 nm with a low-intensity band around 223 nm (Fig. 4A, 2 min trace). This CD pattern is typical for polypeptides with a high content of random (unordered) structure and is in agreement with CD data reported earlier.45,46 Comparison of the time course of changes in CD spectra for amylin alone (Fig. 4A) and amylin in the presence of Cu2+ (Fig. 4B) supports the inhibitory effect of Cu2+ on the amylin aggregation demonstrated in our Th-T assays (Fig. 3A and B) and previously with TEM.44 In the absence of Cu2+ the intensity of the CE at 202 nm decreased within the first 4 hours, showing a slight shift to 204 nm. At 24 h, CD showed broad negative extremum around 220 nm corresponding to a β-sheet-dominated structure. On the contrary, in the presence of Cu2+, not only were the dynamics of decrease in intensity of the CE at 202 nm slower than with amylin alone, but the pattern of the CD spectra time course remained similar to the initial spectrum with the main CE around 202 nm, that never turned to β-sheet-rich structure (Fig. 4B). For clarity, Fig. 4C shows the CD spectra taken at the 4 h time point for 20 μM amylin alone and in the presence of equimolar amount of Cu2+ overlaid to visualize the differences in the CD patterns and intensities. It is noteworthy that the remaining intensity of the CD band at 202 nm for amylin alone is only ~ 35.9%, whereas it is about 80% for the amylin sample containing Cu2+ (Fig. 4C). Further monitoring of amylin alone and Cu2+-containing amylin up to 48 h (Fig. 4D) did not reveal any significant change in comparison with the spectra taken at 24 h, indicating a long-lasting inhibitory effect of Cu2+ on amylin aggregation. Although the intensities of the CD bands at 220 nm (amylin alone) and 201 nm (amylin in the presence of Cu2+) further decreased, there was no change in the patterns typical for each amylin solution. Therefore, in the presence of Cu2+, not only is the amylin amyloid formation retarded in comparison with that of amylin alone, but it is likely to proceed via different route. As for rat amylin, the unchanged time course of the CD spectra with a negative extremum centered near 200 nm are indicative of absence of conformational changes (Fig. S3, ESI‡), consistent with our previously published microscopy data.37,38
Fig. 4.

Cu2+ inhibits human amylin secondary conformational transitions in solution. (A–D) Comparative far-UV CD data for the conformational changes of human amylin (20 μM) in PBS alone or in the presence of equimolar amount of Cu2+. In the presence of Cu2+, human amylin conformational transitions to a β-sheet-rich structure are blocked.
Although Cu2+ abrogated amylin aggregation (Fig. 3 and 4), supplementation with Cu2+ had an opposite, very small and non-additive stimulatory effect on human and rat amylin-evoked H2O2 formation in solution (Fig. S4, ESI‡), indicative of formation of a mildly redox-active metalo-peptide complex. Consistent with this finding, the Cu+-sensitive BCA-based colorimetric assay (Fig. S5, ESI‡) reveals that at a physiologically relevant pH value (pH = 7) neither human nor rat amylin readily reduce Cu2+ to Cu+, a required and preceding step in Cu2+-mediated H2O2 and hydroxyl radical ion production.47,48 These results further support the conclusion that amylin aggregation is not a requirement for amylin-evoked H2O2 production (Fig. 1 and 2, Fig. S1, ESI‡).
Distinct effects of human and rat amylin on Cu2+-catalyzed ROS accumulation in vitro
We also tested modulatory effects of human and rat amylin on Cu2+-evoked H2O2 and hydroxyl radical production in solutions containing the reducing agents glutathione and ascorbate (Fig. 5). In contrast to the negligible stimulatory effect of Cu2+ on amylin-evoked H2O2 production (Fig. S4, ESI‡), interactions of Cu2+ with glutathione and ascorbate produced more than a 5-fold increase in H2O2 in the system (Fig. 5A). Importantly, human amylin reversed this large stimulatory effect of glutathione but not of ascorbate on Cu2+-induced H2O2 production. In marked contrast to human amylin, rat amylin did not alter the production of H2O2 by Cu2+ and glutathione (Fig. 5A), demonstrating differences in redox activities of two metalo-peptide complexes. Interestingly, rat amylin significantly increased levels of H2O2 produced by Cu2+ and ascorbate (Fig. 5A), indicating that a specific combination of ascorbate, Cu2+ and non-amyloidogenic rat amylin may indeed stimulate H2O2 production and accumulation in the system. It is important to note here that human amylin-induced H2O2 production in solution was not significantly elevated under any combination of reducing agents and/or Cu2+. Given that Cu2+ binds to human amylin in a low μM range (Fig. 3C), and taking into account the current results (Fig. 5A), we propose that chelation of Cu2+ by human amylin disrupts copper’s redox cycle catalyzed by glutathione, which, in the absence of amylin, would reduce molecular oxygen and water to H2O2.47,48 It is also possible that a general feature of amyloid peptides to quench ROS (Fig. 2B and C)49,50 may also play an inhibitory role in Cu2+-catalyzed H2O2 production (Fig. 5A).
Fig. 5.

Human and rat amylin oppositely regulate ROS formation in vitro. Human and rat amylin (20 μM) were incubated in the absence or presence of 10 μM CuCl2, 10 μM or 1 mM ascorbate (ASC) and/or 5 mM glutathione (GLU), and the peroxide and hydroxyl radical formations detected using Amplex-Red and TPA fluorescent assays respectively (A and B). (A) Human amylin abrogates Cu2+/ glutathione-catalyzed H2O2 production in solution. In contrast, rat amylin potentiates Cu2+/ascorbate-induced peroxide formation. (B) Rat but not human amylin stimulates Cu2+/ascorbate-catalyzed hydroxyl production in solution. Significance established at *P < 0.05, **P < 0.01 and ***P < 0.001 n = 3–6, ANOVA followed by Newman–Keul post hoc test.
We also tested the effects of these two peptides on metal-catalyzed hydroxyl radical production (Fig. 5B). In the presence of H2O2, either added exogenously or internally produced in reaction with ascorbate, Cu2+ catalyzes hydroxyl radical production (Fig. 5B) through the well-known Haber–Weiss chemistry.47,48 As observed with H2O2 production (Fig. 5A), rat but not human amylin further increased hydroxyl radical production by Cu2+ and ascorbate. Rat and human amylin, as in case of H2O2 formation (Fig. 5A), had no significant stimulatory effect on Cu2+- or Cu2+/glutathione-induced hydroxyl radical formation (Fig. 5B). Due to a strong hydroxyl radical quenching effect of glutathione (Fig. 5B), we noticed a rather small inhibitory effect of human amylin on Cu2+/glutathione-mediated hydroxyl radical formation, as compared to its significant inhibitory effect on H2O2 formation (Fig. 5A). Thus, our results indicate that human amylin, a culprit in T2DM, produces negligible H2O2 concentrations, does not substantially reduce Cu2+, does not stimulate metal-catalyzed ROS production in vitro, and Cu2+/glutathione-induced H2O2 production is reduced significantly (Fig. 5). These results are in contrast to known pro-oxidative features of certain metal-reactive amyloid peptides such as α-synuclein.22 In that respect, human amylin shares similar (in vitro) anti-oxidative properties with other amyloid proteins such as brain β-amyloid and prions.49,50
Lack of stimulatory effect of Cu2+ on amylin-evoked oxidative stress and toxicity in pancreatic cells
Given that Cu2+ and human amylin catalyze H2O2 formation in vitro (Fig. 1A, Fig. S4, ESI‡),32 and considering the elevated levels of human amylin and Cu2+ in diabetics and animal models,15,16 we investigated the extent to which amylin, Cu2+ or their combination affect intracellular redox homeostasis in β-cells. We used a common marker of oxidative stress in cells, H2DCFDA, the fluorescence of which is proportional to the generation and accumulation of free radicals in the cytosol.40 Incubation of RIN-m5F cells with 20 μM human amylin increases H2DCFDA fluorescence by 54 ± 9% relative to controls (non-treated cells), indicative of oxidative stress (Fig. 6). Cu2+, at its pathophysiologically relevant concentrations (10 μM),15 also increases intracellular ROS significantly, matching the pro-oxidative effect of human amylin. Interestingly, co-incubation of β-cells with human amylin and Cu2+ produced the same increase in intracellular ROS levels as compared to amylin alone (Fig. 6), mirroring the slight stimulatory effect of copper on amylin-induced H2O2 formation (Fig. S4, ESI‡). Hence, this result (Fig. 6) indicates formation of human amylin–Cu2+ complexes with low pro-oxidative capacity in the cellular environment, an observation that was confirmed in vitro using other methods (Fig. S4 and S5, ESI‡). Taken together, the above results indicate that amylin–Cu2+ interactions play a rather minor role (if any) in the amylin-induced oxidative stress in β-cells, and that amylin’s pro-oxidative activity in cells (Fig. 6) is likely due its ability to disrupt mitochondrial activity and electron flux.51,52 Hence, we used the well-know MTT reduction assay (Fig. 7A and B) to confirm the effects of human amylin and Cu2+ on mitochondrial activity, which may also indicate changes in β-cell’s viability.39
Fig. 6.
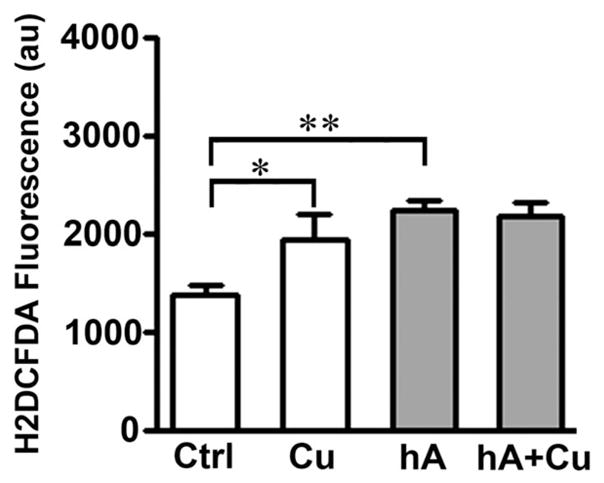
Cu2+-does not increase oxidative stress in cells evoked by human amylin. Human amylin (20 μM) was incubated with cells in the absence or presence of 10 μM CuCl2 for 24 h. Following incubations, relative ROS levels in controls and treatments were determined by measuring changes in fluorescence intensity of oxidative stress marker, H2DCFDA. Cu2+ and human amylin induced oxidative stress in cells in a non-additive manner. Significance established at *P < 0.05 and **P < 0.01, n = 6, ANOVA followed by Newman–Keul post hoc test.
Fig. 7.

Cu2+-inhibits amylin-evoked apoptosis in pancreatic cells. Human amylin (20 μM) was incubated in the absence or presence of 10 μM CuCl2 and corresponding changes on cells’ viability were determined using MTT reduction (A and B) and apoptotic western-blot (C) and microscopy (D) assays. (A) Amylin induced a marked drop in the MTT signal, indicative of metabolic stress in cells. (B) Cu2+ impaired mitochondrial enzymatic activity in a dose-dependent manner. (C) Western blot analysis reveals an inhibitory effect of Cu2+ on caspase-3 and stress kinases activity in cells treated with human amylin. (D) Confocal microscopy analysis of apoptosis in RIN-m5F cells treated with human amylin, in the absence and in the presence of Cu2+. Arrows depict non-apoptotic nuclei (blue) in viable cells, whereas arrowheads depict caspase-3 (green) and annexin-V (red) positive apoptotic cells. Bar, 10 μm. Quantitative analysis of amylin-induced apoptosis in human islets is summarized in a bar graph. Significance established at **P < 0.01, n = 6, ANOVA followed by Newman–Keul post hoc test.
Mitochondria are a major source of ROS in cells and disruption of their respiratory and metabolic functions is linked to a host of ROS-related pathologies including amyloid diseases such as Alzheimer’s disease and T2DM.52 This organelle is also a cross-road for redox-sensitive pro-apoptotic and anti-apoptotic pathways. Human amylin (20 μM) reduced mitochondrial activity by ~ 50% relative to controls (non-treated cells). However, Cu2+, at concentrations found in serum (10 μM),15 only marginally affected mitochondrial activity, evident by ~ 20% decrease in MTT signal (Fig. 7A). Consistent with other results (Fig. 6 and Fig. S4, ESI‡), a minor (~ 10%) although significant further decrease in mitochondrial activity of cells exposed to amylin and Cu2+, as compared to amylin alone, was observed (Fig. 7A). Interestingly, their inhibitory effects on MTT reduction was less than additive, suggesting that Cu2+ and human amylin impede mitochondrial function, at least in part, through a common signaling pathway (Fig. 7A). Although addition of Cu2+ up to 20 μM showed little toxicity, at concentrations greater than 50 μM, it markedly impaired mitochondrial function (Fig. 7B). The important question is whether this minor detrimental effect of Cu2+ on human amylin-induced decrease in mitochondrial activity observed in our study (Fig. 7A), and also reported by Yu and colleagues,53 bears any significance for amylin toxicity. Particularly, is an additional 10% drop in the mitochondrial metabolic activity in the presence of Cu2+ sufficient to potentiate human amylin-evoked apoptosis and loss of α-cell mass, a hallmark of T2DM? To clarify the issue, we used western blot analysis to determine the extent to which human amylin, Cu2+ and its complex modulate protein and phosphorylation levels of a key apoptotic marker (capase-3), central stress-kinases c-jun N-terminal kinase (JNK) and its downstream substrate c-jun, an apoptotic transcription factor. These three signaling molecules are activated by human amylin and are implicated in amylin-induced oxidative stress and apoptosis of islet β-cells.9–11 Consistent with its disrupting effect on mitochondrial activity (Fig. 7A), human amylin increased the levels of a cleaved (activated) caspase-3 form by 1.6-fold (n = 3) as compared to controls (Fig. 7C). Similarly, a 2-fold increase in phosphorylation levels of redox-sensitive stress kinases JNK and c-jun by human amylin, indicative of their catalytic activation, were detected in these cells (Fig. 7C). In line with the MTT data (Fig. 7A), addition of Cu2+ failed to sensitize RIN-m5F cells to the toxic effect of human amylin (Fig. 7C). Surprisingly, in the presence of Cu2+, amylin-induced stress/apoptotic signaling was completely blocked (Fig. 7C). This finding is in contrast with a recent report showing the potentiation of amylin toxicity by Cu2+.53 Hence, we used our published apoptotic caspase-3/ annexin V assay to verify the effect of Cu2+ on amylin toxic properties in our cells.39 In concordance with the biochemical data (Fig. 7C) and based on its ability to stabilize human amylin in a native (non-toxic) conformation (Fig. 4), Cu2+ significantly abrogated amylin-induced apoptosis in pancreatic cells (Fig. 7D). In the absence of Cu2+, human amylin evoked a 4-fold increase in the number of apoptotic cell deaths as compared to controls. This toxicity was completely reversed in Cu2+-containing samples (Fig. 7D). Different cell types and/or experimental conditions may account for the opposing effects of human amylin on cell viability reported in these two studies (Fig. 7).53 However, our findings are in agreement with several studies in the field showing inhibitory effect of Cu2+ on the toxicity of other amyloid proteins such as β-amyloid and prion protein (PrP).19,31,54–56 Consistent with its inability to produce toxic oligomers and aggregates,1,2 rat amylin had no significant effect on mitochondrial, caspase-3 or JNK activities in cells or on apoptosis (data not shown). Given that Cu2+–amylin complex inversely regulates mitochondrial activity and apoptotic signaling (Fig. 7), it could be inferred that Cu2+ inhibits amylin-evoked JNK and caspase-3 activation by inhibiting signals downstream of mitochondria-mediated pro-apoptotic pathway in these cells.
Disturbance of redox and Cu2+ homeostasis exacerbates amylin-induced toxicity in pancreatic insulinoma β-cells
The role of ROS and Cu2+ in amylin-induced toxicity in β-cells was further elucidated by probing intracellular redox and Cu2+ homeostasis using the antioxidant N-acetyl-cysteine (NAC) and bathocuproine, a specific chelator of Cu1+ (Fig. 8). Cells were incubated with CuCl2 and human amylin, in the presence or absence of 5 mM NAC, 50 μM bathocuproine, or combinations of NAC and bathocuproine, followed by the MTT reduction assay (Fig. 8A). For NAC treatment groups, cells were pre-incubated with NAC for 3 hours before various treatments in the continuous presence of NAC. Consistent with its well-known anti-oxidative properties, preincubation of cells with NAC completely protected β-cells from human amylin-induced cytotoxicity (Fig. 8A), as demonstrated previously.11,57 In contrast, the detrimental effect of Cu2+ on mitochondrial function was not attenuated by NAC addition. Paradoxically, NAC pre-treatment resulted in a slight increase in mitochondria dysfunction in Cu2+-treated groups, including human amylin (Fig. 8A). Copper is a redox-active metal that undergoes redox cycling between the oxidized (Cu2+) and reduced (Cu+) states.58 Although NAC is known to be an antioxidant that lowers the oxidation of biological reducing agents such as cholesterol, it can also behave as a pro-oxidant upon interaction with the oxidized forms of metal ions such as Cu2+.59 Based on this fact, we propose that the significant increase in amylin cytotoxicity observed in NAC-pretreated cells, in the presence of Cu2+ (but not in its absence) is a result of two opposing NAC actions: (1) quenching of ROS by NAC,57 and (2) production of H2O2 by NAC and Cu2+.59
Fig. 8.

Copper-chelating and antioxidant agents sensitize pancreatic cells to human amylin toxicity. The roles of redox and copper homeostasis in amylin toxicity were investigated by testing modulatory effects of anti-oxidant NAC and the specific Cu1+-chelator, bathocuproine (BAT), on human amylin-induced mitochondria dysfunction (A) and peroxide formation (B). (A) Amylin alone induced a significant drop in mitochondrial enzymatic activity, which was completely abolished in the presence of NAC. NAC shows opposite (inhibitory) effect on mitochondrial activity in the presence of Cu2+. Similarly, BAT required Cu2+ to potentiate amylin-toxicity. (B) Ascorbate (ASC) but not bathocuproine potentiates Cu2+-induced H2O2 production in solution. Significance established at **P < 0.01, and ***P < 0.001, n = 6, ANOVA followed by Newman–Keul post hoc test.
To further probe the role of Cu2+ in amylin toxicity, bathocuproine, a cell membrane-impermeant chelator of Cu+, was used in conjunction with human amylin. We reasoned that if amylin reduces Cu2+ to produce significant quantities of H2O2 in solution, analogous to α-synuclein/Cu+-catalyzed H2O2 formation in vitro,22 then the chelation of Cu+ by bathocuproine, should reduce amylin/Cu+-induced H2O2 production and attenuate the toxicity of this metalo-peptide complex. Interestingly, bathocuproine showed no significant modulatory effect on H2O2 production elicited by human and rat amylin either in the presence or absence of copper (Fig. 8B). However, in the presence of Cu2+, bathocuproine modestly increased H2O2 levels in solution by ~ 400 nM as compared to Cu2+ or bathocuproine alone, which was an order of magnitude smaller than the stimulatory effect of ascorbate (~ 3.1 μM increase in H2O2, Fig. 8B). Although bathocuproine can reduce Cu2+ to Cu1+, studies show that chelation of its reduced form by bathocuproine prevents metal re-oxidation and hence H2O2 accumulation in solution.60 Thus, in comparison with other reducing (but not chelating) agents such as ascorbate, bathocuproine is rather limited in its ability to stimulate Cu2+-induced ROS production. In contrast to its low pro-oxidative ability, bathocuproine markedly potentiated amylin toxicity, the effect of which required the presence of Cu2+ but not reducing agents such as NAC (Fig. 8A). In the absence of either amylin or copper, bathocuproine did not cause any significant change in mitochondrial activity. Similarly, bathocuproine alone had no significant effect on human amylin toxicity in these cells (Fig. 8A). Given the ability of these cells to efficiently internalize human amylin within 24 hours,39 the current results imply that, under normal situations, bathocuproine does not enter the cells and exert any significant accumulation of H2O2. However, in the presence of human amylin and copper, bathocuproine is likely to be internalized as a complex of amylin–copper–bathocuproine, which in turn would disrupt intracellular copper homeostasis, exacerbating amylin toxicity in β-cells (Fig. 8A).
Discussion
Although β-amyloid and α-synuclein interactions with Cu2+ in neuronal cells have been studied in depth,22,25–31 very little is known about amylin interactions with Cu2+ in β-cells. In the present study, we showed, for the first time, a surprisingly protective role for Cu2+ in amylin-induced cytotoxicity. The presence of Cu2+ affected amylin aggregation in two major ways: it prolonged the lag (nucleation) phase and it reduced the rate of human amylin aggregation by 3 fold (by increasing the activation energy, Ea, Fig. 3B). These data are consistent with inhibition of both fibril nucleation and elongation. We previously demonstrated, along with others, that the process of oligomer and fibril formation accounts for amylin toxicity in cells.1,39,61 Given the inhibitory effect of Cu2+ on amylin aggregation (Fig. 3A and B),44 it is possible that copper’s antitoxic effect, at least in part, stems from its ability to halt amylin’s conformational transition toward β-sheets (Fig. 4), as observed by its impact on the dynamics of amylin oligomerization and preserving initial (native) conformation of soluble amylin. This by itself cannot entirely account for the protective effect of Cu2+ seen in our study given (i) the incomplete blockage of amylin aggregation by Cu2+ (Fig. 3A), and (ii) its opposite effect on mitochondrial function and apoptosis (Fig. 7). In fact Cu2+ is very effective in shutting down apoptotic and stress kinase signaling pathways, apparently acting downstream of amylin-induced mitochondrial dysfunction, which is sensitive to toxic amylin oligomers and aggregates.1 In contrast to its potential pro-oxidative action, Cu2+ had only miniscule and non-significant stimulatory effects on amylin’s pro-oxidative capabilities such as the catalytic (peptide-initiated) production of H2O2 in vitro and in cells. We reconfirmed reports of Allsop and colleagues that copper can slightly enhance H2O2 production (by few nM) evoked by human amylin in solution.32 However, this potentiation was by order of magnitude smaller than the Cu2+ mediated H2O2 production with reducing agents glutathione and ascorbate. Our study provides an explanation for the inability of Cu2+ alone or in complex with human amylin to evoke significant H2O2 production at physiological pH: either in the free form or bound to peptide, and in the absence of reducing agents, Cu2+ retains its valency (Fig. S5, ESI‡). Like brain β-amyloid, human amylin cannot reduce Cu2+ in the absence of reducing species.21 However, the redox potential of β-amyloid–Cu2+ complex is high enough (E0 = 0.295 V vs. NHE) to be reduced by species with a more negative reduction potential such as ascorbate and glutathione.20,21 As conversion of Cu2+ to Cu+ in amyloid complexes is a prerequisite for H2O2 generation,19 the lack of a significant stimulatory effect of Cu2+ on human amylin-evoked H2O2 production, in the absence of reducing species, is understandable. We also show that this catalytic effect is not unique to human amylin, as the non-amyloidogenic rat isoform produced comparable H2O2 concentrations, indicating that amylin aggregation and amyin-induced ROS are un-related processes. In vitro studies raised uncertainty about the ability of the human amylin–copper complex to induce comparable or higher oxidative damage in cells, particularly in the reducing environment of the cytosol, as compared to uncomplexed Cu2+. Supporting this view, an increase in free Cu2+ markedly diminished the viability of β-cells in a dose-dependent manner. Not surprisingly, human amylin diminished the ability of glutathione to reduce copper and produce H2O2. This protective effect was much less apparent in the case of the other cellular reducing agent ascorbate. However, there was no further potentiation of H2O2 production in vitro by Cu2+ and either reductant. Cu2+ also failed to stimulate amylin’s pro-oxidative activity in cells. Binding studies revealed that human and rat amylin interact with Cu2+ to form metalo-peptide complexes with Kd ~ 10−6 M, matching the dissociation constants reported for the β-amyloid–Cu2+ complex.19,23 The lower affinity of human amylin towards Cu2+, as compared to other Cu2+-binding proteins such as human serum albumin (Kd ~ 10−17),24 suggests that human amylin may serve as a secondary metal-binding site in the pancreas, thereby preventing the Cu2+ from freely interacting with reduced cellular species such as glutathione to produce high (μM) concentrations of H2O2, which are harmful to islet β-cells.62 Given the novel findings that human amylin can significantly reduce ROS accumulation in the system (Fig. 5), and that Cu2+ protects cells from amylin toxicity (Fig. 7), the formation of human amylin–Cu2+ complex is not, at least under our experimental conditions, detrimental to the β-cells. Our results (Fig. 2 and 5) are in agreement with increasing number of studies showing a sacrificial quenching of ROS by other amyloid proteins.49,50 Taken together, our results suggest that the human amylin–Cu2+ complex functions as an anti-oxidant rather than pro-oxidant in cells and in a cell-free environment. This conclusion, however, does not preclude the role for radicals in amylin toxicity, as amylin clearly elevates ROS accumulation in β-cells and impairs mitochondrial metabolic activity. These effects are completely ablated by the addition of the antioxidant NAC, reconfirming the view that amylin-induced cytotoxicity is, at least in part, through ROS generation.
Although our findings corroborate the view that antioxidants, such as NAC, could alleviate amylin-induced oxidative stress, they also indicate that NAC, in addition to its antioxidant activity, may paradoxically act as a pro-oxidant under certain conditions, thereby ameliorating its therapeutic efficacy. Thus, in the presence of exogenous copper, NAC can elicit only a limited amount of protection or can even potentiate level of cell death (Fig. 8A). Therefore, these findings imply that NAC can be an effective therapeutic agent for mitigating amylin-induced cytotoxicity but not under elevated copper scenarios, as is often the case with T2DM. We also show that strategies depending on copper’s chelation, particularly chelation of its reduced (Cu+) form, are not the best approach to alleviate amylin toxicity due to the unexpectedly high toxicity of the Cu+–amylin–bathocuproine complex. However, this complex did not produce significant amount of H2O2, suggesting that altered copper homeostasis rather than ROS imbalance accounts for the increased sensitivity of β-cells to the Cu2+–amylin–bathocuproine complex. Our results further show that ROS generated by human amylin induces persistent activation of stress kinases such as JNK as well as activation of the intrinsic caspase cascade, including caspase-3, which are effectively blocked by Cu2+. Previous studies on complex formation between Cu2+ and two other common amyloid proteins, β-amyloid and prions, are consistent with our findings. It has been reported that, similar to human amylin, these two misfolded proteins form a complex with Cu2+ with relatively low affinity.19,23 Chelation of Cu2+ by these two amyloids greatly diminished the production of ROS by Cu2+ and reductants and reduced their toxicity,49,50,55 which further corroborates our hypothesis that amylin–Cu2+ complex may serve a protective role in cells.
In summary, our findings provide a new insight into the nature of amylin–Cu2+ interactions in cells and in vitro. Copper’s intrinsic ability to stabilize human amylin in its native, non-toxic random coil conformation, to block amylin aggregation and amylin-induced apoptosis, and to form a low affinity complex with human amylin featuring low pro-oxidative activity in vitro and in cells may likely be beneficial for amylin-evoked β-cell dysfunction, which in turn could be applied to translational research to develop strategies for the prevention and treatment of the T2DM.
Supplementary Material
Acknowledgments
This work was supported by the GWU CCFF research grant.
Abbreviations
- BAT
Bathocuproine
- Cu
Copper
- HA
Human amylin
- RA
Rat amylin
- NAC
N-Acetyl-cysteine
- NHE
Normal hydrogen electrode
- RIN-m5F
Rat insulinoma cells
- ROS
Reactive oxygen species
- Th-T
Thioflavin-T
- TTDM
Type two diabetes mellitus
- PM
Plasma membranes
Footnotes
This article was submitted as part of a themed issue on biophysical studies on protein misfolding and amyloid diseases. Other papers on this topic can be found in issue 23 of vol. 15 (2013).
Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cp44542a
References
- 1.Haataja L, Gurlo T, Huang CJ, Butler PC. Endocr Rev. 2008;29:303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaikaran ET, Clark A. Biochim Biophys Acta. 2001;1537:179–203. doi: 10.1016/s0925-4439(01)00078-3. [DOI] [PubMed] [Google Scholar]
- 3.Engel MF, Khemtemourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, Hoppener JW. Proc Natl Acad Sci U S A. 2008;105:6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenzo A, Razzaboni B, Weir GC, Yankner BA. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 5.Meier JJ, Kayed R, Lin CY, Gurlo T, Haataja L, Jayasinghe S, Langen R, Glabe CG, Butler PC. Am J Phys. 2006;291:E1317–E1324. doi: 10.1152/ajpendo.00082.2006. [DOI] [PubMed] [Google Scholar]
- 6.Guardado-Mendoza R, Davalli AM, Chavez AO, Hubbard GB, Dick EJ, Majluf-Cruz A, Tene-Perez CE, Goldschmidt L, Hart J, Perego C, Comuzzie AG, Tejero ME, Finzi G, Placidi C, La Rosa S, Capella C, Halff G, Gastaldelli A, DeFronzo RA, Folli F. Proc Natl Acad Sci U S A. 2009;106:13992–13997. doi: 10.1073/pnas.0906471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura S, Okabayashi S, Ageyama N, Koie H, Sankai T, Ono F, Fujimoto K, Terao K. Vet Pathol. 2008;45:67–72. doi: 10.1354/vp.45-1-67. [DOI] [PubMed] [Google Scholar]
- 8.Janciauskiene S, Ahren B. Biochem Biophys Res Commun. 2000;267:619–625. doi: 10.1006/bbrc.1999.1989. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S, Liu J, Dragunow M, Cooper GJ. J Biol Chem. 2003;278:52810–52819. doi: 10.1074/jbc.M308244200. [DOI] [PubMed] [Google Scholar]
- 10.Zhang S, Liu J, MacGibbon G, Dragunow M, Cooper GJ. J Mol Biol. 2002;324:271–285. doi: 10.1016/s0022-2836(02)01044-6. [DOI] [PubMed] [Google Scholar]
- 11.Zraika S, Hull RL, Udayasankar J, Aston-Mourney K, Subramanian SL, Kisilevsky R, Szarek WA, Kahn SE. Diabetologia. 2009;52:626–635. doi: 10.1007/s00125-008-1255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A. Biochemistry. 2008;47:12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirzabekov TA, Lin MC, Kagan BL. J Biol Chem. 1996;271:1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 14.Naka T, Kaneto H, Katakami N, Matsuoka TA, Harada A, Yamasaki Y, Matsuhisa M, Shimomura I. Endocr J. 2013;60:393–396. doi: 10.1507/endocrj.ej12-0342. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka A, Kaneto H, Miyatsuka T, Yamamoto K, Yoshiuchi K, Yamasaki Y, Shimomura I, Matsuoka TA, Matsuhisa M. Endocr J. 2009;56:699–706. doi: 10.1507/endocrj.k09e-051. [DOI] [PubMed] [Google Scholar]
- 16.Uriu-Adams JY, Rucker RB, Commisso JF, Keen CL. J Nutr Biochem. 2005;16:312–320. doi: 10.1016/j.jnutbio.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Ford ES. Am J Epidemiol. 2000;151:1182–1188. doi: 10.1093/oxfordjournals.aje.a010168. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi H, Yano M, Fujita Y, Wakusawa S. Med Mol Morphol. 2006;39:121–126. doi: 10.1007/s00795-006-0326-7. [DOI] [PubMed] [Google Scholar]
- 19.Eskici G, Axelsen PH. Biochemistry. 2012;51:6289–6311. doi: 10.1021/bi3006169. [DOI] [PubMed] [Google Scholar]
- 20.Jiang D, Li X, Liu L, Yagnik GB, Zhou F. J Phys Chem. 2010;114:4896–4903. doi: 10.1021/jp9095375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang D, Men L, Wang J, Zhang Y, Chickenyen S, Wang Y, Zhou F. Biochemistry. 2007;46:9270–9282. doi: 10.1021/bi700508n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang C, Liu L, Zhang L, Peng Y, Zhou F. Biochemistry. 2010;49:8134–8142. doi: 10.1021/bi1010909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maiti NC, Jiang D, Wain AJ, Patel S, Dinh KL, Zhou F. J Phys Chem. 2008;112:8406–8411. doi: 10.1021/jp802038p. [DOI] [PubMed] [Google Scholar]
- 24.Lau SJ, Sarkar B. J Biol Chem. 1971;246:5938–5943. [PubMed] [Google Scholar]
- 25.Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ. J Biol Chem. 2001;276:20466–20473. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- 26.Murray IV, Sindoni ME, Axelsen PH. Biochemistry. 2005;44:12606–12613. doi: 10.1021/bi050926p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sayre LM, Perry G, Harris PL, Liu Y, Schubert KA, Smith MA. J Neurochem. 2000;74:270–279. doi: 10.1046/j.1471-4159.2000.0740270.x. [DOI] [PubMed] [Google Scholar]
- 28.Baruch-Suchodolsky R, Fischer B. Biochemistry. 2008;47:7796–7806. doi: 10.1021/bi800114g. [DOI] [PubMed] [Google Scholar]
- 29.Kontush A. Cell Mol Neurobiol. 2001;21:299–315. doi: 10.1023/A:1012629603390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura M, Shishido N, Nunomura A, Smith MA, Perry G, Hayashi Y, Nakayama K, Hayashi T. Biochemistry. 2007;46:12737–12743. doi: 10.1021/bi701079z. [DOI] [PubMed] [Google Scholar]
- 31.Zou K, Gong JS, Yanagisawa K, Michikawa M. J Neurosci. 2002;22:4833–4841. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masad A, Hayes L, Tabner BJ, Turnbull S, Cooper LJ, Fullwood NJ, German MJ, Kametani F, El-Agnaf OM, Allsop D. FEBS Lett. 2007;581:3489–3493. doi: 10.1016/j.febslet.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 33.Masad A, Tabner BJ, Mayes J, Allsop D. Free Radicals Biol Med. 2011;51:869–875. doi: 10.1016/j.freeradbiomed.2011.05.033. [DOI] [PubMed] [Google Scholar]
- 34.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 35.Opazo C, Huang X, Cherny RA, Moir RD, Roher AE, White AR, Cappai R, Masters CL, Tanzi RE, Inestrosa NC, Bush AI. J Biol Chem. 2002;277:40302–40308. doi: 10.1074/jbc.M206428200. [DOI] [PubMed] [Google Scholar]
- 36.Moreira PI. J Alzheimers Dis. 2012;30(suppl 2):S199–S215. doi: 10.3233/JAD-2011-111127. [DOI] [PubMed] [Google Scholar]
- 37.Cho WJ, Jena BP, Jeremic AM. Methods Cell Biol. 2008;90:267–286. doi: 10.1016/S0091-679X(08)00813-3. [DOI] [PubMed] [Google Scholar]
- 38.Cho WJ, Trikha S, Jeremic AM. J Mol Biol. 2009;393:765–775. doi: 10.1016/j.jmb.2009.08.055. [DOI] [PubMed] [Google Scholar]
- 39.Trikha S, Jeremic AM. J Biol Chem. 2011;286:36086–36097. doi: 10.1074/jbc.M111.240762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eruslanov E, Kusmartsev S. Methods Mol Biol. 2010;594:57–72. doi: 10.1007/978-1-60761-411-1_4. [DOI] [PubMed] [Google Scholar]
- 41.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 42.Yan EB, Unthank JK, Castillo-Melendez M, Miller SL, Langford SJ, Walker DW. J Appl Phys. 2005;98:2304–2310. doi: 10.1152/japplphysiol.00617.2004. [DOI] [PubMed] [Google Scholar]
- 43.Varadarajan S, Kanski J, Aksenova M, Lauderback C, Butterfield DA. J Am Chem Soc. 2001;123:5625–5631. doi: 10.1021/ja010452r. [DOI] [PubMed] [Google Scholar]
- 44.Ward B, Walker K, Exley C. J Inorg Biochem. 2008;102:371–375. doi: 10.1016/j.jinorgbio.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 45.Pappalardo G, Milardi D, Magri A, Attanasio F, Impellizzeri G, La Rosa C, Grasso D, Rizzarelli E. Chemistry. 2007;13:10204–10215. doi: 10.1002/chem.200700576. [DOI] [PubMed] [Google Scholar]
- 46.Rigacci S, Guidotti V, Bucciantini M, Parri M, Nediani C, Cerbai E, Stefani M, Berti A. J Nutr Biochem. 2010;21:726–735. doi: 10.1016/j.jnutbio.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 47.Manevich Y, Held KD, Biaglow JE. Radiat Res. 1997;148:580–591. [PubMed] [Google Scholar]
- 48.Stohs SJ, Bagchi D. Free Radicals Biol Med. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- 49.Nadal RC, Abdelraheim SR, Brazier MW, Rigby SE, Brown DR, Viles JH. Free Radicals Biol Med. 2007;42:79–89. doi: 10.1016/j.freeradbiomed.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 50.Nadal RC, Rigby SE, Viles JH. Biochemistry. 2008;47:11653–11664. doi: 10.1021/bi8011093. [DOI] [PubMed] [Google Scholar]
- 51.Li XL, Chen T, Wong YS, Xu G, Fan RR, Zhao HL, Chan JC. Int J Biochem Cell Biol. 2011;43:525–534. doi: 10.1016/j.biocel.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Lim YA, Rhein V, Baysang G, Meier F, Poljak A, Raftery MJ, Guilhaus M, Ittner LM, Eckert A, Gotz J. Proteomics. 2010;10:1621–1633. doi: 10.1002/pmic.200900651. [DOI] [PubMed] [Google Scholar]
- 53.Yu YP, Lei P, Hu J, Wu WH, Zhao YF, Li YM. Chem Commun. 2010;46:6909–6911. doi: 10.1039/c0cc02141e. [DOI] [PubMed] [Google Scholar]
- 54.Bishop GM, Robinson SR. Brain Pathol. 2004;14:448–452. doi: 10.1111/j.1750-3639.2004.tb00089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stevens DJ, Walter ED, Rodriguez A, Draper D, Davies P, Brown DR, Millhauser GL. PLoS Pathog. 2009;5:e1000390. doi: 10.1371/journal.ppat.1000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoshiike Y, Tanemura K, Murayama O, Akagi T, Murayama M, Sato S, Sun X, Tanaka N, Takashima A. J Biol Chem. 2001;276:32293–32299. doi: 10.1074/jbc.M010706200. [DOI] [PubMed] [Google Scholar]
- 57.Konarkowska B, Aitken JF, Kistler J, Zhang S, Cooper GJ. FEBS J. 2005;272:4949–4959. doi: 10.1111/j.1742-4658.2005.04903.x. [DOI] [PubMed] [Google Scholar]
- 58.Turski ML, Thiele DJ. J Biol Chem. 2009;284:717–721. doi: 10.1074/jbc.R800055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oikawa S, Yamada K, Yamashita N, Tada-Oikawa S, Kawanishi S. Carcinogenesis. 1999;20:1485–1490. doi: 10.1093/carcin/20.8.1485. [DOI] [PubMed] [Google Scholar]
- 60.Ivanov AI, Parkinson JA, Cossins E, Woodrow J, Sadler PJ. J Biol Inorg Chem. 2000;5:102–109. doi: 10.1007/s007750050013. [DOI] [PubMed] [Google Scholar]
- 61.Konarkowska B, Aitken JF, Kistler J, Zhang S, Cooper GJ. FEBS J. 2006;273:3614–3624. doi: 10.1111/j.1742-4658.2006.05367.x. [DOI] [PubMed] [Google Scholar]
- 62.Saxena G, Chen J, Shalev A. J Biol Chem. 2010;285:3997–4005. doi: 10.1074/jbc.M109.034421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.