

Abstract
Otitis media (OM), a common infectious disease in children, is associated with bacterial middle ear (ME) infection. Tolllike receptors (TLRs) are important mediators of innate immune responses, and TLR9 specifically recognizes the unmethylated cytidine-phosphate-guanosine (CpG) motifs in bacterial DNA. Additional sensors of foreign DNA have recently been identified. The role of DNA sensing and TLR9 was investigated in a murine model of OM induced by non-typeable Haemophilus influenzae (NTHi). Expression of genes related to DNA-sensing pathways involved in innate immunity was assessed via DNA microarray, qPCR and immunohistochemistry. Middle ear responses to NTHi were examined in wild-type and TLR9−/− mice by histopathology and bacterial culture. Expression of TLR9 signaling genes was modestly up-regulated during OM, as was TLR9 protein in both ME mucosal cells and infiltrating leukocytes. However, genes known to be regulated by CpG DNA were dramatically up-regulated, as were genes involved in DNA sensing by DIA, Pol-III and AIM2. Toll-like receptor 9 deletion significantly prolonged the inflammatory response induced by NTHi in the ME and delayed bacterial clearance. The results suggest that DNA sensing via TLR9 plays a role in OM pathogenesis and recovery. Alternative forms of DNA sensing may also contribute to OM.
Keywords: CpG DNA, Haemophilus influenzae, otitis media, pathogen DNA sensing, Toll-like receptor
Introduction
Otitis media (OM) is an extremely common pediatric disease.1 While uncomplicated OM typically resolves without treatment, about 10–20% of children develop more serious chronic or recurrent disease requiring antibiotic therapy or surgery.2 The cost of OM, including treatment and lost productivity, is estimated at more than $5 billion annually in the US.
The etiology of OM is complex. Eustachian tube dysfunction, cognate immune status, prior viral infection and allergy are all known to contribute to susceptibility to OM.3,4 However, bacterial infection is the ultimate cause of most acute and/or recurrent OM, with non-typeable Haemophilus influenzae (NTHi) the most commonly isolated pathogen.5 Otitis media is characterized by inflammation of the middle ear (ME), mucosal hyperplasia, the development of effusion, and leukocytic infiltration of the tympanic cavity.6,7
While cognate immunity is critical for the defense of the body against infection, it requires the naïve organism 1–2 wk to mount an effective cognate response to a new pathogen.8 Therefore, the first line of defense against infection must be served by innate immune responses that do not require prior sensitization. Innate immunity is mediated by receptors that recognize molecules produced by pathogens, such as pathogen-associated molecular patterns (PAMPs), including proteins, lipids, lipoproteins and nucleic acids. The best-characterized of these innate immune receptors are the family of Toll-like receptors (TLRs),9,10 but there are many other types of receptors that participate in innate immune defense, including the NOD-like receptors (NLRs).
Non-typeable H. influenzae produces a variety of PAMPs that are recognized by innate immune receptors. These include endotoxin (lipooligosaccharide, similar to LPS) recognized by TLR4, as well as lipopeptides recognized by TLR2.11,12 It is likely that NTHi also produces peptides and lipopeptides that can activate the NLRs,13 and bacterial RNA has been shown to activate both TLRs (including TLR3 and TLR7) and NLRs, especially the NALPs.14 However, bacterial and other forms of non-self DNA are very important PAMPs, and several pathways for their recognition exist. All of these receptors for DNA are intra-cellular, requiring internalization of DNA for recognition. However, internalization of NTHi by respiratory epithelial cells has been well documented.15–17 While this internalization occurs independently of DNA receptors,15,16 it would potentially expose the receptors to bacterial DNA.
Toll-like receptor 9 is localized to endosomes, and recognizes unmethylated cytidine-phosphate-guanosine (CpG) motifs that are common in bacterial, but not mammalian, DNA.18 Access of TLR9 to bacterial DNA is enhanced by the modulator HMGB1.19 Exposure of TLR9 to bacterial DNA results in the recruitment of signaling molecules, leading ultimately to the production of pro-inflammatory cytokines and other target genes.18 Toll-like receptor 9 is also thought to contribute to the development of effective cognate immunity through facilitating development of the inflammasome. In this role, TLR9 has been targeted, employing immunostimulatory sequences of DNA (ISD), to enhance immune responses to pollen allergens for the treatment of allergic rhinitis.20 Other non-self DNA receptors have recently been described, which are cytoplasmic and which activate innate immune effectors such as cytokines and IFNs. These include DNA-dependent activator of IFN regulatory factor (DAI),21 absent in melanoma-2 (AIM2)22,23 and RNA polymerase III.24,25
The signaling cascades activated by receptors for pathogen DNA are illustrated in Figure 1. Like other TLRs, TLR9 utilizes the TLR adaptor molecule MyD88, which leads, via the activation of NF-κB, to the production of TNF-α and other pro-inflammatory cytokines such as IL-6. We have shown previously that MyD88 and TNF-α are important for the resolution of OM caused by NTHi infection.26 However, the role of TLR9 in this disorder has not been well characterized. Toll-like receptor 9 has been found to be a significant mediator of inflammation at other sites, and to synergize with other TLRs that we have found to be involved in OM pathogenesis.27–29 Moreover, in a model of sepsis,30 mortality was reduced by its inhibition or in its absence. In addition, reperfusion injury in the liver has been shown to be decreased by inhibition of TLR9.31 These studies emphasize the potential for TLR9 signaling to contribute to disease and injury pathogenesis.
Figure 1.
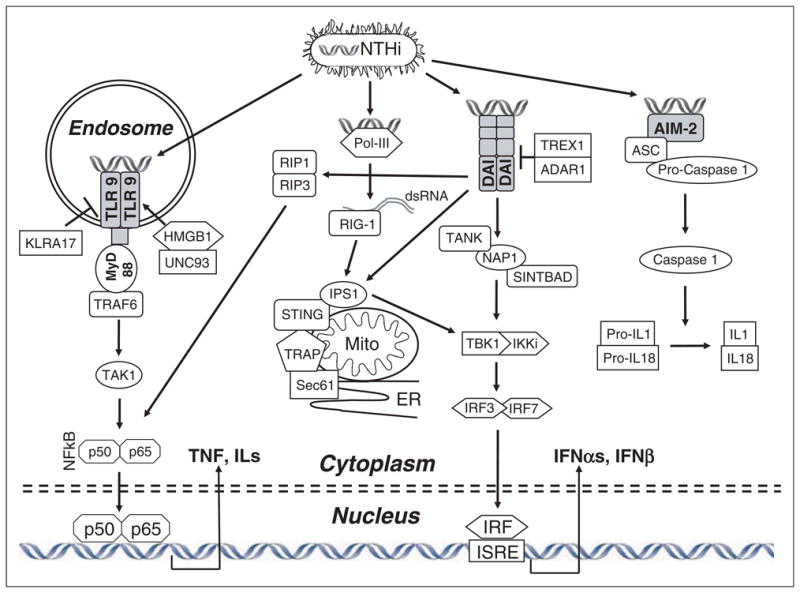
A schematic representation of pathogen DNA sensing. NTHi internalized as intact bacteria or fragments releases DNA, which can interact with specific pathogen DNA receptors. Toll-like receptor 9, located in endosomes, signals primarily via MyD88 to stimulate NF-κB and cytokine production. DAI can activate IRFs, leading to type I IFN production, via several intermediaries. Pol-III can convert bacterial DNA into dsRNA, also activating IRFs. DNA interaction with AIM2 recruits ASC and procaspase1 to form an inflammasome, leading to caspase1 activation and IL-1/IL-18 maturation.
With respect to other pathways able to recognize pathogen DNA, DAI signals via a complex of proteins associated with mitochondria and the endoplasmic reticulum to activate IRF3 and IFN production.21 Pol-III transcribes bacterial DNA into RNA, where it is sensed by RIG-1 and also induces IFN production.24,25 Upon interaction with bacterial DNA, AIM2 complexes with (ASC) and pro-caspase 1 to form an inflammasome, leading to caspase 1 activation and subsequently to maturation of IL-1 and IL-18.22,23 The role of these innate immune DNA sensing pathways in OM also remains unexplored.
Simple injection of bacterial DNA into the ME seemed unlikely to be informative, since all of the receptors noted above are intracellular and entry of DNA into cells is most likely dependent upon other bacterial elements. To determine whether pathogen DNA plays a role in bacterial OM, we characterized, using DNA microarrays, the expression of genes involved in the DNA sensing pathways described above, as well as genes known to be induced by their activation, during the course of OM induced by NTHi in mice. In addition, we evaluated OM in mice that are genetically deficient in TLR9 (the only non-self DNA sensor for which a gene deletion mouse is commercially available) using a well-established model of NTHi inoculation of the mouse ME.32,33
Materials and methods
Animals
C57Bl/6:CB F1 hybrid mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) for gene array analysis. Toll-like receptor 9−/− mice on a C57BL/6 background (6× crossed) were originally generated by Akira and colleagues34,35 and used with their permission, but were generously supplied by Dr Eyal Raz of UCSD. Age-matched C57BL/6 control mice were purchased from Jackson Laboratories.
Bacteria
Haemophilus influenzae strain 3655 (non-typeable, bio-type II; NTHi), a clinical isolate from the ME of an OM patient, was used at a concentration of 105–106/ml to induce acute OM.32
Surgery
For DNA microarrays, 40 C57BL/6:CB F1 hybrid mice per time point were used. The TLR9−/− and C57BL/6 mice were divided into groups of six mice for each experimental time point (3 each for histopathology and bacterial culture). Animals were inoculated with NTHi as described previously.29,32 Uninoculated animals (time 0) served as controls.
DNA microarray
Expression of selected genes involved in DNA sensing was evaluated in wild-type mice by DNA microarray. Forty wild-type ME mucosae were harvested at each of the following intervals: 0 (no treatment), 3 h and 6 h, as well as 1, 2, 3, 5 and 7 d after NTHi inoculation. The tissue was homogenized in TRIzol™ (Invitrogen, Carlsbad, CA, USA) and total RNA was extracted. RNA was labeled and hybridized to two Affymetrix MU430 2.0 microarrays. This procedure was then duplicated for each time point to obtain an independent replication. Gene transcript expression levels were evaluated using variance-modeled posterior inference (VAMPIRE).36 Functional gene families were assessed by gene ontogeny (GO) analysis, and specific genes were assessed at individual time points, after Bonferonni correction for multiple tests, using Genespring GX 7.3 (Agilent).
mRNA quantification by qPCR
Gene expression due to TLR signaling was investigated during NTHi-induced OM in six MEs per time point of C57BL/6 and TLR9−/− mice using real-time PCR (qPCR). The mRNA was extracted using Dynabeads mRNA DirectTM (Invitrogen). Messenger RNA (20 μl) was reverse transcribed using the SuperScript™ First-Strand cDNA Synthesis kit (Invitrogen). Quantitative real-time PCR was performed using 1 μg/μl of cDNA and predeveloped TaqMan qPCR primers (Applied Biosystems) for TNF-α and IL-10 in the ABI Prism 7000 Sequence Detection System (Applied Biosystems). Fold induction was calculated using the comparative threshold cycle (Ct) method as previously described.29 Relative expression of the target gene was normalized to GAPDH and compared to uninfected mucosa.
Histology
Wild-type and TLR9−/− MEs were harvested at 0 (uninoculated), 1, 2, 3, 5, 10 and 14d post inoculation, processed into paraffin, sectioned and stained with H&E. Sections from standardized locations were digitally recorded and mucosal thickness determined morphometrically using NIH Image-Pro image analysis software. The percentage area of the ME lumen occupied by inflammatory cells at these locations was measured using the same software, and computer-averaged to obtain a measure of cellular effusion present within the ME, as described previously.26 This measure is used to quantify cellular inflammation in other tissue spaces.37,38 The numbers of neutrophils and macrophages comprising ME infiltrates were also counted in five randomly selected clusters of cells for each ME (400×) by two independent observers and computer-averaged.
Immunohistochemistry
Four wild-type MEs at each time were harvested 0, 1, 2, 3, 5 or 7 d (because wild-type animals were used, OM is resolved by 7 days) after NTHi inoculation and fixed in 4% PFA. The tissue was decalcified, cryoprotected, frozen and sectioned on a cryostat at 10 μm. Sections were reacted with anti-TLR9 antibody, followed by a fluorescein-labeled secondary antibody, to detect TLR9. The sections were counterstained with DAPI to visualize cell nuclei.
Bacterial clearance
Bacterial presence was assessed from at least six wild-type and six TLR9−/− MEs per time point, by obtaining a 1μl loop sample from each ME for NTHi culture. Culture positivity and degree was classified as described previously.26
Statistical analysis
Except for gene array values as discussed above, data were analyzed with ANOVA using StatView statistics calculation software, as described elsewhere.39 Differences were considered significant at P < 0.05.
All experiments were performed according to NIH guidelines and approved by the IACUC of the San Diego VA Medical Center.
Results
Regulation of DNA-sensing pathway genes during OM
Gene ontogeny analysis of gene array expression patterns during the course of OM identified innate immune and inflammatory signaling as pathways significantly regulated in the ME by exposure to NTHi, including TLR. Since a GO pathway for DNA sensing has not been developed, we individually assessed the genes involved in each of the DNA sensing pathways illustrated in Figure 1, as well as genes that have been identified as being specifically up-regulated by ligands for TLR9 (CpG DNA) and DAI (interferon stimulatory DNA, ISD).
Toll-like receptor 9 signaling genes are only modestly influenced by NTHi, but TLR9 target genes are strongly up-regulated
We evaluated the expression of four genes involved in DNA sensing by TLR9 (see Figure 1) in the ME mucosa of wild-type mice, using DNA microarrays (Figure 2A and Supplementary Table 1). Expression was determined as fold change relative to control MEs.
Figure 2.

Middle ear expression of genes related to DNA sensing in wild-type mice during OM, assessed by DNA microarray. (A) The ME expression of most genes involved in TLR9 signaling (see Figure 1) was minimally affected by NTHi. Exceptions were KLRA17, a negative regulator of TLR9 signaling, which was strongly (18×) up-regulated during OM, and the general TLR adaptor, MyD88, which was up-regulated 6×. (B) In contrast, TLR9 target genes, known to be induced by unmethylated CpG DNA, were highly up-regulated by NTHi. (C,D) mRNA encoding several genes of the DAI/Pol-III signaling pathways was significantly up-regulated during OM, especially from 1-5 d after NTHi inoculation. DAI itself was up-regulated more than 40-fold, 2 d after ME inoculation with NTHi. (E) In contrast, DAI/Pol-III target genes, known to be induced by ISD, were modestly up-regulated early in the response to NTHi. (F) Moderate up-regulation of genes involved in AIM2 signaling from pathogen DNA was observed, although mRNA encoding caspase1 was actually down-regulated from 3–6 h after NTHi inoculation. *P < 0.05, **P < 0.01, ***P < 0.001.
The TLR9 gene itself was up-regulated only 2–3-fold, at 48 h after inoculation. The co-factor UNC93, which is required for TLR9 trafficking to endosomes and for TLR9 function,40 and HMGB1 which supports DNA internalization into endosomes19 were essentially unaffected throughout the course of OM. However, a negative regulator of TLR9 signaling, KLRA17, was significantly up-regulated from 1–3 d after NTHi inoculation, with a peak of 18-fold regulation at 1 d. Of downstream genes involved in signaling from a number of TLR receptors, MyD88 showed moderate up-regulation, up to 6-fold at 1 d after inoculation, while TRAF6 and TAK1 were minimally affected.
In contrast to TLR9 signaling genes, genes that have been shown to be specifically up-regulated by its target PAMP, unmethylated CpG DNA,41,42 were dramatically up-regulated by exposure to NTHi (Figure 2B and Supplementary Table 2). In particular, expression of both CSF3 and CXCL1 were enhanced by 200– 300-fold.
DAI and Pol-III signaling genes are up-regulated during OM more than their target genes
Genes encoding the proteins that mediate DAI and Pol-III DNA signaling (see Figure 1) were strongly up-regulated during OM in wild-type mice (Figure 2C, D and Supplementary Table 1). Expression of DAI itself peaked at more than 40-fold induction, while several additional genes were up-regulated by 5–10-fold. In contrast, genes that are preferentially regulated by a DIA/Pol-III ligand, ISD,42,43 showed no, or only modest, regulation during NTHi-induced OM (Figure 2E and Supplementary Table 2).
Genes involved in AIM2 DNA signaling are up-regulated during OM
The expression of five genes involved in AIM2 DNA sensing was determined (Figure 2F and Supplementary Table 1). AIM2 and the inflammasome targets IL-1β and IL-18 were significantly up-regulated from 3- and 9-fold. Caspase1 showed brief down-regulation followed by modest up-regulation, while ASC was consistently down-regulated.
Toll-like receptor 9 protein is observed in the ME during OM
Little or no TLR9 immunoreactivity was observed in the resting ME mucosa. However, TLR9 was present in both epithelial and inflammatory cells during OM from 6–72 h after inoculation. Toll-like receptor 9-positive cells were observed, scattered through the ME mucosa, and were primarily epithelial cells. Within inflammatory exudates, TLR9 was more prevalent, and was detected primarily in mononuclear leukocytes that had the appearance of macrophages (Figure 3A).
Figure 3.
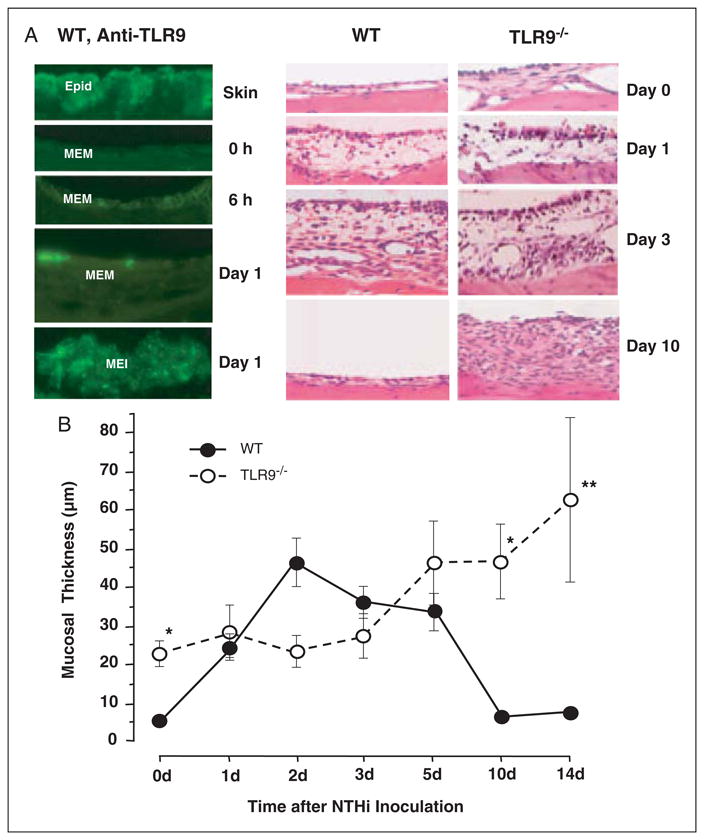
Mucosal response to NTHi in the MEs of wild-type versus TLR9−/− mice. (A) Left. Immunocytochemistry for TLR9 in the wild-type ME during OM. Skin from the external ear canal strongly expressed TLR9 in the epidermal layer (Epid). While little TLR9 was observed in the untreated ME (0h), TLR9 was observed in both the ME mucosa (MEM) and in cells infiltrating the ME lumen (MEI) during OM, as shown for examples at 6 h and 1 d after inoculation; × 100 original magnification. Similar results were observed on days 2 and 3. By days 5 and 7, TLR9 immunoreactivity was no longer present in the ME. Right. Hematoxylin and eosin staining of wild-type and TLR9−/− ME at 0, 1, 3 and 10 d after NTHi challenge; ×40 original magnification. (B) Quantitative evaluation of mucosal thickness in wild-type and TLR9−/− mice during NTHi-induced OM. Inoculation of NTHi into the MEs of TLR9−/− mice resulted in significantly enhanced thickness prior to and by 10–14 d, and full recovery was not observed.
Toll-like receptor 9−/− mice show delayed ME inflammatory response to NTHi and compromised OM recovery
To assess the functional role of TLR9, we investigated the ME response to NTHi inoculation in TLR9−/− mice, as compared to wild-type controls. The wild-type mice exhibited a typical response to NTHi.26 This response included hyperplasia of the ME mucosa, which reached a maximum thickness 2 d after inoculation with NTHi (Figure 3A,B). Mucosal thickness remained elevated at 3 d, and then recovered to baseline by 5 d. Toll-like receptor 9−/− mice exhibited modestly, but significantly, greater thickness of the ME mucosa prior to NTHi administration, compared to wild-type mice. However, in contrast to wild-type mice, NTHi induced no significant increase in mucosal thickness for the first 3 d after inoculation. Thickness then increased from days 5–14, with recovery not complete at the longest post-inoculation time point examined.
The infiltration of cells into the ME lumen of TLR9–/– mice after NTHi inoculation was assessed by measuring the percentage of the ME lumen covered by cells (Figure 4A). Infiltration peaked at 2–3 d in wild-type mice and then declined to low levels. No cells were present in the ME lumina of uninoculated TLR9−/− mice. Moreover, after inoculation, ME cellular infiltration in TLR9−/− mice was not significantly different from that observed in wild-types. However, when individual cell types were assessed, significantly fewer polymorphonuclear leukocytes were observed in the ME exudates of TLR9−/− mice at 1 d and 3 d after inoculation, when compared to wild-type mice (Figure 4B). Macrophage infiltration was not significantly different (Figure 4C). Interestingly, two TLR9−/− MEs showed both neutrophils and macrophages at 14 d after NTHi inoculation, more than one week after these cells had been cleared from all wild-type mouse MEs. In these two cases, the kinetics of both cell types might have been delayed by lack of TLR9.
Figure 4.

Leukocyte infiltration of the ME cavity in wild-type versus TLR9−/− mice. (A) Inflammatory cell infiltration into the ME lumen of wild-type and TLR9−/− mice during OM, measured as a percentage of ME luminal area covered by cells. There was no significant difference in the recruitment of cells to the MEs of TLR9−/− mice during OM. n = 6–8 MEs per time point; bars represent the mean ± SEM. (B) Neutrophils increased significantly less from days 1-3 compared to wild-type mice, but their presence in the ME was prolonged. (C) There was no significant difference in the recruitment of macrophages to the TLR9−/− ME. n = 6–8 MEs per time point; bars represent ±SEM.
Cytokine expression was enhanced in the MEs of TLR9−/− mice
Cytokine expression is often regulated by NF-κB, a primary downstream target of TLR9 via MyD88. Therefore, expression of the classical pro-inflammatory cytokine TNF-α (Figure 5A) and the classical antiinflammatory cytokine IL-10 (Figure 5B), were compared in the MEs of wild-type versus TLR9−/− mice by qPCR. The TNF-α mRNA showed a biphasic response in wild-type mice, with high levels at 1 d and 2 d after NTHi inoculation, a steep decrease, and then modest levels of mRNA at 5 d and 10 d. While the TLR9−/− ME also displayed a biphasic response, significantly more TNF-α mRNA was recovered from TLR9−/− MEs than from wild-type mice 2 d and 10 d after NTHi inoculation. The minimum was observed at 5 d. In contrast, IL-10 mRNA expression peaked at 5 d in the TLR9−/− ME, but was not observed in the wild-type ME.
Figure 5.

Cytokine expression in the ME mucosa of wild-type versus TLR9−/− mice. (A) ME TNF mRNA and (B) IL-10 mRNA during NTHi-induced OM, assessed by qPCR. Target genes were normalized to GAPDH and compared to uninfected wild-type mucosa. n = >6 MEs per/point; mean±SEM.
Middle ear bacterial clearance was delayed in TLR9−/− mice
Bacterial clearance of the ME cavity was examined in TLR9-deficient mice and compared to that observed in wild-type animals (Table 1). In wild-type mice, 4 out of 6 culture plates were positive at day 1, increasing to 6/6 at day 2, and decreasing to 3/6 on day 3. Thereafter, no bacteria were recovered from wild-type MEs. The TLR9−/− MEs were very similar to wild-type MEs from days 1–3 after NTHi inoculation, with 3/6, 4/6 and 2/6 MEs positive for bacterial colonization, respectively. In contrast to wild-type, CFUs could still be detected at 5 d and 10 d, with two culture-positive MEs out of 6 at each time. This observation suggests that bacterial clearance is compromised in the MEs of TLR9−/− mice, compared to wild-type animals. However, TLR9−/− ME cultures were negative at 14 d, thus clearance was delayed, not prevented.
Table 1. Comparison of impaired bacterial clearance in wild-type and TLR9−/− MEs.
| Time after NTHi instillation | C57BL/6 | TLR9−/− |
|---|---|---|
| Day 1 | 4/6 | 3/6 |
| Day 2 | 6/6 | 4/6 |
| Day 3 | 3/6 | 3/6 |
| Day 5 | 0/6 | 2/6 |
| Day 10 | 0/6 | 2/6 |
| Day 14 | 0/6 | 0/6 |
Bacterial colonization of the culture positive plates was evaluated using semi-quantitative analysis of bacterial colonization: 0 indicates no CFUs, 1 indicates one quadrant with CFUs, 2 indicates two quadrants with CFUs, 3 indicates three quadrants with CFUs and 4 indicates four quadrants with CFUs. Data represent positive culture plates out of six.
Discussion
In this investigation, we obtained evidence that detection of DNA by intracellular innate immune receptors contributes to the pathogenesis of NTHi-induced OM in mice, and also plays a role in bacterial clearance and OM recovery. After NTHi infection of wild-type mice, the expression of genes encoding DNA sensing receptors and many associated downstream signaling genes, as well as a number of target genes, was up-regulated. Moreover, animals deficient in TLR9 exhibited altered OM, consisting of prolonged and enhanced morphological signs of mucosal hyperplasia and inflammation, as well as a delay in bacterial clearance and OM recovery.
Interestingly, TLR9−/− mice showed greater baseline thickness of the ME mucosa than wild-type mice, in the absence of overt signs of inflammation such as effusion or leukocyte infiltration. The reasons for this are not clear. It seems unlikely that TLR9 would directly influence mucosal growth. However, it is possible that prior ME infections with bacterial species unrelated to NTHi are responsible.
Genes encoding proteins that participate in the DAI/Pol-III signaling pathway were most strongly up-regulated during OM, suggesting the potential for a significant role for these DNA sensing cascades in this disorder. However, genes that are known to be directly regulated by this pathway, including type I IFN genes, showed only brief, modest up-regulation. Interestingly, genes that negatively regulate these pathways were also up-regulated. This implies that inflammation produced in response to the activation of these pathways is tightly regulated. This may help to explain why DAI/Pol-III target genes were only briefly up-regulated.
The DNA sensing pathway that showed the least gene regulation of its signaling molecules during OM was TLR9, with only one adapter molecule and one negative regulator showing strong up-regulation. In contrast, the expression of genes that are known to be preferentially regulated by the TLR9 ligand CpG DNA41,42 were dramatically enhanced. Moreover, OM in TLR9−/− mice was more severe and prolonged than that in wild-type animals. These disparities may be clarified by the results of immunohistochemistry for TLR9 protein. Little TLR9 protein was detected in the resting ME, and only scattered mucosal cells were positive for TLR9 during OM. However, more TLR9 was present in infiltrating leukocytes, which may bring TLR9 protein into the ME pre-formed.
The small amounts of TLR9 induced in epithelial cells may nevertheless be important. The lower level of neutrophil infiltration observed in the MEs of TLR9−/− mice (Figure 4B) was particularly noteworthy. NTHi up-regulated the neutrophil chemokine, CXCL5, in wild-type mice more than 30-fold at 3, 6 and 34 h after inoculation (Figure 2B and Supplementary Table 2). Since CXCL5 is known to be preferentially induced by CpG DNA via TLR9, it is possible that a reduction in the production of this mediator may contribute to reduced neutrophil chemotaxis in response to NTHi infection in the TLR9−/− ME.
In previous studies, we and others have found that the TLR signaling molecule MyD88 plays a significant role in OM.26,44,45 In fact, this adapter can be required for OM recovery, since OM induced by NTHi in MyD88-null mice persists for weeks, and bacteria can be cultured from some MyD88−/− MEs 6wk after bacterial inoculation.26 While TLR9 primarily employs MyD88 to activate downstream signaling and target gene expression, and MyD88 gene expression was up-regulated during OM, the more severe phenotype observed in MyD88−/− mice reflects the demonstrated role of other receptors that signal via this adaptor, including TLR2 and TLR49 and IL-1 receptors.46 Deletion of TLR2 or TLR4 also prolongs OM.29 Thus TLR9 participates as one component of a broader innate immune response to NTHi that is mediated via MyD88.
Evidence for a complex role of TLR9 is provided by the behavior of cytokines in the ME of TLR9-deficient mice. The expression of cytokine genes is preferentially regulated by NF-κB, which often functions downstream of MyD88. This can include cytokines that are either pro- or anti-inflammatory. We noted significant changes in the expression of mRNA encoding indicator cytokines chosen to represent each category, TNF-α and IL-10, respectively. Initial expression of proinflammatory TNF-α during OM was unchanged in TLR9-null mice, but expression was higher at later times, in a biphasic manner, when compared to wild-type. However, expression of anti-inflammatory IL-10 was also dramatically up-regulated later in OM in the mutants. Interestingly, the peak of IL-10 production in the TLR9−/− ME exactly corresponded with the trough in TNF-α production. These results cannot be explained by a simple model of signaling disruption. They, therefore, underscore the complex interplay of different elements of the innate immune response during the response to NTHi in the ME, and the importance of balance between pro- and anti-inflammatory mediators. The significant delay in bacterial clearance observed in TLR9-null mice indicates that this receptor plays an important role in recovery from OM. However, the strong up-regulation of KLRA17, a negative regulator of TLR9, in wild-type mice suggests that this receptor is tightly regulated, presumably to limit potential damage to tissue.
The existence of parallel DNA signaling pathways in addition to TLR9 further suggests the potential for both redundancy and synergy in the response to pathogen DNA. An example of the latter is provided by IL-1β, the inactive precursor of which is encoded by an important target gene of CpG DNA via TLR9, and is strongly up-regulated during NTHi-induced OM. The simultaneous up-regulation of AIM2 signaling genes which we also observed provides a substrate for formation of the inflammasome, with subsequent cleavage of pro-IL-1 into its active form.
It should be noted that both our gene array and qPCR cytokine data were obtained from homogenates of ME tissue that include a number of mucosal cell types as well as inflammatory cells, and we cannot identify the cellular sources of gene expression. Especially in the case of cytokines, it is likely that infiltrating leukocytes contribute significantly to our data and indeed to ME pathogenesis during OM. However, epithelial cells can also produce all of the cytokines assessed in this study.47
Similarly, while we have discussed DNA signaling in the ME globally, it should be noted that this signaling may vary between different cell types. Given the many different cell types that participate in OM and may be exposed to bacterial DNA, including epithelial and stromal cells as well as many types of leukocytes, the situation in the ME is doubtless complex. Additional research will be required to explore fully the role of bacterial DNA in the pathogenesis and resolution of OM. The development of gene deletion models for other DNA receptors48 will be helpful in this regard.
Conclusions
The results of the present study demonstrate that many genes associated with innate immune responses to pathogen DNA are up-regulated during OM. Thus the substrates for innate immune response to DNA are present in the ME during OM. Moreover, the data also support a significant role for TLR9 activation by NTHi nucleic acids in OM pathogenesis and recovery. Modulation of responses mediated by pathogen DNA may offer new means of treating OM.
Supplementary Material
Acknowledgments
The authors wish to thank Drs Akira and Raz for TLR9−/− mice and Eduardo Chavez for mouse colony maintenance. The authors have no conflict of interest. This work was supported by NIH/NIDCD grants DC000129 and DC006279 and by the VA Research Service. A. Ryan and S. Wasserman contributed equally to the supervision of this work.
Abbreviations
- AIM2
absent in melanoma-2
- ASC
apoptosis-associated speck-like protein containing a caspase recruitment domain
- CFUs
colony-forming units
- DAI
DNA-dependent activator of IFN regulatory factor
- EUs
endotoxin units
- IHC
immunohistochemistry
- IRF
interferon regulatory factor
- ME
middle ear
- MyD88
myeloid differentiation factor 88
- NALP
NACHT-LRR- and pyrin
- NTHi
non-typeable Haemophilus influenzae
- OM
otitis media
- PFA
paraformaldehyde
- Pol-III
RNA polymerase III
- RIG-1
retinoid-inducible gene 1
- TRIF
Tir-domain-containing adaptor inducing interferon β
References
- 1.Infante-Rivard C, Fernandez A. Otitis media in children: frequency, risk factors, and research avenues. Epidemiol Rev. 1993;15:444–465. doi: 10.1093/oxfordjournals.epirev.a036129. [DOI] [PubMed] [Google Scholar]
- 2.Leibovitz E. Acute otitis media in children aged less than 2 years: drug treatment issues. Paediatr Drugs. 2006;8:337–346. doi: 10.2165/00148581-200608060-00002. [DOI] [PubMed] [Google Scholar]
- 3.Klein JO. The burden of otitis media. Vaccine. 2000;19(Suppl 1):S2–S8. doi: 10.1016/s0264-410x(00)00271-1. [DOI] [PubMed] [Google Scholar]
- 4.Seibert JW, Danner CJ. Eustachian tube function and the middle ear. Otolaryngol Clin North Am. 2006;39:1221–1235. doi: 10.1016/j.otc.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Leibovitz E, Jacobs MR, Dagan R. Haemophilus influenzae: a significant pathogen in acute otitis media. Pediatr Infect Dis J. 2004;23:1142–1152. [PubMed] [Google Scholar]
- 6.Luong A, Roland PS. The link between allergic rhinitis and chronic otitis media with effusion in atopic patients. Otolaryngol Clin North Am. 2008;41:311–323. vi. doi: 10.1016/j.otc.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Mandel EM, Doyle WJ, Winther B, Alper CM. The incidence, prevalence and burden of OM in unselected children aged 1–8 years followed by weekly otoscopy through the ‘common cold’ season. Int J Pediatr Otorhinolaryngol. 2008;72:491–499. doi: 10.1016/j.ijporl.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nossal GJ. Kinetics of antibody formation and regulatory aspects of immunity. Acta Endocrinol Suppl (Copenh) 1975;194:96–116. doi: 10.1530/acta.0.080s096. [DOI] [PubMed] [Google Scholar]
- 9.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 10.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 11.Leake ER, Holmes K, Lim DJ, DeMaria TF. Peptidoglycan isolated from nontypeable Haemophilus influenzae induces experimental otitis media in the chinchilla. J Infect Dis. 1994;170:1532–1538. doi: 10.1093/infdis/170.6.1532. [DOI] [PubMed] [Google Scholar]
- 12.DeMaria TF, Apicella MA, Nichols WA, Leake ER. Evaluation of the virulence of nontypeable Haemophilus influenzae lipooligosaccharide htrB and rfaD mutants in the Chinchilla model of otitis media. Infect Immun. 1997;65:4431–4435. doi: 10.1128/iai.65.11.4431-4435.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenstiel P, Jacobs G, Till A, Schreiber S. NOD-like receptors: ancient sentinels of the innate immune system. Cell Mol Life Sci. 2008;65:1361–1377. doi: 10.1007/s00018-008-7502-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creagh EM, O'Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Ketterer MR, Shao J, Hornick D, Buscher B, Bandi V, Apicella M. Infection of primary human bronchial epithelial cells by Haemophilus influenzae: macropinocytosis as a mechanism of airway epithelial cell entry. Infect Immun. 1999;67:4161–4170. doi: 10.1128/iai.67.8.4161-4170.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swords WE, Buscher B, Ver Steeg I, et al. Non-typeable Haemophilus influenzae adhere to and invade human bronchial epithelial cells via an interaction of lipooligosaccharide with the PAF receptor. Mol Microbiol. 2000;37:13–27. doi: 10.1046/j.1365-2958.2000.01952.x. [DOI] [PubMed] [Google Scholar]
- 17.Hotomi M, Arai J, Billal D, et al. Nontypeable Haemophilus influenzae isolated from intractable acute otitis media internalized into cultured human epithelial cells. Auris Nasus Larynx. 2010;37:137–1344. doi: 10.1016/j.anl.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 18.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 19.Ivanov S, Dragoi AM, Wang X, et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110:1970–1981. doi: 10.1182/blood-2006-09-044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Creticos PS, Schroeder JT, Hamilton RG, et al. Immunotherapy with a ragweed-Toll-like receptor 9 agonist vaccine. N Engl J Med. 2006;355:1445–1450. doi: 10.1056/NEJMoa052916. [DOI] [PubMed] [Google Scholar]
- 21.Takaoka A, Wang Z, Choi MK, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 22.Bürckstümmer T, Baumann C, Blüml S, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 23.Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065–1072. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez M, Leichtle A, Pak K, et al. Myeloid differentiation primary response gene 88 is required for the resolution of otitis media. J Infect Dis. 2008;198:1862–1869. doi: 10.1086/593213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson AC, Heinzel FP, Diaconu E, et al. Activation of Tolllike receptor (TLR)2, TLR4, and TLR9 in the mammalian cornea induces MyD88-dependent corneal inflammation. Invest Ophthalmol Vis Sci. 2005;46:589–595. doi: 10.1167/iovs.04-1077. [DOI] [PubMed] [Google Scholar]
- 28.De Nardo D, De Nardo CM, Nguyen T, Hamilton JA, Scholz GM. Signaling crosstalk during sequential TLR4 and TLR9 activation amplifies the inflammatory response of mouse macrophages. J Immunol. 2009;183:8110–8118. doi: 10.4049/jimmunol.0901031. [DOI] [PubMed] [Google Scholar]
- 29.Leichtle A, Hernandez M, Pak K, et al. TLR4-mediated induction of TLR2 signaling is critical in the pathogenesis and resolution of otitis media. Innate Immun. 2009;14:205–215. doi: 10.1177/1753425909103170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plitas G, Burt BM, Nguyen HM, Bamboat ZM, DeMatteo RP. Toll-like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J Exp Med. 2008;205:1277–1283. doi: 10.1084/jem.20080162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology. 2010;51:621–632. doi: 10.1002/hep.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melhus A, Ryan AF. A mouse model for acute otitis media. Apmis. 2003;111:989–994. doi: 10.1034/j.1600-0463.2003.1111012.x. [DOI] [PubMed] [Google Scholar]
- 33.Ryan AF, Ebmeyer J, Furukawa M, et al. Mouse models of induced otitis media. Brain Res. 2006;1091:3–8. doi: 10.1016/j.brainres.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Hoshino K, Takeuchi O, Kawai T, et al. Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 35.Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 36.Hsiao A, Ideker T, Olefsky JM, Subramaniam S. VAMPIRE microarray suite: a web based platform for the interpretation of gene expression data. Nucleic Acids Res. 2005;33:W627–W632. doi: 10.1093/nar/gki443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goddard CM, Allard MF, Hogg JC, Walley KR. Myocardial morphometric changes related to decreased contractility after endotoxin. Am J Physiol. 1996;270:H1446–H1452. doi: 10.1152/ajpheart.1996.270.4.H1446. [DOI] [PubMed] [Google Scholar]
- 38.Kurose I, Argenbright LW, Anderson DC, et al. Reperfusion-induced leukocyte adhesion and vascular protein leakage in normal and hypercholesterolemic rats. Am J Physiol. 1997;273:H854–H860. doi: 10.1152/ajpheart.1997.273.2.H854. [DOI] [PubMed] [Google Scholar]
- 39.Furukawa M, Ebmeyer J, Pak K, et al. Jun N-terminal protein kinase enhances middle ear mucosal proliferation during bacterial otitis media. Infect Immun. 2007;75:2562–2571. doi: 10.1128/IAI.01656-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol. 2007;177:265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao JJ, Diesl V, Wittmann T, et al. Bacterial LPS and CpG DNA differentially induce gene expression profiles in mouse macrophages. J Endotoxin Res. 2003;9:237–243. doi: 10.1179/096805103225001431. [DOI] [PubMed] [Google Scholar]
- 42.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 43.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jono H, Xu H, Kai H, et al. Transforming growth factor-beta-Smad signaling pathway negatively regulates nontypeable Haemophilus influenzae-induced MUC5AC mucin transcription via mitogen-activated protein kinase (MAPK) phosphatase-1-dependent inhibition of p38 MAPK. J Biol Chem. 2003;278:27811–27819. doi: 10.1074/jbc.M301773200. [DOI] [PubMed] [Google Scholar]
- 45.Lee HY, Takeshita T, Shimada J, et al. Induction of beta defensin 2 by NTHi requires TLR2 mediated MyD88 and IRAK-TRAF6-p38MAPK signaling pathway in human middle ear epithelial cells. BMC Infect Dis. 2008;8:87. doi: 10.1186/1471-2334-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- 47.Stadnyk AW. Cytokine production by epithelial cells. FASEB J. 1994;8:1041–1047. doi: 10.1096/fasebj.8.13.7926369. [DOI] [PubMed] [Google Scholar]
- 48.Jones JW, Kayagaki N, Broz P, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci USA. 2010;107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.