

Abstract
The SHAPE (Screened Health Assessment and Pacer Evaluation) trial was a 24 month randomized multicenter placebo-controlled study to determine the efficacy of an implantable gastric stimulator (IGS) for weight loss. This report is an investigator-initiated sub-study at one site designed to assess whether IGS affects plasma levels of ghrelin and peptide YY (PYY). The device was implanted in all subjects but was activated in the Treatment group (n = 7, BMI = 41.5 ± 2.0 kg/m2) and remained inactive in the Control (n = 6, BMI = 39.5 ± 1.7 kg/m2) during the first 12 months. IGS was activated in both groups during months 12–24. Fasting venous blood was drawn at months 0, 12, and 24 and an oral glucose tolerance test (OGTT) was performed at month 12. Although there was no difference in weight loss at 6 months (Control: −6.6 ± 1.5% vs. Treatment: −6.2 ± 1.4%), at 24 months the Control group exhibited weight gain from baseline (+2.2 ± 1.5%) that was significantly different from the weight loss in the Treatment group (−1.9 ± 1.4%; P < 0.05). At 12 months, fasting ghrelin was significantly increased (P < 0.05) in the Treatment group (285 ± 35 to 336 ± 35 pg/ml; weight change, −4.9 ± 1.4%), but not in the Control (211 ± 36 to 208 ± 35 pg/ml; weight change, −3.4 ± 1.5%). No significant change was observed in postprandial suppression of plasma ghrelin or in fasting and postprandial PYY levels. In conclusion, IGS does not prevent the increase in fasting plasma ghrelin levels associated with weight loss. Further studies are needed to determine whether changes in technology can improve weight loss and maintenance, perhaps using gut hormones as biomarkers of possible efficacy.
INTRODUCTION
The epidemic of overweight and obesity is a global dilemma, affecting more than 68% of adults in the United States alone (1). Traditional approaches to weight loss, including lifestyle modification and US Food and Drug Administration approved pharmacotherapy, have limited long-term efficacy (1–4). Bariatric procedures yield significant and long-term maintenance of weight loss, up to 10 years after surgery (2). However, not all individuals have access to surgery or desire invasive intervention due to perioperative morbidity and mortality and sequelae from restrictive or malabsorptive physiology (1). A novel device that maintains gastric anatomy, the implantable gastric stimulator (IGS), has been developed as an alternative to bariatric surgery (5).
Data on efficacy of weight loss with this procedure has been variable. European trials including the multicenter nonrandomized LOSS (Laparoscopic Obesity Stimulation Survey) study demonstrated safety and reported 25–40% excess weight loss. Maintenance of weight loss was seen up to 5 years after implantation (6–9). However, the nonrandomized DIGEST (Dual-lead Implantable Gastric Electrical Stimulation Trial) trial, and the randomized US O-01 trial failed to show significant weight reduction (10,11). Some of the differences in outcomes between studies and individual subjects have been attributable to technical difficulties and patient selection. The more recent large randomized multicenter placebo-controlled SHAPE (Screened Health Assessment and Pacer Evaluation) trial, which used prescreening algorithms to select patients with successful weight loss in prior trials, also failed to show significant differences in weight loss after 1 year between Treatment and Control groups (12).
The proposed mechanisms by which IGS activation could induce weight loss have also been debated. Activation of electrodes implanted into the gastric wall theoretically disrupts intrinsic control of gastric motility leading to increased sensation of fullness (5,13–15). In addition to mechanical effects, another hypothesis implicates altered expression of gastrointestinal peptides known to affect feeding behavior and energy metabolism such that the pattern of secretion would favor satiation rather than hunger. Contrary to this hypothesis though, Cigaina and Hirschberg (2003) demonstrated that peripheral levels of satiety hormones after IGS mediated weight loss showed a significant reduction in meal-related cholecystokinin and somatostatin response and basal levels of glucagon-like peptide-1 and leptin (16). However, interpretation of this study is difficult due to the lack of an appropriate control group without gastric pacing.
In addition to satiation peptides, the gut produces signals that increase appetite (17). Ghrelin is a peptide secreted mainly from X/A-like cells in the gastric mucosa. Ghrelin mediates both short-term and long-term effects on energy balance. Plasma levels peak before a meal and decrease once nutrients reach the duodenum (18,19). Ghrelin levels also increase over time with diet induced weight loss (20). This rise in ghrelin after weight reduction is considered to be a counter-regulatory signal to induce hunger and maintain body fat. In fact, it is the absence of an increase in fasting ghrelin levels reported in most studies that is one of the presumed mechanisms responsible for the long-term efficacy of Roux-en-Y gastric bypass surgery (21,22). While the regulation of ghrelin secretion is multifactorial and not entirely understood, the autonomic nervous system—especially the parasympathetic system—plays an important role (23). Excitation of the vagus nerve stimulates ghrelin secretion. Varied results have been reported on the effects of vagotomy in rats on ghrelin secretion ranging from no effect, an acute decrease, a chronic increase or prevention of food deprivation-induced elevation in levels (23–25). It has been shown that IGS impairs physiological gastric electrical activity particularly causing a reduction in signaling from the vagal afferent pathway (5). A potential mechanism for sustained weight loss in IGS treated patients, therefore, may be a failure of ghrelin to increase after weight loss and/or enhancement of meal-induced suppression of ghrelin through a reduction in vagal stimulation. Since electric signals that induce expansion of the fundus may release satiety signals, it is also possible that pacing stimulates secretion of peptide YY (PYY) which is regulated in part by neural stimulation (26,27).
To our knowledge, this is the first study to examine the effects of IGS on peripheral levels of ghrelin and PYY in humans. Specifically, we addressed whether IGS implantation affects levels of plasma ghrelin and PYY in a subset of patients from a single study site in the SHAPE trial. Although the primary endpoint of improved weight loss in the parent study was not achieved, the unique randomized design of this study with a placebo control allowed the examination of hormone levels in patients both with and without IGS activation.
METHODS AND PROCEDURES
Study design
The study design for the multicenter SHAPE trial has been published (12). Briefly, SHAPE was a randomized, placebo-controlled, double-blinded study. Subjects included in the study were 18–65 years of age, with a BMI between 35 and 55 kg/m2. Subjects were asked to undertake the BaroScreen screening algorithm (Medtronic, Minneapolis, MN), a measure developed from data collected during previous studies to select patients who might be most likely to lose ≥15% excess body weight within 12 months. Subjects who met the initial eligibility criteria were given the BaroScreen, and if selected, were considered for enrollment in the study. All subjects signed the Research Subject Information and Consent Form approved by the Columbia University Institutional Review Board.
The device used in this study was the Transneuronix Implantable Gastric Stimulation System. It consists of the Transcend II IGS, Model 8848 Transender Programming System, Model 8898 Software Application Card, and two Transcend Model 9107 Gastric Stimulation Leads. The IGS was implanted in a subcutaneous pocket in the abdomen, while the two leads were implanted in parallel fashion 2.5 cm from the lesser curvature. Fourteen days following IGS implantation, there was a double-blind randomization of all subjects to one of two study groups—the Control (IGS OFF for 12 months) and the Treatment (IGS ON for 12 months) group. All devices were set to a stimulation frequency of 40 Hz, and a standardized burst rate or ON and OFF cycle of ON for 2 seconds and OFF for 3 seconds. For each patient, a stimulation assessment was performed whereby the device settings were systematically increased from an initial voltage of 10.5 volts, until accompanying symptoms such as bloating, nausea, cramping, or the perception of tingling or stimulation were elicited. The voltage was then lowered in 1 volt decrements until the patient became asymptomatic, just under the threshold for being able to sense the device. Although the standard setting for the pulse width was 450 μs, programmers had the option of adjusting the pulse width in addition to the voltage, in order to elicit a response from the patient. Upon the conclusion of the stimulation assessment, the unblinded programmer then turned off the devices for patients randomized to the Control group. Both groups were asked to follow a 500 kcal/day deficit diet, participate in monthly support group meetings and return for monthly interrogation of the IGS device for the first 12 months post-randomization. For the second 12 months, all devices were activated and patients continued to be followed on a monthly basis. A schematic representation of the 2-year study design is presented in Figure 1.
Figure 1.
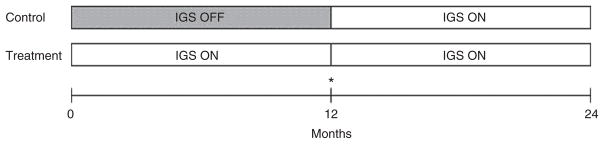
Schematic representation of study design. After implantation, the IGS was activated in Treatment group, but remained inactive in Control. After 12 months, the device was activated in Control. Fasting venous blood was drawn at months 0, 12, and 24. *OGTT was performed at 12 months prior to IGS activation in Control group.
Hormone assays
Hormonal evaluation was performed as a sub-study on consenting patients from the New York-Presbyterian Hospital site of the SHAPE trial. Venous blood was collected in EDTA tubes after an overnight fast of 10–12 h at 0, 12, and 24 months. At 12 months subjects also underwent a 75 g oral glucose tolerance test (OGTT) during which venipuncture was performed at time 0, 30, 60, 90, and 120 min. Blood samples were centrifuged at 4 °C and plasma was stored at −70 °C until assayed in duplicate. Total plasma immunoreactive ghrelin was measured by an RIA kit (Phoenix Pharmaceuticals, Belmont, CA) using 125I-iodinated ghrelin tracer and a rabbit polyclonal antibody against full-length, octanoylated human ghrelin that recognizes the acylated and des-acyl forms of the hormone. The lower limit of detection for this assay was 20 pg/ml, the coefficient of variation was 8.5% within assays and 11.3% between assays. Total plasma levels of PYY were measured using a commercial enzyme-linked immunosorbent assay (Diagnostic Systems Laboratories, Webster, TX) that measures PYY(1–36) and PYY(3–36). The lower limit of detection was 12 pg/ml and the coefficients of variation were 10.1% within and 10.3% between assays. PYY levels are not available for the 24-month follow-up due to discontinuation of this assay kit. Plasma insulin was measured with the Immulite Analyzer with the lower limit of detection of 2 μIU/ml. Plasma leptin was measured with a human RIA kit (LINCO Research, St. Charles, MO) using a 125I-iodinated human leptin tracer.
Statistical analyses
Differences in the distribution of sex between treatment groups was tested with Fisher’s Exact test and between-group differences in continuous variables at baseline were tested with Student’s independent t-test. Longitudinal changes from baseline were tested with linear mixed models with fixed effects for group and time and the group by time interaction; and a compound symmetry within-subject covariance structure. OGTT excursion difference between groups was also assessed with linear mixed models with fixed effect of group, and group by sample time interaction with a spatial power covariance structure for repeated measures. Within-group comparisons between times and between-groups comparisons at specific times were made using model-estimated mean differences and simultaneous 95% confidence intervals. No adjustment was made for multiple comparisons. The strength of association between variables were estimated using Pearson’s correlations. All tests were two-tailed with P values <0.05 considered statistically significant. The area under the curve (AUC) was calculated using the trapezoidal rule. Statistical model-estimated means and standard errors are presented.
RESULTS
Baseline characteristics of subjects randomized to IGS OFF (Control; n = 6) or IGS ON (Treatment; n = 7) are presented in Table 1. The mean age, weight and BMI were similar between groups. Change in body weight and mean percent weight loss at 6, 12, 18, and 24 months are shown in Figure 2. In the Control group, there was a statistically significant percent decrease in weight at 6 (−6.6% ± 1.5%; P < 0.05) and 12 months (−3.4 ± 1.5%; P < 0.05) compared to baseline. Despite IGS activation at month 12, there was continued weight regain for the Control group during the second year such that the decrease in weight at 18 months (−0.9 ± 1.5%) was no longer significant compared to baseline, and in fact there was a small, although not statistically significant, increase in weight from baseline by 24 months (2.2 ± 1.5%). In patients randomized to the Treatment group, there was also significant (P < 0.05) weight loss at 6 (−6.2 ± 1.4%) and 12 (−4.9 ± 1.4%) months. In contrast to the Control group, weight loss was maintained to a greater degree such that at 24 months there was a significant difference in weight change from baseline between the Control (2.2 ± 1.5%) and Treatment groups (−1.9 ± 1.4%; P < 0.05).
Table 1.
Baseline characteristics of study subjects
| Control (IGS OFF) | Treatment (IGS ON) | |
|---|---|---|
| N (M/F) | 2/4 | 1/6 |
| Age (year) | 47.7 ± 1.2 | 49.7 ± 3.7 |
| Wt (kg) | 112.3 ± 6.3 | 113.7 ± 8.5 |
| BMI (kg/m2) | 39.5 ± 1.7 (35–45) | 41.5 ± 2.0 (36–48) |
Data are represented as mean ± SEM. The range of BMI is presented in parentheses.
Figure 2.
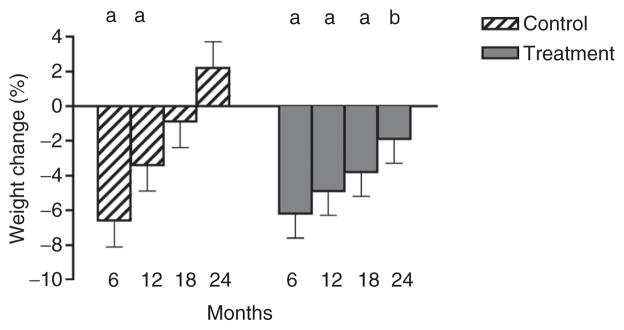
Mean percent change in body weight at 0, 6, 12, 18, and 24 months. aP < 0.05 compared with baseline within group. bP < 0.05 comparing the same time point between groups.
Fasting ghrelin levels at baseline were similar between groups (P = 0.14) and did not change significantly throughout the 2-year study period in the Control group (Table 2). In the Treatment group, fasting ghrelin levels were significantly greater at 12 months when compared both to baseline value within the group (336 ± 35 vs. 283 ± 35 pg/ml, P < 0.05) and to the 12 month time point in the Control group (208 ± 35 pg/ml, P < 0.05). At 12 months, other hormones that could potentially affect ghrelin levels such as insulin (24.3 ± 7.9 vs. 17.9 ± 8.3 μU/ml) and leptin (29.3 ± 5.6 vs. 25.6 ± 5.6 ng/ml) were similar in the fasted state in the Control and Treatment groups, respectively. By month 24, fasting ghrelin levels were not different from baseline in either group. At month 12, the ghrelin response to a 75 g OGTT was not significantly different between the control and treatment groups, respectively: AUC(0–120 min) ghrelin (25.6 ± 4.4 vs. 35.3 ± 4.4 pg × min/ml × 1,000, P = 0.15); and percent postprandial suppression (7.4 ± 3.8 vs. 13.8 ± 3.8%, P = 0.26). Fasting PYY levels were similar between groups throughout the study and were not changed significantly at 12 months in either group (Table 2). The AUC(0–120 min) PYY response to the OGTT was similar in the Control and Treatment groups (27.9 ± 6.8 vs. 26.7 ± 6.8 pg × min/ml × 1,000, P = 0.9).
Table 2.
Fasting plasma ghrelin and PYY levels over time
| 0 m | 12 m | 24 m | |
|---|---|---|---|
| Fasting ghrelin (pg/ml) | |||
| Control | 211 ± 36 | 208 ± 35 | 222 ± 35 |
| Treatment | 283 ± 35 | 336 ± 35ab | 313 ± 35 |
| Fasting PYY (pg/ml) | |||
| Control | 110 ± 56 | 139 ± 54 | na |
| Treatment | 113 ± 52 | 154 ± 52 | |
Values represent the mean ± SEM.
P < 0.05 compared with month 0 within the same group.
P < 0.05 compared with the same time point between groups. na = not available (due to discontinuation of assay kit by manufacturer).
DISCUSSION
In this study, we examined a subset of patients recruited to the New York-Presbyterian Hospital site of the SHAPE trial in order to determine whether IGS activation affects plasma ghrelin and PYY levels. The main end point of the parent study was to determine whether gastric pacing resulted in greater weight loss at 1 year compared with subjects with an implanted device that was not activated. In our subjects at 6 and 12 months, there were no significant differences in weight loss between the Control and Treatment groups, suggesting that life style intervention rather than device activation was the main factor in inducing weight loss over this relatively short-term period. Moreover, the 12–24-month data for the control group, showing progressive weight gain despite IGS activation, again suggests the lack of a weight loss effect from this system.
By month 24, there was an increase in weight from baseline in the control group that was significantly different from the decrease in weight in the treatment group. Indeed, the overall pattern of weight change in the control group over 24 months mimics trends seen in traditional obesity programs addressing lifestyle changes (3). Although it is conceivable that greater maintenance of weight loss at 24 months in the Treatment group may have been due to a longer period of IGS activation, it is also likely that confounding variables, such as differences in adherence to life style interventions, contributed to different outcomes due to small sample size.
The hypothesis of this hormonal sub-study was that IGS would mimic the predicted effects of chronic vagal inhibition on fasting and meal-related gut hormone secretion. Specifically, we anticipated that weight loss with chronic stimulation would not be associated with an increase in fasting ghrelin levels and would be associated with exaggerated nutrient-induced secretion of PYY. However, in contrast to this hypothesis, with significant weight loss in the first 12 months of IGS activation there was also a significant increase in fasting ghrelin levels, indicating that IGS activation does not inhibit ghrelin secretion. It is unlikely that IGS activation could have actually caused the observed increase in ghrelin levels, given that ghrelin levels trended back toward baseline with some weight regain during continued stimulation in the second year. Furthermore, there was no increase in ghrelin levels in the Control group upon activation of IGS. No effect was observed with activation on fasting or postprandial levels of PYY.
Assessment of ghrelin response after OGTT measured by AUC and ghrelin suppression also failed to show significant differences, suggesting lack of effect from IGS activation. Our results are in agreement with a shorter-term study showing an increase in fasting ghrelin levels that correlated with weight loss after 6 months of IGS activation (28), but are not in agreement with results from a second study showing unchanged ghrelin levels (8). The latter study, however, measured only the acylated form of ghrelin which may have been subject to cleavage by esterases in the absence of specific inhibitors at the time of sample collection (29).
Results from this small sub-study suggest that IGS activation does not augment weight loss and are in agreement with the parent SHAPE trial that reported weight loss up to a period of 1 year. Results from the second year of the parent study have not been published. Of note, settings of the gastric pacemaker in the SHAPE study were adjusted in each individual to just below parameters that caused nausea or bloating. Another potential approach to gastric pacing would be intermittent or meal-related pacing as opposed to chronic stimulation. It has been shown that the pattern of brain activity measured by positron emission tomography and labeled glucose in patients with chronic IGS activation differs from that reported with acute vagal stimulation by gastric distention via balloon inflation (30). Therefore, intermittent as opposed to chronic pacing may result in different effects regarding both brain activation and gut hormone release. Perhaps in the future, physiological markers such as a decrease in ghrelin or an increase in PYY plasma concentrations could be considered to help guide optimal sites for lead placement and parameters for stimulation to ultimately improve clinical outcomes for what still may be a promising weight loss treatment.
Acknowledgments
This report was an investigator-initiated sub-study funded by the NIH/NIDDK (RO1DK 072011 to J.K.). The investigators would like to acknowledge Carmen Taveras for research assistance and Irene M. Conwell for technical assistance.
Footnotes
DISCLOSURE
The SHAPE trial was sponsored by Medtronic/Transneuronix.
References
- 1.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 2.Bult MJ, van Dalen T, Muller AF. Surgical treatment of obesity. Eur J Endocrinol. 2008;158:135–145. doi: 10.1530/EJE-07-0145. [DOI] [PubMed] [Google Scholar]
- 3.Franz MJ, VanWormer JJ, Crain AL, et al. Weight-loss outcomes: a systematic review and meta-analysis of weight-loss clinical trials with a minimum 1-year follow-up. J Am Diet Assoc. 2007;107:1755–1767. doi: 10.1016/j.jada.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 4.Freedman DS, Khan LK, Serdula MK, Galuska DA, Dietz WH. Trends and correlates of class 3 obesity in the United States from 1990 through 2000. JAMA. 2002;288:1758–1761. doi: 10.1001/jama.288.14.1758. [DOI] [PubMed] [Google Scholar]
- 5.Chen J. Mechanisms of action of the implantable gastric stimulator for obesity. Obes Surg. 2004;14 (Suppl 1):S28–S32. doi: 10.1007/BF03342135. [DOI] [PubMed] [Google Scholar]
- 6.Cigaina V. Long-term follow-up of gastric stimulation for obesity: the Mestre 8-year experience. Obes Surg. 2004;14 (Suppl 1):S14–S22. doi: 10.1007/BF03342133. [DOI] [PubMed] [Google Scholar]
- 7.D’Argent J. Gastric electrical stimulation as therapy of morbid obesity: preliminary results from the French study. Obes Surg. 2002;12 (Suppl 1):21S–25S. doi: 10.1381/096089202762552638. [DOI] [PubMed] [Google Scholar]
- 8.De Luca M, Segato G, Busetto L, et al. Progress in implantable gastric stimulation: summary of results of the European multi-center study. Obes Surg. 2004;14 (Suppl 1):S33–S39. doi: 10.1007/BF03342136. [DOI] [PubMed] [Google Scholar]
- 9.Miller K, Hoeller E, Aigner F. The Implantable Gastric Stimulator for Obesity: An Update of the European Experience in the LOSS (Laparoscopic Obesity Stimulation Survey) Study. Treat Endocrinol. 2006;5:53–58. doi: 10.2165/00024677-200605010-00006. [DOI] [PubMed] [Google Scholar]
- 10.Shikora SA. Implantable gastric stimulation for the treatment of severe obesity. Obes Surg. 2004;14:545–548. doi: 10.1381/096089204323013596. [DOI] [PubMed] [Google Scholar]
- 11.Shikora SA. “What are the yanks doing?” the U.S. experience with implantable gastric stimulation (IGS) for the treatment of obesity - update on the ongoing clinical trials. Obes Surg. 2004;14 (Suppl 1):S40–S48. doi: 10.1007/BF03342137. [DOI] [PubMed] [Google Scholar]
- 12.Shikora SA, Bergenstal R, Bessler M, et al. Implantable gastric stimulation for the treatment of clinically severe obesity: results of the SHAPE trial. Surg Obes Relat Dis. 2009;5:31–37. doi: 10.1016/j.soard.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 13.Cigaina V. Gastric pacing as therapy for morbid obesity: preliminary results. Obes Surg. 2002;12 (Suppl 1):12S–16S. doi: 10.1007/BF03342141. [DOI] [PubMed] [Google Scholar]
- 14.Cigaina VV, Pinato G, Rigo VV, et al. Gastric Peristalsis Control by Mono Situ Electrical Stimulation: a Preliminary Study. Obes Surg. 1996;6:247–249. doi: 10.1381/096089296765556845. [DOI] [PubMed] [Google Scholar]
- 15.Yin J, Chen JD. Retrograde gastric electrical stimulation reduces food intake and weight in obese rats. Obes Res. 2005;13:1580–1587. doi: 10.1038/oby.2005.194. [DOI] [PubMed] [Google Scholar]
- 16.Cigaina V, Hirschberg AL. Gastric pacing for morbid obesity: plasma levels of gastrointestinal peptides and leptin. Obes Res. 2003;11:1456–1462. doi: 10.1038/oby.2003.195. [DOI] [PubMed] [Google Scholar]
- 17.Berthoud HR. Vagal and hormonal gut-brain communication: from satiation to satisfaction. Neurogastroenterol Motil. 2008;20 (Suppl 1):64–72. doi: 10.1111/j.1365-2982.2008.01104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castañeda TR, Tong J, Datta R, Culler M, Tschöp MH. Ghrelin in the regulation of body weight and metabolism. Front Neuroendocrinol. 2010;31:44–60. doi: 10.1016/j.yfrne.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 19.Cummings DE, Overduin J. Gastrointestinal regulation of food intake. J Clin Invest. 2007;117:13–23. doi: 10.1172/JCI30227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy KG, Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature. 2006;444:854–859. doi: 10.1038/nature05484. [DOI] [PubMed] [Google Scholar]
- 21.Korner J, Inabnet W, Febres G, et al. Prospective study of gut hormone and metabolic changes after adjustable gastric banding and Roux-en-Y gastric bypass. Int J Obes (Lond) 2009;33:786–795. doi: 10.1038/ijo.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vincent RP, le Roux CW. Changes in gut hormones after bariatric surgery. Clin Endocrinol (Oxf) 2008;69:173–179. doi: 10.1111/j.1365-2265.2007.03164.x. [DOI] [PubMed] [Google Scholar]
- 23.Yin X, Li Y, Xu G, An W, Zhang W. Ghrelin fluctuation, what determines its production? Acta Biochim Biophys Sin (Shanghai) 2009;41:188–197. doi: 10.1093/abbs/gmp001. [DOI] [PubMed] [Google Scholar]
- 24.Williams DL, Grill HJ, Cummings DE, Kaplan JM. Vagotomy dissociates short- and long-term controls of circulating ghrelin. Endocrinology. 2003;144:5184–5187. doi: 10.1210/en.2003-1059. [DOI] [PubMed] [Google Scholar]
- 25.Hosoda H, Kangawa K. The autonomic nervous system regulates gastric ghrelin secretion in rats. Regul Pept. 2008;146:12–18. doi: 10.1016/j.regpep.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Lin HC, Taylor IL. Release of peptide YY by fat in the proximal but not distal gut depends on an atropine-sensitive cholinergic pathway. Regul Pept. 2004;117:73–76. doi: 10.1016/j.regpep.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 27.Onaga T, Zabielski R, Kato S. Multiple regulation of peptide YY secretion in the digestive tract. Peptides. 2002;23:279–290. doi: 10.1016/s0196-9781(01)00609-x. [DOI] [PubMed] [Google Scholar]
- 28.Cigaina V, Hirschberg AL. Plasma ghrelin and gastric pacing in morbidly obese patients. Metabolism. 2007;56:1017–1021. doi: 10.1016/j.metabol.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Prudom CE, Nass R, et al. Novel ghrelin assays provide evidence for independent regulation of ghrelin acylation and secretion in healthy young men. J Clin Endocrinol Metab. 2008;93:1980–1987. doi: 10.1210/jc.2007-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang GJ, Yang J, Volkow ND, et al. Gastric stimulation in obese subjects activates the hippocampus and other regions involved in brain reward circuitry. Proc Natl Acad Sci USA. 2006;103:15641–15645. doi: 10.1073/pnas.0601977103. [DOI] [PMC free article] [PubMed] [Google Scholar]