

1. Introduction
Vascular-endothelial growth-factor (receptor) (VEGF)(R)-inhibiting agents – sunitinib [1–3], sorafenib [4,5], pazopanib [2,6], bevacizumab [7,8], axitinib [5] and tivozanib [9,10] – have changed the therapeutic landscape in metastatic renal-cell carcinoma (mRCC). Five out of six agents have been approved for either first-line (sunitinib, pazopanib and bevacizumab + interferon-alpha) or second-line (sorafenib, axitinib) treatment of metastatic or advanced RCC. With these novel strategies, the median overall survival of patients has increased considerably, often, however, at the expense of chronic side-effects. Common treatment-related side-effects include: (1) general symptoms such as fatigue and asthenia, (2) gastrointestinal symptoms such as diarrhoea and stomatitis, (3) skin toxicities, (4) cardiovascular toxicities and (5) a variety of laboratory abnormalities. Some of these side-effects are clinically highly relevant because they may jeopardise the patient’s safety or quality of life, while others may have little clinical relevance. Treating physicians need to be aware of potential side-effects that may occur, how to prevent and/or manage them, and the clinical implications for the ongoing treatment. This is of paramount importance since dose reductions and treatment discontinuations may significantly affect the outcome [11].
2. Incidence of toxicities associated with VEGF inhibitors
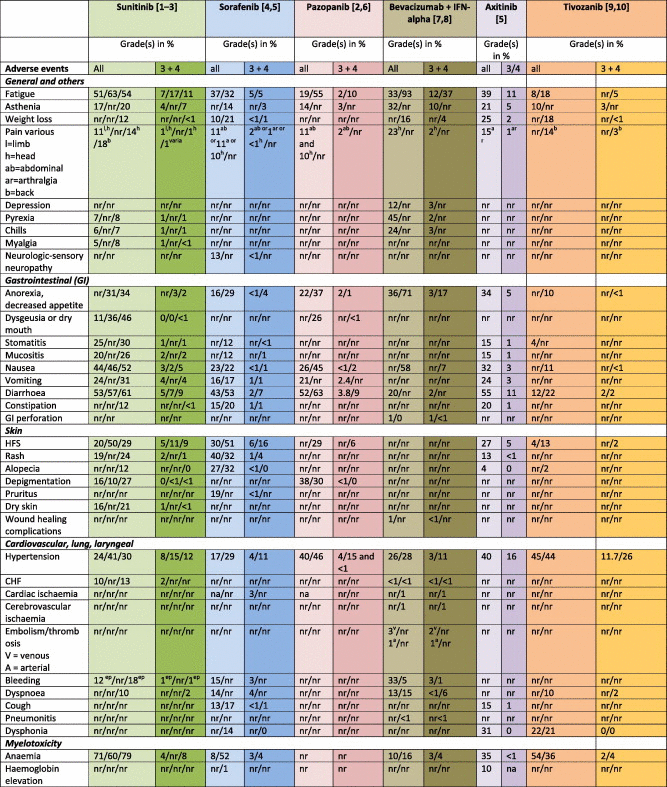
Toxicities reported from VEGFR inhibitors in mRCC are outlined in Table 1. Among general symptoms, fatigue (and/or asthenia) has been most commonly reported for sunitinib (up to 63% all grades; grade ⩾3: 17%), followed by axitinib (all grades 39%, grade ⩾3: 11%) and sorafenib (all grades 37%; grade ⩾3: 5%). A high incidence of fatigue has also been reported from the combination of bevacizumab + interferon-alpha (IFNα). However, the incidence of fatigue appears to be low in patients being treated with bevacizumab alone [12,13]; thus, this side-effect may be attributed to IFNα rather than bevacizumab. Interestingly, the newest VEGFR–tyrosine kinase inhibitor (TKI) tivozanib appears to have little effect on fatigue levels (all grades: up to 18%, grade ⩾3: 5%).
Table 1.
Toxicities reported from phase III trials (%).
 |
 |
Gastrointestinal side-effects are extremely common in patients on VEGFR–TKI treatment. In particular, sunitinib and axitinib were shown to cause reduced appetite and/or anorexia in up to 34% of the patients. In the case of sunitinib, this may be caused partly by the high incidence of stomatitis and/or dysgeusia (30% and 46%, respectively). Diarrhoea is another frequent gastrointestinal toxicity: high incidences of all grades of diarrhoea were reported from patients on sunitinib (61%), sorafenib (53%), pazopanib (63%) and axitinib 55%, again with quite a favourable profile for tivozanib (22%).
The most common skin toxicities caused by VEGFR inhibitors are hand–foot syndrome (HFS), skin- and/or hair-depigmentation and rash. The highest incidence of all grades of HFS has been reported from sorafenib (51%) and sunitinib patients (50%), with a higher grade 3 + 4 HFS incidence in sorafenib patients (16%). Similarly, sorafenib was shown to cause rash in up to 32% of patients (all grades). Hair and/or skin depigmentation is commonly observed in patients on pazopanib (up to 38%) and sunitinib (up to 27%).
Among the group of cardiovascular, lung and laryngeal side-effects, hypertension is the most common (up to 46%). Hypertension has been observed with all of these agents and has been considered a fairly reliable biomarker for response, progression-free survival (PFS) and overall survival (OS) [14]. The highest incidence of grades 3 + 4 hypertension has been observed with tivozanib (26%). Cardiac side-effects include congestive heart failure (sunitinib: 13%) and ischaemia or myocardial infarction (sorafenib: 3%; bevacizumab + interferon-alpha 1%). Bleeding events, most commonly epistaxis, have been observed in patients treated with bevacizumab + IFN, sunitinib and sorafenib (33%, 18% and 15%, respectively). While dyspnoea is a common side-effect of mTOR-inhibitors, the incidence is low in patients with VEGFR inhibitors. No direct effect of these agents on lung tissue has been reported so far; thus, the occurrence of dyspnoea might be a secondary event due to lung metastases or oedema as a result of high-grade hypertension or congestive heart failure. In contrast, dysphonia is a common side-effect of new-generation TKIs such as axitinib (31%) and tivozanib (22%).
The incidence of grade 3 + 4 myelotoxicity is low with VEGFR inhibitors when compared to classical cancer treatment such as chemotherapy. Nevertheless, multikinase inhibitors, particularly sunitinib, may induce grade 3 + 4 anaemia (8%), neutropenia (18%), thrombocytopenia (9%) and lymphopaenia (18%). Infections, however, have not been reported yet.
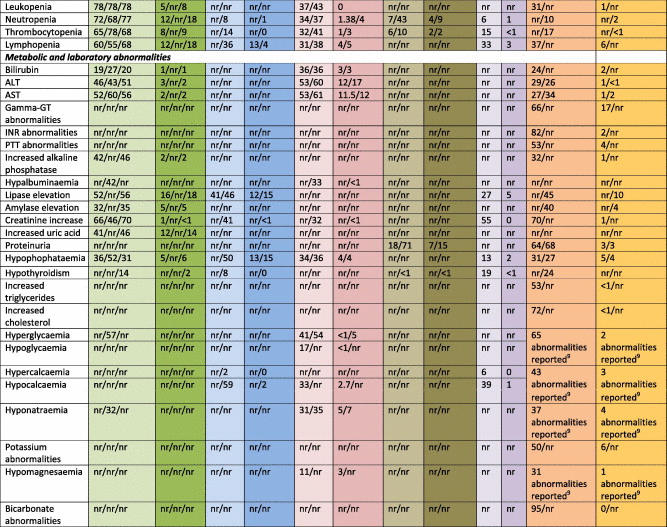
Various metabolic and laboratory abnormalities have been shown to occur in patients treated with VEGFR inhibitors. These include renal and electrolyte abnormalities such as creatinine increase (up to 70%), proteinuria (71%), abnormalities in sodium, potassium, magnesium and calcium levels in up to 37%, 50%, 31% and 59%, respectively. In addition, pazopanib, sunitinib and tivozanib in particular were shown to cause increased levels of bilirubin, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in up to 36%, 60% and 61% of the patients, respectively. In the case of tivozanib, international normalised ratio (INR) and partial thromboplastin time (PTT) abnormalities were also reported (82% and 53%, respectively). Sunitinib, sorafenib and tivozanib were shown to increase amylase and lipase levels in approximately 50% of patients. Finally, the induction of hypothyroidism is an observation that may have been underreported in the pivotal trials; in later analyses, up to 36% of patients was shown to develop hypothyroidism [15]. As with hypertension, the occurrence of hypothyroidism has been linked to a better outcome [15,17].
3. The severity of side-effects and their impact on outcome
The occurrence of grade 3 + 4 toxicities, and to some extent also toxicities of lower grades, may tempt clinicians to reduce the dose, or to interrupt or discontinue treatment. Table 2 outlines the incidence of dose reductions and interruptions, the rate and most common reasons for treatment discontinuations, as well as the most common toxicities that have led to death. Dose reductions occurred in up to 51% of sunitinib patients, 52% of sorafenib patients, 44% of pazopanib patients, 31% of axitinib patients and 14% of tivozanib patients, while no dose reductions of bevacizumab were permitted in the Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial (AVOREN) and CALGB bevacizumab trials. Similarly, dose interruptions and treatment delays were required in 38% of sunitinib patients, 80% sorafenib patients, 62% bevacizumab + IFN patients and 77% of axitinib patients. In contrast, the number of patients with dose interruptions was considerably lower in tivozanib patients (18%). Both dose reductions and treatment interruption may help to prevent or manage treatment-associated side-effects. However, several authors have shown that higher relative dose intensities were associated with better outcome. A pharmacodynamic/pharmakokinetic analysis including six sunitinib trials revealed that response rates, time to progression and overall survival increased with the mean daily exposure to sunitinib [11]. Similarly, intra-patient dose-escalated sorafenib was shown to exert promising antitumour activity and led to a complete/partial response (CR–PR) rate of 48%, with eight out of 44 patients achieving complete remission [18]. Finally, Rini et al. could demonstrate in a randomised phase II study that axitinib dose titration significantly improved overall response rates when compared to placebo in patients eligible for dose titration [19]. The challenge in the management of mRCC patients is to have a balanced approach to maintaining both dose intensity and safety of the patient.
Table 2.
Dose reductions and treatment discontinuation due to adverse events.
| Sunitinib [1–3] | Sorafenib [4,5] | Pazopanib [2,6] | Bevacizumab [7,8] | Axitinib [5] | Tivozanib [9,10] | |
|---|---|---|---|---|---|---|
| Dose reduction due to AEs (%) | 32 [1] 51 [2] 50 [3] |
13 [4] 52 [5] |
44 [2] | Not permitted for bevacizumab | 31 | 8 [9] 13.9 [10] |
| Dose interruptions (treatment delays) | 38 [1] | 21 [4] for dermatological events, 80 for GI events [5] | nr | nr [7] 61.7 [8] |
77 | 4 [9] 18 [10] |
| Discontinuation due to AEs | 8 [1] 19 [2] |
10 [4] 8 [5] |
12 [6] 24 [2] |
28 [7] any 19 [7] bevacizumab 23 [8] |
4 | 9 [9] 4 [10] |
| Most common reason for discontinuation | Cytopaenia [2] | Constitutional [4], gastrointestinal [4], dermatologic [4], respiratory tract symptoms [4] HFS [5], diarrhoea [5], asthenia [5] |
Liver events [2] | nr | Fatigue, and transient ischaemic attack | nr [9,10] |
| AEs leading to hospitalisation | 34 [4] | Nr | nr | nr | nr [9,10] | |
| TX-related death or non-PD related deaths | Renal failure [1]n = 1 Gastric haemorrhage [1]n = 1 Respiratory failure [1]n = 1 Sudden death [1]n = 1 |
2 death from cardiac ischaemia/infarction [4] 2 death [5] from tumour necrosis causing retroperitoneal bleeding and n = 1 GI bleeding |
4 [6] ischaemic stroke, hepatic failure, rectal haemorrhage, peritonitis and bowel perforation |
2 [7] bleeding events n = 2, GI perforation n = 1, myocardial infarction n = 1 atrial fibrillation n = 1 pneumonia n = 1 hepatic failure (history of active hepatitis B) n = 3 [8] |
0 |
n[9]=8/272 ischaemic stroke n = 2, acute coronary syndrome, acute respiratory failure, cerebral vascular accident, hypotension and pulmonary embolism all n = 1 nr [10] |
nr, not reported.
4. Reasons for and incidence of dose adjustments and treatment discontinuation by agent
4.1. Sunitinib
In the final analysis of sunitinib versus IFN-alpha [3], 51% and 19% of sunitinib patients were reported to have required dose reductions or treatment discontinuation, respectively, because of adverse events. Causes of death apart from disease progression included acute renal failure (n = 1), gastric haemorrhage (n = 1), respiratory failure (n = 1) and sudden death (n = 1). In the COMPARZ trial, the most common reason for sunitinib discontinuation was cytopaenia (3%).
4.2. Pazopanib
In the pivotal pazopanib trial [6] 33% and 4% of patients experienced grade 3 and 4 toxicities, respectively. The adverse event (AE) profile was similar in treatment-naïve and cytokine-pretreated patients, although discontinuation rates because of AEs were higher in the cytokine-pretreated (19%) compared with the treatment-naïve patients (12%). Arterial thromboembolic events occurred in 3% of pazopanib patients – myocardial infarction or ischaemia 2%, cerebrovascular accident <1% and transient ischaemic attack (TIA) <1% – compared with none in the placebo arm. The incidence of all-grade haemorrhagic events was 13% in the pazopanib arm versus 5% in the placebo arm. Deaths from adverse events were reported in 4% of pazopanib patients and in 3% in the placebo arm. Four pazopanib patients (1%) had fatal adverse events, including ischaemic stroke, abnormal hepatic function, rectal haemorrhage and peritonitis/bowel perforation. In the COMPARZ trial [2] dose reductions and treatment discontinuations occurred in 44% and 24% of patients, respectively. The most common reasons for pazopanib discontinuation were liver events (6%).
4.3. Sorafenib
In the TARGET trial [4] 10% of sorafenib patients had to discontinue the treatment, mostly because of constitutional, gastrointestinal, dermatological or pulmonary upper respiratory tract symptoms. Dose reductions occurred in 13% of sorafenib patients versus 3% of placebo patients (P < 0.001), and dose interruptions because of adverse events occurred in 21% of sorafenib patients versus 6% of placebo patients (P < 0.001). Dose interruptions were mostly because of HFS, rash or diarrhoea. Cardiac ischaemia occurred in 12 sorafenib patients (3%) and two patients in the placebo groups (<1%), (P = 0.01). Bleeding was more frequent in sorafenib than in placebo patients (15% versus 8%, respectively). In the axitinib phase III trial [5], the most frequent grade-3 and -4 adverse events associated with sorafenib were HFS, hypophosphataemia, lipase elevation and hypertension. Two treatment-related deaths occurred in sorafenib patients and were caused by necrosis with retroperitoneal bleeding and gastrointestinal haemorrhage.
4.4. Bevacizumab + IFN
In the AVOREN trial [7], serious adverse events occurred in 29% of patients who received bevacizumab versus 16% of those who did not. Similarly, AEs requiring treatment discontinuation were more frequent in bevacizumab-treated patients versus placebo patients (28% versus 12%). Grade 3 and 4 AEs in patients who received bevacizumab included four gastrointestinal perforations (1%; grade 4: n = 3) and 10 thromboembolic events (3%; grade 4, n = 4). Moreover, seven (2%) and 5% of bevacizumab patients discontinued treatment because of hypertension and proteinuria, respectively. Deaths due to AEs were reported in 2% of patients who received bevacizumab and 2% who did not. Three deaths (<1%) among the patients who received bevacizumab (n = 2 bleeding events and n = 1 gastrointestinal perforation) were thought to be associated with treatment. The other causes of death of bevacizumab-treated patients included myocardial infarction, atrial fibrillation, pneumonia, hepatic failure (in a patient with a history of hepatitis B) and staphylococcal sepsis.
In the CALGB trial [8] 80% of patients receiving bevacizumab experienced grade ⩾3 toxicities compared with 63% IFN patients (P < 0.001). Bevacizumab resulted in significantly more grade ⩾3 hypertension (11% versus 0), anorexia (17% versus 8%), fatigue (37% versus 30%) and proteinuria (15% versus <1%). There were four treatment-related deaths in the IFN arm and three in the bevacizumab arm.
4.5. Axitinib
In the axitinib phase III trial [5], the most common AEs of grade ⩾3 were hypertension, diarrhoea and fatigue; 32% of axitinib patients had elevations in thyroid-stimulating hormone (TSH) and 27% required initiation or dose adjustments of thyroid hormone replacement. No treatment-related deaths occurred in the axitinib arm.
4.6. Tivozanib
In the phase II randomised discontinuation trial on tivozanib, the most common treatment-related toxicities were hypertension and dysphonia which occurred in 45% and 22% of patients, respectively. Grade 3 and 4 toxicities were rare and included hypertension (grade 3: 11%, grade 4: 1%) and laboratory abnormalities (5%). Dose reductions and interruptions were deemed necessary in 8% and 4% of patients, respectively. Treatment discontinuation because of adverse events occurred in 9% of patients. Causes of death that were not attributed to disease progression included ischaemic stroke (n = 2), coronary syndrome, respiratory failure, cerebral vascular accident, hypotension and embolism (n = 1 each); however, none of these were associated with tivozanib treatment.
Understanding the pathophysiology of individual toxicities, and developing proactive strategies for their prevention and treatment are important aspects of disease management and may avoid dose reductions and treatment discontinuation.
5. Pathophysiology of selected side-effects (involved targets) and management
Targeted agents differ regarding their side-effect profile, and these differences may be attributed to the mode of action; the incidence and severity of side-effects may depend on the number of inhibited targets (single versus multikinase inhibitors), the type of inhibited target (VEGF inhibition versus platelet-derived growth factor, PDGF, inhibition versus Flt-3 inhibition, etc.) and the strength of target inhibition (affinity to the tyrosine kinase, ‘on- and off-target’ toxicities). While some toxicities have been linked to the inhibition of a specific target, e.g. hypertension and VEGF, the association of other side-effects with a particular target is less clear (e.g. stomatitis, diarrhoea). Moreover, the incidence and severity of side-effects may differ between patient populations depending on single-nucleotide polymorphisms.
5.1. Fatigue, asthenia
5.1.1. Mechanisms
Fatigue and to a slight extent asthenia are frequent symptoms in patients undergoing treatment with VEGF inhibitors. In 1998, Cella and colleagues described fatigue as a ‘subjective state of overwhelming and sustained exhaustion and decreased capacity for physical and mental work that is not relieved by rest’ [20]. Fatigue induced by VEGF inhibitors may indeed include more symptoms than tiring easily. Patient’s description of what their fatigue involves includes a loss of social interest, reduced voluntariness for physical activity, cognitive disorders, reduced appetite and depressive symptoms. Together, these symptoms have been described as sickness behaviour, a common status in patients with acute or chronic diseases [21]. In patients treated with VEGF inhibitors, many factors appear to contribute to fatigue and sickness behaviour. These include several biological processes related to the patient and/or the disease in which inflammation appears to have a major role. Moreover, treatment-related side-effects may contribute to fatigue and asthenia.
The underlying mechanisms appear to involve many aspects: (1) the individual genome of the patient, (2) disease-related factors, either biological or as a result of behavioural and psychological factors that may arise during the disease, (3) cancer treatment itself, which may induce fatigue either on the basis of the specific mode of action or secondarily by leading to side-effects that may be associated with the symptom of fatigue.
Several studies have investigated the relationship between genomic markers and fatigue. An association was found between functional interleukin-6 (IL-6) polymorphism and fatigue in patients and their relatives [22], while Aouizerat and colleagues found evidence for a genetic association between tumour necrosis factor alpha (TNFα) and the severity of sleep disturbances and morning fatigue [23]. Furthermore, single-nucleotide polymorphisms (SNPs) of several cytokines – including Interleukin-1β, IL-1RN and IL-10 – showed associations with fatigue levels in lung cancer patients [24]. So far, no genomic markers for fatigue have been identified in patients undergoing VEGF inhibitor treatment.
Fatigue has also been strongly correlated with depression in cancer patients [25], and increased stress-induced inflammatory responses were observed in patients with major depression and stress [26]. Moreover, acute psychological stress was shown to influence pro-inflammatory cytokines [27]. Finally, physical inactivity and increased body mass index have been associated with fatigue in patients with breast cancer [28]. Again, no data have been obtained so far with respect to patients treated with VEGF inhibitors.
Many tumour types have been shown to promote progression though inflammatory cells [29]. Chemokines and cytokines were shown to attract immune cells to tumour cells [30], thereby leading to immune-cell dysfunction [31]. Research on neuro-immune signalling has linked pro-inflammatory cytokines with the development of fatigue. For instance, sickness has been shown to be triggered by IL-1α and β, TNFα and IL-6 [32–34]. Although no studies have been undertaken in patients undergoing anti-VEGF-treatment, IL-6 levels have been linked to disease progression in patients with mRCC [35].
Cancer treatment itself is known to induce fatigue. Induction of chronic inflammation has also been recognised as a treatment-related factor that may induce fatigue through pro-inflammatory cytokines produced by monocytes [36]. Wang et al. measured markers of inflammation in patients with gastrointestinal cancers undergoing chemotherapy and their relationship with fatigue. They found that serum concentrations of TNF-R1 were associated with the severity of fatigue [37]. Similar findings have been reported by various other authors with regards to C-reactive protein (CRP) levels, IL-6, etc. It is currently unknown as to whether VEGFR inhibitors can trigger fatigue through inflammation.
Various side-effects associated with VEGF inhibitors may contribute to fatigue and sickness behaviour; persistent fatigue is a common symptom of patients with hypothyroidism [38] and several VEGF inhibitors have been shown to induce hypothyroidism (Table 1). Similarly, hypophosphataemia is frequently observed in patients treated with VEGF inhibitors. Although a low serum phosphate level does not necessarily correlate with clinically relevant total body phosphate depletion, it should be considered that hypophosphataemia may cause muscle weakness [39]. Muscular dysfunction has also been shown to involve the heart [40] by causing myocardial changes and a reversible reduced sensitivity to catecholamines [40]. Muscle weakness may also be explained by an impaired muscle glucose uptake. Muscle activity involves an increased rate of glucose uptake in the contracting muscle. This is enabled by glucose transporter recruitment [41]. Multikinase inhibitors may interfere with signals required for this process, such as protein kinase C, nitric oxide, etc. [42]. Furthermore, TKI-induced hypoglycaemia may also contribute to muscular weakness. Sunitinib, for instance, was shown to decrease blood glucose levels [43]. In a clinical study that sought to distinguish between a central and peripheral cause of fatigue, Yavuszen et al. [44] found that patients with cancer-related fatigue had a greater central fatigue, indicated by shorter endurance time. Central fatigue has been linked to a loss of voluntarily activated muscles because of mechanisms proximal to the neuromuscular junction. On the other hand, muscle fatigue may have a peripheral cause, e.g. due to metabolic changes within the muscles. Antoun et al. [45] could demonstrate that treatment with sorafenib exacerbates excessive muscle loss and that this loss increased during the course of treatment. A possible explanation for this phenomenon is that VEGF inhibition leads to downstream inhibition of AKT and mTOR, which are of paramount importance for skeletal muscle hypertrophy and muscle protein synthesis [46].
Finally, malnutrition as a result of VEGF-inhibitor-induced anorexia, anaemia and dehydration caused by diarrhoea may account for the high incidence of fatigue, asthenia and sickness behaviour in patients undergoing VEGF inhibitor treatment.
5.1.2. Management
Effective treatment for fatigue includes pharmaceutical and non-pharmaceutical interventions.
5.1.2.1. Non-pharmacological interventions
Patterson and colleagues [47] recently reviewed studies that looked at the impact of eight different types of intervention on fatigue levels. These interventions included (1) psycho-education, (2) cognitive behavioural therapy, (3) exercise combined with education and support, (4) exercise alone, (5) acupressure, (6) energy conservation and activity management, (7) relaxation breathing exercises and (8) distraction. With the exception of cognitive behavioural therapy, all of the above-mentioned non-pharmacological interventions were found to effectively reduce fatigue in patients with various diseases. In particular the impact of exercise has been well studied in patients with cancer-related fatigue. A Cochrane analysis [48] recently revealed that among 56 studies including 4068 participants aerobic exercise significantly reduced fatigue while resistance training and alternative forms of exercise did not. In contrast, Strasser et al. [49] performed a meta-analysis on the impact of resistance training in cancer survivors and found an association between resistance training and positive effects on muscular function and body composition. Finally, a meta-analysis on exercise programmes for cancer patients revealed an impact of these interventions on physical functioning and quality of life [50].
5.1.2.2. Pharmacological interventions
The finding that fatigue is linked to the activation of pro-inflammatory cytokines has led to the hypothesis that agents inhibiting these cytokines may reduce fatigue levels. In a randomised placebo-controlled trial [51] the immunosuppressant etanercept, a recombinant TNFα receptor fusion protein, enabled a significant improvement in fatigue (mean FACIT-F improvement 5.5 versus 1.9; P < 0.0001; 95% CI 1.6–4.5) in patients with psoriasis. Similarly, the monoclonal antibody infliximab was shown to significantly reduce fatigue levels in breast cancer survivors with persistent fatigue [52]. Other pharmacological interventions may include psychostimulants such as methyphenidate [53]. The relationship between inflammation and depression has also generated the hypothesis that the essential amino acid tryptophan might have a role. Tryptophan is a precursor for serotonin, and patients receiving immunotherapy were shown to have a fall in tryptophan plasma levels [54]. Another strategy that might interfere positively with inflammation-induced fatigue is the administration of thyrotropin-releasing hormone (TRH). TRH was shown to be involved in the biological processes of cytokine-induced sickness behaviour [55], and the administration of TRH has been associated with a significant improvement in fatigue levels, sleep disturbances and quality of life. These effects were accompanied by a decrease in CRP levels and an improvement in energy levels [55]. Attempts to interfere with muscle wasting have been made with l-carnitine supplementation, which was shown to have beneficial effects by improving nitrogen balance via increased protein synthesis or reduced protein degradation, inhibition of myonuclear apoptosis and interference with inflammation. Finally, any intervention that reduces the incidence and severity of fatigue-inducing side-effects may secondarily help to reduce fatigue levels: this includes control of anaemia, diarrhoea, hypothyroidism, hypophosphataemia, congestive heart failure and malnutrition.
5.2. Hypertension
5.2.1. Mechanism
Hypertension is a common and dose-dependent side-effect of all VEGF inhibitors. Classical known risk factors for hypertension failed to predict the development of hypertension in patients undergoing anti-VEGF treatment [56]. The development of hypertension in patients undergoing anti-VEGF treatment has mechanistically been linked to the pathophysiology of pre-eclampsia in pregnant women. In both cases, a deficient production of the vasodilator nitric oxide (NO) from endothelial cells and/or decreased NO levels seems to play a central role [57,58]. VEGF activates endothelial NO synthase through AKT [59], and a VEGF antibody has been shown to inhibit this process leading to a decrease in NO levels [58]. Inhibition of NO may cause vasoconstriction and hypertension [56,60]. An additional aspect of impaired NO functioning is related to its role in the control of renal function and salt sensitivity. NO was found to act as a regulator of pressure-natriuresis and plays an important role in the regulation of blood flow to the renal medulla and in the tubular regulation of sodium excretion [56]. It has therefore been concluded that inhibition of NO synthase may result in hypertension through its role in the control of renal water and sodium excretion. Finally, other mechanisms that may contribute to hypertension have been described. High salt intake has been shown to increase lymphatic vessel growth through increased VEGF-C production. Inhibition of VEGF-C by pan-VEGF inhibitors may therefore decrease lymphatic vessel density and increase blood pressure [61]. Apart from the impaired NO production, prostacyclins may contribute to the development of hypertension under anti-VEGF treatment. VEGF has been shown to activate the production of the vasodilator prostacyclin (PGI2) [62], and reduced levels of PGI2 metabolites were found in patients with pre-eclampsia, suggesting a role for decreased VEGF levels. Another mechanism that appears to trigger hypertension is vascular rarefaction. Inhibition of VEGF signalling was found to cause a loss of endothelial fenestration followed by regression of tumour vessels [63]. Similarly, normal capillaries in healthy tissues were shown to regress after VEGF inhibition [64]. Thus, a reduction in tissue microvessel density may increase blood pressure through an increase in after-load. An involvement of an up-regulated renin–angiotensin–axis resulting from glomerular ischaemia has also been discussed as a potential mechanism for the development of hypertension. However, Kappers and colleagues demonstrated that a sunitinib-induced increase in blood pressure is accompanied by a decrease in renin [65]. The same group also showed that sunitinib was associated with a considerable rise in endothelin-1 levels. In a rat model, pretreatment with atrasentan, an endothelin-A receptor antagonist, completely prevented a TKI-induced rise in blood pressure [66].
5.2.2. Management
Patients undergoing VEGF inhibitor treatment should be checked for existing blood pressure and informed about the importance of monitoring and treating blood pressure. Home monitoring has been recommended as a reasonable method to closely monitor blood pressure. Recommendations regarding the frequency of measurements vary between three times daily [67] to once weekly [68]. Accepted thresholds for initiating antihypertensive treatment are blood pressures of ⩾140/90 mmHg and 130/80 in patients with diabetes or chronic renal failure [67,69]. The selection of a specific antihypertensive drug should be based on the general cardiovascular status of the patient, as assessed by electrocardiogram (ECG) and echocardiography before treatment [67]. According to the practice guidelines for the management of arterial hypertension [70], the choice of antihypertensive treatment should be based on the underlying cardiovascular co-morbidity. Other recommendations for optimal treatment may be based on considerations of the underlying pathological mechanisms. As VEGF inhibitors also induce proteinuria, angiotensin-converting enzyme (ACE) inhibitors may be considered appropriate: ACE inhibitors were shown to improve nephrin expression and to improve endothelial function [71]. Angiotensin inhibitors as well as angiotensin-receptor blockers may have an additional advantage since they were shown to exert antitumour activity (also discussed in section on proteinuria). Calcium-channel blockers such as nifedipine may offer the (vascular) advantage of VEGF secretion from coronary smooth muscles cells [72]. In contrast, non-dihydropyridine calcium-channel blockers such as verapamil or diltiazem should be avoided: being Cytochrome P540 3A4 (CYP3A4) inhibitors, they may interfere with the tumour drug metabolism. Finally, it is important to inform the patient to withhold antihypertensive medication in off-treatment periods, where blood pressure mostly normalises.
5.3. Proteinuria
5.3.1. Mechanism
Although proteinuria has been understood as a classical on-target side-effect, such as hypertension, little has been reported from the pivotal RCC studies. This could be due either to the lack of systematic assessment of proteinuria during the trials or to a low incidence, leading to a drop-out from the toxicity tables. No data on proteinuria have been published in the randomised trials of sunitinib, sorafenib, bevacizumab + IFNα and axitinib; in contrast, proteinuria has been noted as a common side-effect in the pazopanib and tivozanib trials trials, with an all-grade incidence up to 71% and a grade 3 + 4 incidence up to 15%. As proteinuria has been linked to VEGF inhibition, it is likely that all of these agents induce proteinuria to a considerable extent. Several mechanisms have been summarised in a comprehensive review on proteinuria induced by VEGF signalling inhibition by Izzedine et al. [73], and include (1) inhibition of VEGF on podocytes which results in loss of endothelial fenestrations in glomerular capillaries, endotheliosis, loss of podocytes and proteinuria [74,75], (2) an anti-VEGF treatment-induced glomerular endothelial cell detachment and hypertrophy [76], (3) a subacute glomerular thrombotic microangiopathy [73] and (4) an adaptive hyperfiltration response to nephrectomy [73].
5.3.2. Management
Any abnormal proteinuria may not only trigger loss of kidney function, it also represents a considerable risk for renal disease and cardiovascular morbidities. In patients with chronic renal disease, structural and functional cardiac changes have been linked to persistent pressure and volume overload [77]. Moreover, strong association between proteinuria and subsequent risk of coronary artery disease have been confirmed in a meta-analysis involving 169,949 patients; in this analysis, the presence of proteinuria was associated with a 50% increased risk for coronary artery events [78]. VEGF inhibitors have enabled many patients to live long enough to experience such additional drug-induced diseases. Thus, both regular monitoring for proteinuria as well as thorough management of proteinuria appears mandatory in patients under chronic VEGF inhibitor treatment. It has been recommended that patients should be assessed for existing kidney disease prior to the start of the treatment [73], and that a dipstick analysis and/or quantitative protein test analysis be performed before each cycle of VEGF inhibitor treatment. In the case of isolated proteinuria of <1 g/L, anti-VEGF treatment might be continued along with continued monitoring. In the case of proteinuria ⩾1 g/L or proteinuria with microscopic haematuria, a nephrologist should be involved who may decide on whether to perform biopsy. Treatment recommendations include ACE inhibitors or angiotensin-2 receptor antagonists which were both shown to reduce proteinuria [79,80]. Additional measures may include salt restriction and the use of dexamethasone, which may stabilise the podocyte cytoskeleton [81].
5.4. Cardiac toxicities
5.4.1. Mechanism
Cardiac toxicities occurring under VEGFR inhibitors may be the result of both on-target and off-target inhibition. Several kinases inhibited by multikinase inhibitors such as VEGF, Hypoxia-inducible factor (HIF), PDGF and KIT are physiologically highly relevant for the heart, and the inhibition of these kinases may impair compensatory mechanisms [82–84]. HIF inhibition, for instance, may cause cardiac toxicity since in the cardiovascular system HIF-related gene products are understood as mediators of myocardial response to acute or chronic ischaemia, myocardial remodelling, peri-infarct vascularisation and vascular permeability. Thus, inhibition of these kinases may impair the myocardial response to acute or chronic ischaemia [82–85]. Disruption of PDGF–platelet derived growth factor receptor (PDGFR) signalling may lead to apoptosis and necrosis of the cardiac myocyte. PDGFR is expressed on cardiac myocytes and endothelial cells. Sunitinib-induced inhibition of S6 kinase may lead to the release of pro-apoptotic factor Apoptosis-regulator (BCL2) anatagonist of cell death, BCL2-associated X protein activation and cytochrome C release and activation of the intrinsic apoptotic pathway and cell death [86]. Disruption of KIT signalling may cause cardiac damage by inhibiting repair mechanisms. KIT is expressed on endothelial progenitor cells, and functioning of the KIT receptor might be necessary for the mobilisation of endothelial progenitor cells to sites of injury. Inhibition of KIT was shown to aggravate myocardial remodelling and prevent repair [87–89]. Disruption of VEGF–VEGFR signalling in the heart may induce cardiac dysfunction by preventing compensatory hypertrophy; VEGF is relevant to capillary density in the myocardium and critical for stem-cell differentiation into cardiomyocytes [90,91]. In the murine model, disruption of VEGF–VEGFR signalling during imposition of the pressure load was shown to reduce capillary density, which in turn was associated with contractile dysfunction, fibrosis and heart failure. Thus, inhibition of VEGF–VEGFR signalling in the heart may become relevant to patients with poorly controlled hypertension. Finally, TKI-induced changes to the thyroid function may cause cardiac toxicity. Triiodothyronine has a direct effect on the cardiomyocytes. Any T3 depletion could cause changes at the nuclear level of the myocyte level by influencing T3-regulated transcription of genes that encode Ca2+-ATPase exchanger, Na+/K+-ATPase and voltage-gated potassium channels. Moreover, T3 exerts important non-nuclear functions on the myocyte which include ion channels for sodium, potassium and calcium [92]. Finally, low serum T3 levels have been shown to be the single and the most significant predictor of cardiovascular and all-cause mortalities in adults with heart disease [93]. Triiodothyronine also directly affects vascular smooth muscle cells, promoting relaxation [94]. Hypothyroidism was shown to increase vascular resistance [94,95] and to exert endothelial dysfunction due to reduced nitric oxide availability [96,97].
5.4.2. Management
The management of cardiac toxicities may vary among patients and may depend on the drug that has been used, potential existing co-morbidities and concomitant medications that may trigger cardiac events. The clinical presentation may vary as well: a decrease in left ventricular ejection fraction was noticed in 13% of the patients in the sunitinib phase III study [3]. In the target trial on sorafenib versus placebo, 3% of patients experienced cardiac ischaemia or myocardial infarction. Other investigators noticed arrhythmias and conduction disturbances, ST-segment or T-wave changes [84]. Based on the variety of clinical manifestations of cardiac toxicities, no general recommendation can be made. However, this side-effect clearly requires a multidisciplinary approach between oncologist and cardiologist. The risk of developing a cardiac event during TKI treatment should be assessed prior to treatment. On the other hand, patients with existing cardiac co-morbidities should not be deprived of effective cancer treatment. Early management of hypertension and proteinuria appears mandatory to reduce the risk of a cardiac event during TKI treatment. Congestive heart failure that develops under TKI treatment is generally completely reversible but requires treatment interruption and effective management [98]. TKI treatment can usually be resumed after recovery, but should be initiated carefully with close monitoring of the patient. Cardiac ischaemia and myocardial infarction also require treatment interruption and cardiological care. Upon recovery of the patient, the oncologist and cardiologist need to discuss the conditions under which the patient might be able to resume RCC treatment. The concomitant use of aspirin and/or clopidrogel should not necessarily represent a contraindication; however, increased risks for haemorrhage need to be carefully considered. In patients who have experienced either congestive heart failure or myocardial ischaemia or infarction, regular echocardiograms and electrocardiograms should be obtained; the value of cardiac troponin T, Creatine kinase (CK)-MB pro-B-type Natriuretic Peptide (BNP) is questionable. Many patients on TKI smay have increased serum levels of one or all of these markers; however, for various reasons, not all of them are clinically relevant. In asymptomatic patients with increased cTNT, CK-MB and pro-BNP treatment discontinuation should be avoided. Cardiac events under TKI treatment may not require a different approach to that offered to a patient without cancer. The only difference is that: (1) oncological treatment should be temporarily interrupted, and (2) treatment should be resumed upon recovery along with permanent cardiac co-medication and close cardiac monitoring of the patient. In this context, oncologists need to be aware that some agents, particularly anti-arrhythmic agents, may exert additive toxicities: e.g. some patients may receive amiodarone after a cardiac event. Amiodarone and sotalol are agents that prolong QT intervals, thus increasing the risk for torsades de pointes. Sunitinib is also an agent with a possible risk of torsades de pointes. Given together, the risk for torsades might be higher than with each drug alone. Changes in toxicity or activity profile may also result from concomitant cardiac medication. Calcium channel blockers such as diltiazem or verapamil and again amiodarone may be CYP3A4 inhibitors; concomitant use of these agents with sunitinib may require reduction in the dose of sunitinib. In most cases, patients can be effectively treated for both the cardiac event and the oncological condition. It is of paramount importance to inform the cardiologist about the necessity of these agents for the patient and about the change these agents have made to the prognosis of mRCC patients. Discontinuing RCC treatment should be regarded as the worst-case scenario.
5.5. Diarrhoea
5.5.1. Mechanism
In contrast to pure VEGF inhibitors such as bevacizumab, diarrhoea is a frequent side-effect of multikinase inhibitors. The underlying mechanism has not been elucidated so far and may require systematic bowel biopsies and stool analyses from patients treated with VEGF inhibitors. As both highly selective VEGFR-TKIs as well as less selective TKIs were shown to induce diarrhoea; it is also unclear as to whether this toxicity should be attributed to VEGFR inhibition or off-target inhibition (e.g. PDGFR, KIT, etc.). VEGF and VEGF receptors were shown to be highly expressed in adult organs, including intestines [99]. Thus, VEGF inhibition may indeed induce diarrhoea. This assumption is further supported by data showing that the addition of VEGF(R) inhibitors significantly reduced the capillaries network in pancreatic islets and intestinal villi [100]. All of these findings suggest that VEGF Inhibition may impair the function of digestive organs such as intestines and the pancreatic gland. VEGF inhibitors may cause changes in the bowel mucosa, thereby leading to diarrhoea. In the intestinal mucosa, even small perturbations of blood flow can lead to rapid metabolic changes characteristic of ischaemia and hypoxia [101]. Epithelial hypoxia is clinically associated with diarrhoea [102], and changes in the bowel mucosa are consistent with ischaemic colitis [103]. Other possible mechanisms include changes induced by VEGF inhibitors in the exocrine pancreas, where VEGF and VEGFR are highly expressed [99]. Patients with strong VEGFR inhibitor treatment frequently report on fatty stools, and VEGFR inhibitors were shown to decrease the zymogen granules in the pancreas (observed in animals under axitinib [2,104]) and to reduce pancreatic islets capillaries [100]. Targets other than VEGF may be involved as well. For instance, sunitinib is also a c-kit inhibitor, and KIT is expressed by interstitial cells of Cajal, the pacemaker cells of the intestine [105]. Cajal cells are adjacent to the nerve fibres of the myenteric plexus and regulate rhythmic contractions in the muscle layer. KIT inhibition in interstitial cells of the Cajal could be a potential mechanism for diarrhoea induced by KIT inhibitors such as sunitinib and imatinib [106].
5.5.2. Management
The management of TKI-induced diarrhoea includes dietary measures, probiotics and drugs. Among dietary measures is the avoidance of food and drinks that may cause bowel movements, such as raw fruits, lactose-containing foods, spicy foods, foods high in fibre and an increase in bananas, rice, potatoes, etc. [107]. Another dietary measure is the increased consumption of grated oxidised apples. A randomised double-blinded trial conducted in children revealed that the oxidised apples significantly reduced stool frequency in the treatment group compared to the control group [108]. Probiotics have been shown to prevent diarrhoea in inflammatory bowel disease [109]. Preclinical data yielded a similar efficacy in chemotherapy-induced diarrhoea [110,111]. In the clinical setting, Lactobacillus rhamnosus and fibre were shown to significantly reduce the incidence of grade 3/4 diarrhoea (37% versus 22%) in a randomised study in patients with colorectal cancer and chemotherapy [109]. While the use of probiotics has never been investigated in TKI patients, individual patients report considerable benefits.
Although several medical strategies have been established to manage diarrhoea in general, none of these have been investigated with regard to TKI-induced diarrhoea. One of the most commonly used agents is loperamide, which slows transit by decreasing the tone of the longitudinal muscles and increasing the tone of circular smooth muscles of the intestinal wall [112]. This increases the time substances remain in the intestines, allowing for more water to be absorbed. Loperamide also decreases colonic movements and suppresses the gastrocolic reflux. In mRCC treatment, patient’s satisfaction with this therapeutic measure has never been investigated and appears to vary. In clinical practice, some patients report that loperamide successfully controls higher grades of diarrhoea, while others complain that in the case of watery stools a slower transit is perceived as a larger burden than an increased stool frequency. Other medical strategies to manage or prevent diarrhoea include the use of budesonide, a topical corticosteroid which was shown to reduce bowel inflammation in patients with chemotherapy-induced diarrhoea. Budesonide was shown to reduce the grade of diarrhoea in >50% in loperamide-refractory patients treated with chemotherapy [113,114]. There are no data of budesonide in patients on TKI-induced diarrhoea. Another agent with unknown benefit in TKI patients is octreotide, a synthetic somatostatin that is approved for the treatment of diarrhoea related to vasoactive intestinal peptide (VIP)-secreting tumours and symptoms due to carcinoid syndrome. Octreotide was shown to decrease the secretion of VIP to prolong intestinal transit time and to reduce secretion and increase the absorption of fluid and electrolytes [115]. In patients with colorectal cancer receiving 5-Fluorouracil (FU)-based chemoradiation, no difference from placebo was found [116]. Finally, in patients who complain of bowel movements during meals or right after, a pancreatic insufficiency induced by the VEGFR inhibitor might be considered. In this case, treatment with pancreatin might be helpful (5 meals per day, 25,000 U pancreatin with each meal).
5.6. Anorexia
5.6.1. Mechanisms
As with fatigue, anorexia is regarded as a common and multifactorial phenomenon in tumour patients. Anorexia is part of a syndrome often referred to as anorexia–cachexia and involves metabolic and behavioural factors [117]. Anorexia–cachexia has been strongly linked to an interplay between cytokines, tumour products that induce lipolysis and/or protein degradation and neuropeptides [117–119]. TNFα was shown to induce lipid depletion in white adipose tissue [120]. Moreover, TNFα induces IL-6 secretion, which was found to be significantly elevated in patients reporting weight loss [121]. In a murine model, the cytokine IL-1 was shown to induce body weight loss [122]. These cytokines appear to induce anorexia by both their peripheral as well as their central effects [123]. In addition, several neuropeptide dysregulations have been associated with anorexia. Body weight was shown to be regulated by interactions between various orexigenic and anorexigenic central and peripheral neuropeptides [119]. Whether these interactions are influenced by VEGF inhibitors has not yet been elucidated.
5.6.2. Management
Several strategies have been investigated regarding their impact on anorexia–cachexia. These include medroxyprogesterone acetate, eicosapentanoic acid, l-carnitine and thalidomide. A randomised trial that aimed to identify the most effective among these strategies revealed the greatest benefits with a combination of all [124]. Medroxyprogesterone acetate (MPA) for instance was shown to increase body weight and appetite in patients with the cachexia–anorexia syndrome [125]. The underlying mechanism may involve a down-regulation of high serum levels of IL-6 and TNFα [124]. As MPA has been shown to be increased upon the occurrence of resistance to TKI treatment in mRCC patients [124], the use of this agent may potentially act synergistically with TKIs by preventing or delaying resistance.
5.7. Stomatitis
5.7.1. Mechanisms and presentation
Patients on targeted agents may frequently report changes in the oral mucosa. The symptoms typically differ from chemotherapy-induced stomatitis. It also appears that changes differ between VEGF-TKIs and mTOR inhibitors. In sunitinib patients ulcers, taste alterations and cheilitis have been described [126]. In contrast, oral changes induced by mTOR inhibitors appear differently as superficial ulcers similar to aphthous stomatitis [127].
Dysgeusia or aguesia is quite common in patients undergoing sunitinib treatment. This is a taste disorder where e.g. the taste of meat may be perceived as sweet or a salty taste is not sensed at all. Other VEGFR-TKI patients may complain of oral burning with or without visible signs of inflammation [2,128,129]. Although stomatitis is completely reversible and more or less harmless, it is considered as clinically highly relevant since it often impairs the patient’s quality of life. Moreover, permanent stomatitis or dysgeusia may contribute to chronic refusal of food intake, thereby leading to malnutrition, fatigue and anorexia. As stomatitis resolves rapidly once the drug is withheld or dose-reduced [126], physicians and patients might be tempted to accept treatment delays, dose modifications or even a change of treatment. However, such strategies may affect the outcome. Little is known of the mechanism of stomatitis induced by VEGF inhibitors. Apart from a reduction in the capillary network of the tongue, other mechanisms may contribute to this AE. Interestingly, oral changes – e.g. burning mouth syndrome (BMS) – have also been linked to hypothyroidism [130]. BMS has been characterised by oral burning with or without inflammation, frequently affecting women. In their study, Femiano and colleagues revealed that 85 patients with BMS had thyroid alterations when compared to 13 patients in the control group. Apparently, patients with BMS are affected by dysgeusia, a phenomenon that occurs frequently with tyrosine-kinase inhibitors [131]. Thyroid hormones have been shown to influence the maturation and specialisation of the taste buds [132], and it has been speculated that hypothyroidism could therefore lead to a reduction in taste. Other investigators [133] have suggested a dysfunction of the nigrostriatal dopaminergic pathway that may account for the development of BMS. In a study on patients with BMS, Lauria and colleagues [134] detected a lower density of epithelial nerve fibres and axonal degeneration on biopsy of the tongue and suggested that BMS is caused by a trigeminal small-fibre sensory neuropathy. In a randomised placebo-controlled study, the topical administration of clonazepam improved symptoms in two thirds of BMS patients [135]. Finally, based on the assumption that BMS involves a dysfunction of the dopaminergic central nervous system, anti-epileptic drugs have been investigated [136]. Lopez and colleagues reported on a considerable improvement in BMS after treatment with pregabalin. Other mechanisms that have been discussed include shifts in the oral mucosa due to myelosupression [137], shifts in the ecological balance of oral and gut flora [138], an up-regulation of pro-inflammatory cytokines following cancer treatment [139] followed by NF-κB and cyclooxygenase-2 up-regulation. It remains unclear whether and in what way VEGFR inhibitors are involved in various processes that have been linked to stomatitis.
5.7.2. Management
Recommendations on how to treat or prevent stomatitis most commonly stem from experiences made in patients undergoing chemotherapy. General recommendations include, among others, the avoidance of spicy food, etc., the use of soft toothbrushes and appropriate dental hygiene [140]. No general recommendation exists for the prevention or management of dysgeusia. A review on drug-related taste disturbances in the elderly [141] revealed that zinc replacement might be helpful to enhance taste sensation for sweet, bitter and salty flavours. Patients with dysgeusia may benefit from niacin and vitamin A, and the use of mints, sugarless chewing gums and bicarbonate mouthwashes has been recommended as a palliative measure.
A meta-analysis on prophylactic agents to prevent stomatitis [142] identified 10 interventions that have positive effects on preventing or reducing mucositis. These included amifostine, Chinese herbal mixtures, hydrolytic enzymes such as trypsin, chymotrypsin, wobe-mugo and pepsin. Moreover, a recommendation has been made for ice chips. In patients with haematological malignancies undergoing high-dose chemotherapy, the use of keratinocyte growth factor-1 (palifermin) has been recommended [140] however, no data have been published with regard to VEGF inhibitors. The same expert panel also recommended the use of benzydamine for the prevention of radiation-induced mucositis in patients with head and neck cancer receiving radiotherapy. Stomatitis induced by mTOR inhibitors appears to be different since it involves immune mechanisms. The management might therefore be different, and corticosteroids might be helpful [127].
Treatment of stomatitis may also include mouthwashes with doxycline and/or sucralfat dissolved in water [143]. Patients who complain of inflammatory lesions may benefit from local triamcinolonacetonide. As the management of stomatitis can prove challenging, changes in treatment schedules might be considered. Several authors have reported on different modified sunitinib schedules: e.g. from 4 weeks on/2 weeks off to 2 weeks on/1 week off. While such schedules may help to prevent side-effects such as stomatitis, no changes in efficacy were observed [144–146].
5.8. Gastrointestinal perforation
5.8.1. Mechanism and clinical presentation
Gastrointestinal perforations have been rarely reported in patients with renal cell carcinoma [7]. VEGF has been shown to be highly important for the integrity of the intestinal mucosa. Vasoactive agents such as prostaglandins and NO, which are critical for mucosal defence mechanisms, are activated by VEGF [147]. Thus, VEGF has been considered a survival factor for endothelial and epithelial cells in the intestines [148]. VEGF inhibition on capillary beds of intestinal villi may directly contribute to perforation by inducing the regression of normal blood vessels [100,149]. The occurrence of gastrointestinal perforations with VEGF inhibitors has been linked to the presence of bowel pathologies [148]. Diffuse abdominal carcinomatosis is associated with a risk of bowel obstruction, increased pressure on weakened bowel areas and microperforations [150]. Other risk factors include ulcer, bowel tumour necrosis, diverticulosis, colitis and prior abdominal or pelvic radiotherapy [151] (Gentech Inc., Avastin prescribing information, June 2006). Finally, a reduction in blood flow to the splanchnic vasculature by thrombosis or vasoconstriction may further increase the risk of bowel infarction and perforation [152]. Presentation of gastrointestinal perforation during VEGF inhibitor treatment varies in type and severity, from free air on the abdominal x-ray which resolves without treatment to colonic perforation with abdominal abscess and fatal outcome.
5.8.2. Management
Patients with risk factors should be carefully monitored for clinical signs of perforation, such as abdominal pain, obstipation, fever, vomiting and leucocytosis [149]. In patients under suspicion of an increased risk of gastrointestinal perforation frequent radiographic evaluations for free peritoneal air, extraluminal contrast and abscess formation may be reasonable [151]. Physicians should also be aware of potential risks associated with co-medications such as non-steroidal anti-inflammatory drugs (NSAIDS). These increase the ratio of endostatin to VEGF and may further contribute to the occurrence of gastrointestinal perforations [153]. In patients who experience gastrointestinal perforation with VEGF inhibitors, treatment discontinuation has mostly been recommended.
5.9. Hypothyroidism
5.9.1. Mechanism
Mechanisms of hypothyroidism induced by VEGFR inhibitors may include both on- and off-target inhibition. VEGFR is expressed on thyroid cells and endothelial cells of the thyroid gland which are also able to synthesise VEGF [58,154–160]. Thus, VEGF inhibitors may induce capillary regression in the thyroid [64,100], leading to the destruction of normal thyroid cells. In addition, sunitinib was shown to induce hypothyroidism by inhibiting iodine uptake [161] and peroxidase activity [162]. It remains unclear whether off-target(s) inhibition may also contribute to hypothyroidism. Multikinase inhibitors such as sunitinib were shown to strongly inhibit RET/PTC signalling, thus being potentially beneficial in the management of thyroid cancer.
5.9.2. Management
Patients treated with VEGF inhibitors should be monitored for hypothyroidism before and at regular intervals during treatment. Both clinically overt and subclinical hypothyroidism may occur. According to the clinical practice guidelines for hypothyroidism in adults [163], the standard treatment is replacement with L-thyroxine in patients with persistent TSH levels >10 mIU/L. In patients with subclinical hypothyroidism (defined as TSH <10 mIU/L), 92% would be considered for hormone replacement. These guidelines have been established to prevent the long-term damage caused by hypothyroidism in otherwise healthy patients. How relevant are these recommendations in patients with mRCC, and what are the clinical implications for the management of TKI-induced hypothyroidism? This is particularly of interest since several authors have reported on an antitumour effect of hypothyroidism. Hypothyroidism was shown to inhibit tumour cell proliferation in various cancer cells and animal models [164–167]. Moreover, hypothyroidism was shown to inhibit neoangiogenesis and to improve outcome in patients with head and neck cancer [168,169]. Thus, the question arises as to whether we should tolerate TKI-induced hypothyroidism to some extent. Physicians need to be aware that hypothyroidism has considerable effects on cardiac function, including impaired relaxation and ventricular filling, increase in peripheral vascular resistance and increased diastolic blood pressure as well as reduced ejection at exercise [170]. Therefore, hormone replacement appears to be mandatory in the majority of patients. In this context it is important to note that triiodothyronine (T3) is the relevant hormone for the cardiac myocyte. Interestingly, T3 supplementation was shown to be 50 times less proliferative and less pro-angiogenic than T4, the ‘bad guy’ among thyroid hormones [171]. An advantage of T3 replacement would also be that it reduces T4 levels; however, T3 replacement is difficult in clinical practice due to the short half-life of available formulations. This problem could potentially be solved by the use of a combination of T3 and T4. It has been stated recently that combined T3 and T4 replacement may represent a more personalised approach to treat hypothyroidism [172].
5.10. Hand–foot syndrome
5.10.1. Presentation and mechanism
HFS has been reported to occur between days 14 and 28 of VEGF inhibitor treatment [173]. According to the Common Terminology Criteria for Adverse Events (CTCAE Version 4.0, 2009), patients with grade 1 HFS present with minimal skin changes or dermatitis (erythema, oedema or hyperkeratosis) without pain. In contrast, patients with grade 2 HFS complain of painful skin changes (peeling, blisters, bleeding, oedema and hyperkeratosis) that may limit activities of daily living. Finally, grade 3 HFS has been defined as the presence of severe skin changes associated with severe pain and limited self-care.
Several histopathological changes have been described by Yang and colleagues [174]. The most common include intracytoplasmic eosinophilic bodies reflecting keratinocyte damage, keratinocyte vacuolar degeneration and confluent keratinocyte necrosis associated with intraepidermal cleavage. In addition, an accelerated epidermal cell replication and increased keratinocyte proliferation has been described by the authors. So far, the exact mechanism of HFS with multikinase inhibitors has not been elucidated. The severity of clinical presentation appears to be correlated with drug exposure. Discussed mechanisms include: (1) an increased drug concentration in the capillaries at the papillary dermis, (2) interference by VEGF–PDGFR inhibition associated with pericyte-mediated endothelial survival mechanisms, leading to damage of the capillary endothelium in hands and feet [82], (3) an impaired vascular repair leading to keratinocyte apoptosis and inflammation and (4) a direct effect of the drug in eccrine sweat glands.
5.10.2. Management
The management of HFS in patients treated with VEGF inhibitors has been reviewed by Anderson and colleagues [175]. Prophylactic measures include pedicure before treatment to remove hyperkeratosis, emollients, topical exfoliating products (urea-based and salicylic-acid-based), protection of pressure-sensitive areas (e.g. shoes with soft insoles) and perhaps systemic administration of pyridoxine, glucocorticosteroids and cycloogygease-2 inhibitors. The authors also highlight the importance of frequent and early collaborations between oncologists and dermatologists. Dose reductions and treatment interruptions may be temporarily required. The authors recommend a dose reduction at first occurrence of grade 2 until HFS resolves to grade 0–1 and to increase the dose afterwards; if no improvement to grade 0–1 occurs, treatment interruption for 7 days may be necessary. The dose may then be escalated depending on the HFS grade. In the case of grade 3 HFS, recommendations regarding dosing include the interruption of TKI treatment for 7 days (until toxicity resolves to grade 0–1) and to resume treatment at a reduced dose. If toxicity is maintained at grade 0–1 at reduced dose, dose escalation may be recommended. In the case of recurrent grade 3 HFS, treatment should be resumed at a reduced dose after recovery without further dose escalations. According to the authors, combinations of cortisone creams and topical antibiotics might be recommended in cases of severe HFS. These recommendations have been made for patients treated with sorafenib. Although they may also apply to patients with other VEGFR–TKIs, individual modifications according to the clinical presentation, the type of drug and the drug schedule may be reasonable.
5.11. Myelotoxicity
5.11.1. Mechanisms
Myelotoxicity of tyrosine kinase inhibitors has been linked to their ability to inhibit various targets. (1) They inhibit KIT signalling: KIT receptors are expressed on haematopoietic progenitor cells and are involved in their growth and differentiation. Sunitinib was shown to inhibit phosphorylation of the KIT receptor and cell proliferation [176]. (2) VEGF inhibition may also account for myelotoxicity. Gerber and colleagues [177] described a regulatory loop by which VEGF controls survival of haematopoietic stem cells. Interestingly, ligands selective for VEGF and VEGFR-2 as well as VEGFR-1 agonists were shown to rescue survival of VEGF-deficient haematopoietic stem cells. Moreover, VEGF was shown to be involved in the formation of myeloid and erythroid colonies from progenitor cells [178]. (3) Inhibition of FLT-3 on haematopoietic stem cells and PDGFR signalling has also been linked to myelotoxicity [179,180]. (4) Finally, it has been suggested that thrombocytopenia might be the result of hypertension [181] or may be caused by drug-induced immune thrombocytopenia [182].
5.11.2. Management
In caucasian populations, myelotoxicity is seldom a dose- or treatment-limiting toxicity. In the case of grade 2 neutropenia or thrombocytopenia, dose adjustments are rarely necessary. The occurrence of grade ⩾3 myelotoxicity has been reported to occur more frequently in Asian patients [183]. In the case of grade 3 neutropenia or thrombocytopenia temporary treatment interruptions may be required. In the case of sunitinib, dose modifications may depend on the day on which grade 3 myelotoxicity is observed. If observed on day 28 of treatment, prior to the 2-week rest, patients may not necessarily require dose reduction in the next course because neutropenia and thrombocytopenia are usually short-lived and tend to resolve during the 2 weeks off treatment; blood cell counts should be repeated on day 1 of the next course, and if neutrophils and thrombocytes return to normal levels, careful continuation at the same dose level might be possible [107]. Blood cell counts should be obtained every 2 weeks, and in the case of repeated grade 3 myelotoxicity, treatment should be withheld for a few days until toxicity is grade 2 or less. In the case of recurring grade 3 myelotoxicity, dose reduction should be recommended after recovery [184].
6. Toxicities as biomarkers for successful outcome
Several retrospective studies have identified specific side-effects to be strongly associated with outcome. Table 3 summarises these findings. The most common side-effect that has been associated with outcome is hypertension. Additional toxicities that were shown to correlate with the outcome are myelotoxicity [185,186], HFS [186] and fatigue/asthenia [187]. What is the biological basis for this correlation? The toxicity may reflect that (1) the mechanism of action may be appropriate in the individual patient, (2) the chosen drug has a high selectivity and adequate potency to hit the target, (3) the tumour is dependent on the inhibited pathway and (4) the drug exposure is appropriate; this may also be influenced by the presence or absence of specific single-nucleotide polymorphisms that influence pharmacokinetic and pharmacodynamic processes [188,189].
Table 3.
Toxicity as a biomarker for outcome.
| Agent | Side-effect | Correlation with outcome |
|---|---|---|
| Bevacizumab [197] | Hypertension >2 | DCR: 91% versus 48% and TTP: 8.1 versus 4.2 |
| Bevacizumab + interferon [8] | Hypertension >2 | RR: 13 versus 9; OS:41.6 versus 16.2 |
| Sunitinib [14] | Hypertension SBP >140, DBP >90 | RR: systolic: 55 versus 10; diastolic 57 versus 25% |
| Sorafenib [198] | Hypertension all | Shrinkage: 90 versus 33 |
| Axitinib [199] | Diastol BP | PFS |
| Sunitinib [16] | Hypothyroidism | PFS: 10.3 versus 3.6 OS: 18.2 versus 6.6 |
| Sunitinib [200] | Hypothyroidism | PFS: 575 versus 481 days |
| Sunitinib [201] | Hypothyroidism | PFS: 8.55 versus 7.03 mo |
| Sunitinib + sorafenib [15] | Hypothyroidism | PFS: 17 versus 10.8; OS: nr versus 13.9 |
| SUN [14] | Hypertension | ORR: 54.8 versus 8.7% PFS: 12.5 versus 3.8; OS: 30.9 versus 7.2 |
DCR, disease control rate; TTP, time to progression; RR, response rate; SBP, systolic blood pressure; DBP, diastolic blood pressure; BP, blood pressure; PFS, progression-free survival; OS, overall survival; ORR, objective response rate.
The potential association of toxicities and outcome has several clinical implications and raises three major questions. (1) Should we treat the toxicity, or would this impair the outcome? In this context, correction of hypertension has been well studied. While the occurrence of hypertension appears to be predictive, treating hypertension does not appear to impair the outcome. In a retrospective analysis on hypertension as a predictive factor for outcome with sunitinib treatment, Szmit and colleagues [190] reported that patients who required at least three antihypertensive agents had the longest PFS. Thus, managing hypertension is not only mandatory for the patient’s safety, but it also does not appear to affect the outcome. These findings may, however, vary depending on the toxicity observed. As hypothyroidism was shown to be associated with the inhibition of angiogenesis [168,169] and cell proliferation [164], maintaining a state of (preferably) T4-hypothyroidism may to some extent be beneficial for the outcome. In this context, TSH levels above the upper limit of normal and below the threshold for cardiac impairments (>10 mmol/L) may be acceptable. (2) Should we adjust the dose until toxicity is observed (treating according to toxicity)? In the axitinib dose-titration trial, patients with dose titration and those who did not require dose titration as assessed by the occurrence of hypertension had a better outcome when compared to patients without dose titration [19]. Fig. 1 shows the computed tomography (CT) scans of a female mRCC-patient who did not experience either hypertension (or other dose-limiting toxicities) or remission with axitinib 5 mg bid. Only upon dose adjustment to 7 mg bid did the patient develop hypertension and a reduction in the size of metastasis. These findings suggest that we may consider a potential benefit of the ‘treat to toxicity’ approach. Naturally, such strategies should only be considered in the absence of other dose-limiting toxicities and require careful monitoring. (3) What is the role of agents given to manage the toxicity? Do these agents modify the outcome? We cannot rule out that agents given against the toxicity may have additional benefits against tumour progression. For instance, some antihypertensive agents were shown to exert interesting antitumour properties. Beta-blockers, for example, were shown to induce apoptosis in endothelial cells [191] and have been established as standard of care for infantile haemangiomas [192]. Moreover, several reports have demonstrated that angiotensin II stimulates growth and migration of cancer cell lines and induces angiogenesis through up-regulation of VEGF; interestingly, this effect can be inhibited by angiotensin-receptor blockers (ARBs) [193]. Losartan, an ARB, was shown to stimulate pro-apoptotic signalling pathways in various tumour types [194,195]. Finally, calcium-channel blockers have been shown to reduce the proliferation and migration of glioma cells [196].
Fig. 1.
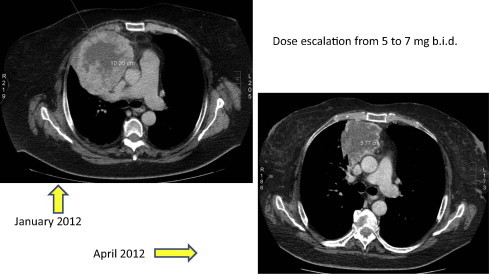
Female patient after 3 months on axitinib (no response, no hypertension) and after dose escalation with onset of hypertension.
7. Conclusions
VEGF inhibitors have substantially improved the outcome of patients with metastatic renal-cell carcinoma. Incidence and severity of side-effects may vary between agents and depend on the mode of action of the chosen drug as well as on individual patient-related factors. Physicians need to be aware of both patient- and agent-related risks that may occur during treatment in order to choose the best individual treatment and maintain the patient’s safety and quality of life. It should be considered that the majority of side-effects are manageable with proactive supportive measures and close monitoring of the patient. Dose reductions, treatment interruptions and discontinuation should be avoided whenever possible.
Conflict of interest
Honoraria for lectures or advisory role from Pfizer, Bayer, Roche, Astellas, GSK, Novartis; Research grants from GSK, Pfizer, Novartis.
References
- 1.Motzer R.J., Hutson T.E., Tomczak P. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 2.Motzer RJ, Hutson TE, Reeves J, et al. Randomized open-label phase III trial of pazopanib versus sunitinib in first-line treatment of patients with metastatic renal cell carcinoma (MRCC): results of the COMPARZ trial. 2012 ESMO Congress; 2012.
- 3.Motzer R.J., Hutson T.E., Tomczak P. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584–3590. doi: 10.1200/JCO.2008.20.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Escudier B., Eisen T., Stadler W.M. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 5.Rini B.I., Escudier B., Tomczak P. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378:1931–1939. doi: 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- 6.Sternberg C.N., Davis I.D., Mardiak J. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 7.Escudier B., Pluzanska A., Koralewski P. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103–2111. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 8.Rini B.I., Halabi S., Rosenberg J.E. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol. 2010;28:2137–2143. doi: 10.1200/JCO.2009.26.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nosov D.A., Esteves B., Lipatov O.N. Antitumor activity and safety of tivozanib (AV-951) in a phase II randomized discontinuation trial in patients with renal cell carcinoma. J Clin Oncol. 2012;30:1678–1685. doi: 10.1200/JCO.2011.35.3524. [DOI] [PubMed] [Google Scholar]
- 10.Motzer R.J. Tivozanib versus sorafenib as initial targeted therapy for patients with advanced renal cell carcinoma: results from a phase III randomized, open-label, multicenter trial. J Clin Oncol. 2012;30(Suppl.) doi: 10.1200/JCO.2012.47.4940. [abstr. 4501] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houk B.E., Bello C.L., Poland B. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol. 2010;66:357–371. doi: 10.1007/s00280-009-1170-y. [DOI] [PubMed] [Google Scholar]
- 12.Yang J.C., Haworth L., Sherry R.M. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bukowski R.M., Kabbinavar F.F., Figlin R.A. Randomized phase II study of erlotinib combined with bevacizumab compared with bevacizumab alone in metastatic renal cell cancer. J Clin Oncol. 2007;25:4536–4541. doi: 10.1200/JCO.2007.11.5154. [DOI] [PubMed] [Google Scholar]
- 14.Rini B. Hypertension (HTN) as a biomarker of efficacy in patients (pts) with metastatic renal cell carcinoma (mRCC) treated with sunitinib. ASCO GU. 2010 doi: 10.1093/jnci/djr128. [abstr. 312] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidinger M., Vogl U.M., Bojic M. Hypothyroidism in patients with renal cell carcinoma: blessing or curse? Cancer. 2011;117:534–544. doi: 10.1002/cncr.25422. [DOI] [PubMed] [Google Scholar]
- 16.Wolter P., Stefan C., Decallonne B. The clinical implications of sunitinib-induced hypothyroidism: a prospective evaluation. Br J Cancer. 2008;99:448–454. doi: 10.1038/sj.bjc.6604497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makita N., Miyakawa M., Fujita T., Iiri T. Sunitinib induces hypothyroidism with a markedly reduced vascularity. Thyroid. 2010;20:323–326. doi: 10.1089/thy.2009.0414. [DOI] [PubMed] [Google Scholar]
- 18.Amato R., Zhai J., Willis J., Saxena S., DeFoe M. A phase II trial of intrapatient dose-escalated sorafenib in patients with metastatic renal cell carcinoma. Clin Genitourin Cancer. 2012;10:153–158. doi: 10.1016/j.clgc.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Rini B.I., Gruenwald V., Fishman M.N. Axitinib with or without dose titration for first-line metastatic renal cell carcinoma (mRCC): unblinded results from a randomized phase II study. J Clin Oncol. 2013;31(Suppl. 6) [abstr. LBA349] [Google Scholar]
- 20.Cella D., Peterman A., Passik S., Jacobsen P., Breitbart W. Progress toward guidelines for the management of fatigue. Oncology (Williston Park) 1998;12:369–377. [PubMed] [Google Scholar]
- 21.Dantzer R., Kelley K.W. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21:153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miaskowski C., Dodd M., Lee K. Preliminary evidence of an association between a functional interleukin-6 polymorphism and fatigue and sleep disturbance in oncology patients and their family caregivers. J Pain Symptom Manage. 2010;40:531–544. doi: 10.1016/j.jpainsymman.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aouizerat B.E., Dodd M., Lee K. Preliminary evidence of a genetic association between tumor necrosis factor alpha and the severity of sleep disturbance and morning fatigue. Biol Res Nurs. 2009;11:27–41. doi: 10.1177/1099800409333871. [DOI] [PubMed] [Google Scholar]
- 24.Rausch S.M., Clark M.M., Patten C. Relationship between cytokine gene single nucleotide polymorphisms and symptom burden and quality of life in lung cancer survivors. Cancer. 2010;116:4103–4113. doi: 10.1002/cncr.25255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacobsen P.B., Donovan K.A., Weitzner M.A. Distinguishing fatigue and depression in patients with cancer. Semin Clin Neuropsychiatry. 2003;8:229–240. [PubMed] [Google Scholar]
- 26.Pace T.W., Mletzko T.C., Alagbe O. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry. 2006;163:1630–1633. doi: 10.1176/ajp.2006.163.9.1630. [DOI] [PubMed] [Google Scholar]
- 27.Steptoe A., Hamer M., Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun. 2007;21:901–912. doi: 10.1016/j.bbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 28.Donovan K.A., Small B.J., Andrykowski M.A., Munster P., Jacobsen P.B. Utility of a cognitive-behavioral model to predict fatigue following breast cancer treatment. Health Psychol. 2007;26:464–472. doi: 10.1037/0278-6133.26.4.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whiteside T.L. The role of immune cells in the tumor microenvironment. Cancer Treat Res. 2006;130:103–124. doi: 10.1007/0-387-26283-0_5. [DOI] [PubMed] [Google Scholar]
- 30.Whiteside T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whiteside T.L. Inhibiting the inhibitors: evaluating agents targeting cancer immunosuppression. Expert Opin Biol Ther. 2010;10:1019–1035. doi: 10.1517/14712598.2010.482207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hart B.L. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–137. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- 33.Dantzer R., Kelley K.W. Stress and immunity: an integrated view of relationships between the brain and the immune system. Life Sci. 1989;44(26):1995–2008. doi: 10.1016/0024-3205(89)90345-7. [DOI] [PubMed] [Google Scholar]
- 34.Dantzer R. Cytokine-induced sickness behavior: where do we stand? Brain Behav Immun. 2001;15:7–24. doi: 10.1006/brbi.2000.0613. [DOI] [PubMed] [Google Scholar]
- 35.Negrier S., Perol D., Menetrier-Caux C. Interleukin-6, interleukin-10, and vascular endothelial growth factor in metastatic renal cell carcinoma: prognostic value of interleukin-6 – from the Groupe Francais d’Immunotherapie. J Clin Oncol. 2004;22:2371–2378. doi: 10.1200/JCO.2004.06.121. [DOI] [PubMed] [Google Scholar]
- 36.Ma Y., Kepp O., Ghiringhelli F. Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin Immunol. 2010;22:113–124. doi: 10.1016/j.smim.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Wang X.S., Shi Q., Williams L.A. Inflammatory cytokines are associated with the development of symptom burden in patients with NSCLC undergoing concurrent chemoradiation therapy. Brain Behav Immun. 2010;24:968–974. doi: 10.1016/j.bbi.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Louwerens M., Appelhof B.C., Verloop H. Fatigue and fatigue-related symptoms in patients treated for different causes of hypothyroidism. Eur J Endocrinol. 2012;167:809–815. doi: 10.1530/EJE-12-0501. [DOI] [PubMed] [Google Scholar]
- 39.Aubier M., Murciano D., Lecocguic Y. Effect of hypophosphatemia on diaphragmatic contractility in patients with acute respiratory failure. N Engl J Med. 1985;313:420–424. doi: 10.1056/NEJM198508153130705. [DOI] [PubMed] [Google Scholar]
- 40.Zazzo J.F., Troche G., Ruel P., Maintenant J. High incidence of hypophosphatemia in surgical intensive care patients: efficacy of phosphorus therapy on myocardial function. Intensive Care Med. 1995;21:826–831. doi: 10.1007/BF01700966. [DOI] [PubMed] [Google Scholar]
- 41.Richter E.A., Derave W., Wojtaszewski J.F. Glucose, exercise and insulin: emerging concepts. J Physiol. 2001;535:313–322. doi: 10.1111/j.1469-7793.2001.t01-2-00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fabian M.A., Biggs W.H., III, Treiber D.K. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 43.Billemont B., Medioni J., Taillade L. Blood glucose levels in patients with metastatic renal cell carcinoma treated with sunitinib. Br J Cancer. 2008;99:1380–1382. doi: 10.1038/sj.bjc.6604709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yavuzsen T., Davis M.P., Ranganathan V.K. Cancer-related fatigue: central or peripheral? J Pain Symptom Manage. 2009;38:587–596. doi: 10.1016/j.jpainsymman.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Antoun S., Birdsell L., Sawyer M.B. Association of skeletal muscle wasting with treatment with sorafenib in patients with advanced renal cell carcinoma: results from a placebo-controlled study. J Clin Oncol. 2010;28:1054–1060. doi: 10.1200/JCO.2009.24.9730. [DOI] [PubMed] [Google Scholar]
- 46.Bodine S.C., Stitt T.N., Gonzalez M. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 47.Patterson E, Wan YW, Sidani S. Nonpharmacological nursing interventions for the management of patient fatigue: a literature review. J Clin Nurs 2013 May 9. http://dx.doi.org/10.1111/jocn.12211. [DOI] [PubMed]
- 48.Cramp F., Byron-Daniel J. Exercise for the management of cancer-related fatigue in adults. Cochrane Database Syst Rev. 2012;11:CD006145. doi: 10.1002/14651858.CD006145.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strasser B, Steindorf K, Wiskemann J, Ulrich CM. Impact of resistance training in cancer survivors: a meta-analysis. Med Sci Sports Exerc 2013 May 10. [DOI] [PubMed]
- 50.McClellan R. Exercise programs for patients with cancer improve physical functioning and quality of life. J Physiother. 2013;59:57. doi: 10.1016/S1836-9553(13)70150-4. [DOI] [PubMed] [Google Scholar]
- 51.Tyring S., Gottlieb A., Papp K. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29–35. doi: 10.1016/S0140-6736(05)67763-X. [DOI] [PubMed] [Google Scholar]
- 52.Bower J.E., Ganz P.A., Irwin M.R., Arevalo J.M., Cole S.W. Fatigue and gene expression in human leukocytes: increased NF-kappaB and decreased glucocorticoid signaling in breast cancer survivors with persistent fatigue. Brain Behav Immun. 2011;25:147–150. doi: 10.1016/j.bbi.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minton O., Richardson A., Sharpe M., Hotopf M., Stone P.C. Psychostimulants for the management of cancer-related fatigue: a systematic review and meta-analysis. J Pain Symptom Manage. 2011;41:761–767. doi: 10.1016/j.jpainsymman.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 54.Dantzer R., O’Connor J.C., Freund G.G., Johnson R.W., Kelley K.W. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamath J. Cancer-related fatigue, inflammation and thyrotropin-releasing hormone. Curr Aging Sci. 2012;5:195–202. doi: 10.2174/1874609811205030005. [DOI] [PubMed] [Google Scholar]
- 56.Robinson E.S., Matulonis U.A., Ivy P. Rapid development of hypertension and proteinuria with cediranib, an oral vascular endothelial growth factor receptor inhibitor. Clin J Am Soc Nephrol. 2010;5:477–483. doi: 10.2215/CJN.08111109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lowe D.T. Nitric oxide dysfunction in the pathophysiology of preeclampsia. Nitric oxide. 2000;4:441–458. doi: 10.1006/niox.2000.0296. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y., Fei D., Vanderlaan M., Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7:335–345. doi: 10.1007/s10456-004-8272-2. [DOI] [PubMed] [Google Scholar]
- 59.Horowitz J.R., Rivard A., van der Zee R. Vascular endothelial growth factor/vascular permeability factor produces nitric oxide-dependent hypotension. Evidence for a maintenance role in quiescent adult endothelium. Arterioscler Thromb Vasc Biol. 1997;17:2793–2800. doi: 10.1161/01.atv.17.11.2793. [DOI] [PubMed] [Google Scholar]
- 60.Facemire C.S., Nixon A.B., Griffiths R., Hurwitz H., Coffman T.M. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54:652–658. doi: 10.1161/HYPERTENSIONAHA.109.129973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Machnik A., Neuhofer W., Jantsch J. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 62.Wheeler-Jones C., Abu-Ghazaleh R., Cospedal R. Vascular endothelial growth factor stimulates prostacyclin production and activation of cytosolic phospholipase A2 in endothelial cells via p42/p44 mitogen-activated protein kinase. FEBS Lett. 1997;420:28–32. doi: 10.1016/s0014-5793(97)01481-6. [DOI] [PubMed] [Google Scholar]
- 63.Inai T., Mancuso M., Hashizume H. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165:35–52. doi: 10.1016/S0002-9440(10)63273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baffert F., Le T., Sennino B. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol. 2006;290:H547–H559. doi: 10.1152/ajpheart.00616.2005. [DOI] [PubMed] [Google Scholar]
- 65.Kappers M.H., van Esch J.H., Sluiter W. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. 2010;56:675–681. doi: 10.1161/HYPERTENSIONAHA.109.149690. [DOI] [PubMed] [Google Scholar]
- 66.Banfor P.N., Franklin P.A., Segreti J.A. ETA receptor blockade with atrasentan prevents hypertension with the multitargeted tyrosine kinase inhibitor ABT-869 in telemetry-instrumented rats. J Cardiovasc Pharmacol. 2009;53:173–178. doi: 10.1097/FJC.0b013e3181993493. [DOI] [PubMed] [Google Scholar]
- 67.Schmidinger M., Bergler-Klein J. Therapy management of cardiovascular adverse events in the context of targeted therapy for metastatic renal cell carcinoma. Int J Urol. 2012;19:796–804. doi: 10.1111/j.1442-2042.2012.03033.x. [DOI] [PubMed] [Google Scholar]
- 68.Wu S., Chen J.J., Kudelka A., Lu J., Zhu X. Incidence and risk of hypertension with sorafenib in patients with cancer: a systematic review and meta-analysis. Lancet Oncol. 2008;9:117–123. doi: 10.1016/S1470-2045(08)70003-2. [DOI] [PubMed] [Google Scholar]
- 69.Chobanian A.V., Bakris G.L., Black H.R. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42:1206–1252. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- 70.Mancia G., De B.G., Dominiczak A. Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC) Eur Heart J. 2007;28:1462–1536. doi: 10.1093/eurheartj/ehm236. [DOI] [PubMed] [Google Scholar]
- 71.Blanco S., Bonet J., Lopez D., Casas I., Romero R. ACE inhibitors improve nephrin expression in Zucker rats with glomerulosclerosis. Kidney Int Suppl. 2005;93:S10–S14. doi: 10.1111/j.1523-1755.2005.09303.x. [DOI] [PubMed] [Google Scholar]
- 72.Miura S., Fujino M., Matsuo Y., Tanigawa H., Saku K. Nifedipine-induced vascular endothelial growth factor secretion from coronary smooth muscle cells promotes endothelial tube formation via the kinase insert domain-containing receptor/fetal liver kinase-1/NO pathway. Hypertens Res. 2005;28:147–153. doi: 10.1291/hypres.28.147. [DOI] [PubMed] [Google Scholar]
- 73.Izzedine H., Massard C., Spano J.P. VEGF signalling inhibition-induced proteinuria: mechanisms, significance and management. Eur J Cancer. 2010;46:439–448. doi: 10.1016/j.ejca.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 74.Eremina V., Sood M., Haigh J. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schrijvers B.F., Flyvbjerg A., De Vriese A.S. The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney Int. 2004;65:2003–2017. doi: 10.1111/j.1523-1755.2004.00621.x. [DOI] [PubMed] [Google Scholar]
- 76.Garovic V.D., Wagner S.J., Petrovic L.M. Glomerular expression of nephrin and synaptopodin, but not podocin, is decreased in kidney sections from women with preeclampsia. Nephrol Dial Transplant. 2007;22:1136–1143. doi: 10.1093/ndt/gfl711. [DOI] [PubMed] [Google Scholar]
- 77.Paoletti E., Bellino D., Cassottana P., Rolla D., Cannella G. Left ventricular hypertrophy in nondiabetic predialysis CKD. Am J Kidney Dis. 2005;46:320–327. doi: 10.1053/j.ajkd.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 78.Perkovic V., Verdon C., Ninomiya T. The relationship between proteinuria and coronary risk: a systematic review and meta-analysis. PLoS Med. 2008;5:e207. doi: 10.1371/journal.pmed.0050207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peterson J.C., Adler S., Burkart J.M. Blood pressure control, proteinuria, and the progression of renal disease. The modification of diet in renal disease study. Ann Intern Med. 1995;123:754–762. doi: 10.7326/0003-4819-123-10-199511150-00003. [DOI] [PubMed] [Google Scholar]
- 80.de Z.D., Remuzzi G., Parving H.H. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004;65:2309–2320. doi: 10.1111/j.1523-1755.2004.00653.x. [DOI] [PubMed] [Google Scholar]
- 81.Xing C.Y., Saleem M.A., Coward R.J., Ni L., Witherden I.R., Mathieson P.W. Direct effects of dexamethasone on human podocytes. Kidney Int. 2006;70:1038–1045. doi: 10.1038/sj.ki.5001655. [DOI] [PubMed] [Google Scholar]
- 82.Kido M., Du L., Sullivan C.C. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol. 2005;46:2116–2124. doi: 10.1016/j.jacc.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 83.Parisi Q., Biondi-Zoccai G.G., Abbate A. Hypoxia inducible factor-1 expression mediates myocardial response to ischemia late after acute myocardial infarction. Int J Cardiol. 2005;99:337–339. doi: 10.1016/j.ijcard.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 84.Schmidinger M., Zielinski C.C., Vogl U.M. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008;26:5204–5212. doi: 10.1200/JCO.2007.15.6331. [DOI] [PubMed] [Google Scholar]
- 85.Waltenberger J. Modulation of growth factor action: implications for the treatment of cardiovascular diseases. Circulation. 1997;96:4083–4094. doi: 10.1161/01.cir.96.11.4083. [DOI] [PubMed] [Google Scholar]
- 86.Force T., Krause D.S., Van Etten R.A. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7:332–344. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]
- 87.Ayach B.B., Yoshimitsu M., Dawood F. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:2304–2309. doi: 10.1073/pnas.0510997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fazel S., Cimini M., Chen L. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. 2006;116:1865–1877. doi: 10.1172/JCI27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang C.H., Anderson N., Li S.H. Stem cell factor deficiency is vasculoprotective: unraveling a new therapeutic potential of imatinib mesylate. Circ Res. 2006;99:617–625. doi: 10.1161/01.RES.0000243210.79654.fd. [DOI] [PubMed] [Google Scholar]
- 90.Shiojima I., Sato K., Izumiya Y. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Izumiya Y., Shiojima I., Sato K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–893. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klein I., Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501–509. doi: 10.1056/NEJM200102153440707. [DOI] [PubMed] [Google Scholar]
- 93.Iervasi G., Pingitore A., Landi P. Low-T3 syndrome: a strong prognostic predictor of death in patients with heart disease. Circulation. 2003;107:708–713. doi: 10.1161/01.cir.0000048124.64204.3f. [DOI] [PubMed] [Google Scholar]
- 94.Ripoli A., Pingitore A., Favilli B. Does subclinical hypothyroidism affect cardiac pump performance? Evidence from a magnetic resonance imaging study. J Am Coll Cardiol. 2005;45:439–445. doi: 10.1016/j.jacc.2004.10.044. [DOI] [PubMed] [Google Scholar]
- 95.Faber J., Petersen L., Wiinberg N., Schifter S., Mehlsen J. Hemodynamic changes after levothyroxine treatment in subclinical hypothyroidism. Thyroid. 2002;12:319–324. doi: 10.1089/10507250252949450. [DOI] [PubMed] [Google Scholar]
- 96.Lekakis J., Papamichael C., Alevizaki M. Flow-mediated, endothelium-dependent vasodilation is impaired in subjects with hypothyroidism, borderline hypothyroidism, and high-normal serum thyrotropin (TSH) values. Thyroid. 1997;7:411–414. doi: 10.1089/thy.1997.7.411. [DOI] [PubMed] [Google Scholar]
- 97.Taddei S., Caraccio N., Virdis A. Impaired endothelium-dependent vasodilatation in subclinical hypothyroidism: beneficial effect of levothyroxine therapy. J Clin Endocrinol Metab. 2003;88:3731–3737. doi: 10.1210/jc.2003-030039. [DOI] [PubMed] [Google Scholar]
- 98.Schmidinger M., Larkin J., Ravaud A. Experience with sunitinib in the treatment of metastatic renal cell carcinoma. Ther Adv Urol. 2012;4:253–265. doi: 10.1177/1756287212454933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fan L., Iseki S. Immunohistochemical localization of vascular endothelial growth factor in the endocrine glands of the rat. Arch Histol Cytol. 1998;61:17–28. doi: 10.1679/aohc.61.17. [DOI] [PubMed] [Google Scholar]
- 100.Kamba T., Tam B.Y., Hashizume H. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290:H560–76. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- 101.Zheng W., Kuhlicke J., Jackel K. Hypoxia inducible factor-1 (HIF-1)-mediated repression of cystic fibrosis transmembrane conductance regulator (CFTR) in the intestinal epithelium. FASEB J. 2009;23:204–213. doi: 10.1096/fj.08-110221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sreenarasimhaiah J. Diagnosis and management of ischemic colitis. Curr Gastroenterol Rep. 2005;7:421–426. doi: 10.1007/s11894-005-0013-1. [DOI] [PubMed] [Google Scholar]
- 103.Lordick F., Geinitz H., Theisen J., Sendler A., Sarbia M. Increased risk of ischemic bowel complications during treatment with bevacizumab after pelvic irradiation: report of three cases. Int J Radiat Oncol Biol Phys. 2006;64:1295–1298. doi: 10.1016/j.ijrobp.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 104.Axitinib Investigator’s Brochure. Pfizer Inc. 2012.
- 105.Sanders K.M., Koh S.D., Ward S.M. Interstitial cells of cajal as pacemakers in the gastrointestinal tract. Annu Rev Physiol. 2006;68:307–343. doi: 10.1146/annurev.physiol.68.040504.094718. [DOI] [PubMed] [Google Scholar]
- 106.Deininger M.W. Cytogenetic studies in patients on imatinib. Semin Hematol. 2003;40(2 Suppl. 2):50–55. doi: 10.1053/shem.2003.50043. [DOI] [PubMed] [Google Scholar]
- 107.Schmidinger M., Bellmunt J. Plethora of agents, plethora of targets, plethora of side effects in metastatic renal cell carcinoma. Cancer Treat Rev. 2010;36:416–424. doi: 10.1016/j.ctrv.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 108.Becker B., Kuhn U., Hardewig-Budny B. Double-blind, randomized evaluation of clinical efficacy and tolerability of an apple pectin-chamomile extract in children with unspecific diarrhea. Arzneimittelforschung. 2006;56:387–393. doi: 10.1055/s-0031-1296739. [DOI] [PubMed] [Google Scholar]
- 109.Osterlund P., Ruotsalainen T., Korpela R. Lactobacillus supplementation for diarrhoea related to chemotherapy of colorectal cancer: a randomised study. Br J Cancer. 2007;97:1028–1034. doi: 10.1038/sj.bjc.6603990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Von Bultzingslowen I., Adlerberth I., Wold A.E., Dahlen G., Jontell M. Oral and intestinal microflora in 5-fluorouracil treated rats, translocation to cervical and mesenteric lymph nodes and effects of probiotic bacteria. Oral Microbiol Immunol. 2003;18:278–284. doi: 10.1034/j.1399-302x.2003.00075.x. [DOI] [PubMed] [Google Scholar]
- 111.Bowen J.M., Stringer A.M., Gibson R.J. VSL#3 probiotic treatment reduces chemotherapy-induced diarrhea and weight loss. Cancer Biol Ther. 2007;6:1449–1454. doi: 10.4161/cbt.6.9.4622. [DOI] [PubMed] [Google Scholar]
- 112.Placidi E., Marciani L., Hoad C.L. The effects of loperamide, or loperamide plus simethicone, on the distribution of gut water as assessed by MRI in a mannitol model of secretory diarrhoea. Aliment Pharmacol Ther. 2012;36:64–73. doi: 10.1111/j.1365-2036.2012.05127.x. [DOI] [PubMed] [Google Scholar]
- 113.Karthaus M., Ballo H., Abenhardt W. Prospective, double-blind, placebo-controlled, multicenter, randomized phase III study with orally administered budesonide for prevention of irinotecan (CPT-11)-induced diarrhea in patients with advanced colorectal cancer. Oncology. 2005;68:326–332. doi: 10.1159/000086971. [DOI] [PubMed] [Google Scholar]
- 114.Lenfers B.H., Loeffler T.M., Droege C.M., Hausamen T.U. Substantial activity of budesonide in patients with irinotecan (CPT-11) and 5-fluorouracil induced diarrhea and failure of loperamide treatment. Ann Oncol. 1999;10:1251–1253. doi: 10.1023/a:1008390308416. [DOI] [PubMed] [Google Scholar]
- 115.Dueno M.I., Bai J.C., Santangelo W.C., Krejs G.J. Effect of somatostatin analog on water and electrolyte transport and transit time in human small bowel. Dig Dis Sci. 1987;32(10):1092–1096. doi: 10.1007/BF01300194. [DOI] [PubMed] [Google Scholar]
- 116.Zachariah B., Gwede C.K., James J. Octreotide acetate in prevention of chemoradiation-induced diarrhea in anorectal cancer: randomized RTOG trial 0315. J Natl Cancer Inst. 2010;102:547–556. doi: 10.1093/jnci/djq063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tisdale M.J. Molecular pathways leading to cancer cachexia. Physiology (Bethesda) 2005;20:340–348. doi: 10.1152/physiol.00019.2005. [DOI] [PubMed] [Google Scholar]
- 118.Hirai K., Hussey H.J., Barber M.D., Price S.A., Tisdale M.J. Biological evaluation of a lipid-mobilizing factor isolated from the urine of cancer patients. Cancer Res. 1998;58:2359–2365. [PubMed] [Google Scholar]
- 119.Wood S.C. Biological oxygenation: circulation, respiration, and metabolism. Science. 1986;234:491–492. doi: 10.1126/science.234.4775.491. [DOI] [PubMed] [Google Scholar]
- 120.Ryden M., Arvidsson E., Blomqvist L. Targets for TNF-alpha-induced lipolysis in human adipocytes. Biochem Biophys Res Commun. 2004;318:168–175. doi: 10.1016/j.bbrc.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 121.Maltoni M., Fabbri L., Nanni O. Serum levels of tumour necrosis factor alpha and other cytokines do not correlate with weight loss and anorexia in cancer patients. Support Care Cancer. 1997;5:130–135. doi: 10.1007/BF01262570. [DOI] [PubMed] [Google Scholar]
- 122.Moldawer L.L., Andersson C., Gelin J., Lundholm K.G. Regulation of food intake and hepatic protein synthesis by recombinant-derived cytokines. Am J Physiol. 1988;254:G450–6. doi: 10.1152/ajpgi.1988.254.3.G450. [DOI] [PubMed] [Google Scholar]
- 123.Sternberg E.M. Neural–immune interactions in health and disease. J Clin Invest. 1997;100:2641–2647. doi: 10.1172/JCI119807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mantovani G., Maccio A., Madeddu C. Randomized phase III clinical trial of five different arms of treatment in 332 patients with cancer cachexia. Oncologist. 2010;15:200–211. doi: 10.1634/theoncologist.2009-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Testa A., Gebbia V., Borsellino N. Usefulness of oral medroxyprogesterone acetate in the management of cancer-related cachexia–anorexia syndrome. Oncol Rep. 1996;3:493–496. doi: 10.3892/or.3.3.493. [DOI] [PubMed] [Google Scholar]
- 126.Kollmannsberger C., Soulieres D., Wong R. Sunitinib therapy for metastatic renal cell carcinoma: recommendations for management of side effects. Can Urol Assoc J. 2007;1(2 Suppl.):S41–S54. doi: 10.5489/cuaj.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sonis S., Treister N., Chawla S., Demetri G., Haluska F. Preliminary characterization of oral lesions associated with inhibitors of mammalian target of rapamycin in cancer patients. Cancer. 2010;116:210–215. doi: 10.1002/cncr.24696. [DOI] [PubMed] [Google Scholar]
- 128.Rosenthal D.I., Lewin J.S., Eisbruch A. Prevention and treatment of dysphagia and aspiration after chemoradiation for head and neck cancer. J Clin Oncol. 2006;24:2636–2643. doi: 10.1200/JCO.2006.06.0079. [DOI] [PubMed] [Google Scholar]
- 129.National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 4.02, 2009.
- 130.Femiano F., Lanza A., Buonaiuto C. Burning mouth syndrome and burning mouth in hypothyroidism: proposal for a diagnostic and therapeutic protocol. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;105:e22–7. doi: 10.1016/j.tripleo.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 131.Bartoshuk L.M., Snyder D.J., Grushka M. Taste damage: previously unsuspected consequences. Chem Senses. 2005;30(Suppl. 1):i218–9. doi: 10.1093/chemse/bjh192. [DOI] [PubMed] [Google Scholar]
- 132.Brosvic G.M., Doty R.L., Rowe M.M., Harron A., Kolodiy N. Influences of hypothyroidism on the taste detection performance of rats: a signal detection analysis. Behav Neurosci. 1992;106:992–998. doi: 10.1037//0735-7044.106.6.992. [DOI] [PubMed] [Google Scholar]
- 133.Jaaskelainen S.K., Forssell H., Tenovuo O. Abnormalities of the blink reflex in burning mouth syndrome. Pain. 1997;73:455–460. doi: 10.1016/S0304-3959(97)00140-1. [DOI] [PubMed] [Google Scholar]
- 134.Lauria G., Majorana A., Borgna M. Trigeminal small-fiber sensory neuropathy causes burning mouth syndrome. Pain. 2005;115:332–337. doi: 10.1016/j.pain.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 135.Gremeau-Richard C., Woda A., Navez M.L. Topical clonazepam in stomatodynia: a randomised placebo-controlled study. Pain. 2004;108:51–57. doi: 10.1016/j.pain.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 136.Lopez V., Alonso V., Marti N., Calduch L., Jorda E. Marked response of burning mouth syndrome to pregabalin treatment. Clin Exp Dermatol. 2009;34:e449–50. doi: 10.1111/j.1365-2230.2009.03493.x. [DOI] [PubMed] [Google Scholar]
- 137.Frei E., III, Levin R.H., Bodey G.P., Morse E.E., Freireich E.J. The nature and control of infections in patients with acute leukemia. Cancer Res. 1965;25:1511–1515. [PubMed] [Google Scholar]
- 138.Stringer A.M., Gibson R.J., Bowen J.M., Keefe D.M. Chemotherapy-induced modifications to gastrointestinal microflora: evidence and implications of change. Curr Drug Metab. 2009;10:79–83. doi: 10.2174/138920009787048419. [DOI] [PubMed] [Google Scholar]
- 139.Logan R.M., Gibson R.J., Bowen J.M. Characterisation of mucosal changes in the alimentary tract following administration of irinotecan: implications for the pathobiology of mucositis. Cancer Chemother Pharmacol. 2008;62:33–41. doi: 10.1007/s00280-007-0570-0. [DOI] [PubMed] [Google Scholar]
- 140.Keefe D.M., Schubert M.M., Elting L.S. Updated clinical practice guidelines for the prevention and treatment of mucositis. Cancer. 2007;109:820–831. doi: 10.1002/cncr.22484. [DOI] [PubMed] [Google Scholar]
- 141.Arcavi L., Shahar A. Drug related taste disturbances: emphasis on the elderly. Harefuah. 2003;142 446–50, 485, 484. [PubMed] [Google Scholar]
- 142.Worthington H.V., Clarkson J.E., Bryan G. Interventions for preventing oral mucositis for patients with cancer receiving treatment. Cochrane Database Syst Rev. 2011;(4):CD000978. doi: 10.1002/14651858.CD000978.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bensinger W., Schubert M., Ang K.K. NCCN Task Force Report: prevention and management of mucositis in cancer care. J Natl Compr Canc Netw. 2008;6(Suppl. 1):S1–21. [PubMed] [Google Scholar]
- 144.Buti S., Donini M., Lazzarelli S., Passalacqua R. A new modified schedule of sunitinib for metastatic renal cell carcinoma: a retrospective analysis. Acta Biomed. 2012;83:88–94. [PubMed] [Google Scholar]
- 145.Neri B., Vannini A., Brugia M. Biweekly sunitinib regimen reduces toxicity and retains efficacy in metastatic renal cell carcinoma: a single-center experience with 31 patients. Int J Urol. 2013;20:478–483. doi: 10.1111/j.1442-2042.2012.03204.x. [DOI] [PubMed] [Google Scholar]
- 146.Neri B., Vannini A., Tassi R. The efficacy and tolerability of a sunitinib 3-week administration schedule in metastatic renal cell carcinoma patients: report of three cases. Oncol Res. 2012;20:259–264. doi: 10.3727/096504013x13589503482851. [DOI] [PubMed] [Google Scholar]
- 147.Gyires K. Gastric mucosal protection: from prostaglandins to gene-therapy. Curr Med Chem. 2005;12:203–215. doi: 10.2174/0929867053363478. [DOI] [PubMed] [Google Scholar]
- 148.Roodhart J.M., Langenberg M.H., Witteveen E., Voest E.E. The molecular basis of class side effects due to treatment with inhibitors of the VEGF/VEGFR pathway. Curr Clin Pharmacol. 2008;3:132–143. doi: 10.2174/157488408784293705. [DOI] [PubMed] [Google Scholar]
- 149.Saif M.W., Elfiky A., Salem R.R. Gastrointestinal perforation due to bevacizumab in colorectal cancer. Ann Surg Oncol. 2007;14:1860–1869. doi: 10.1245/s10434-006-9337-9. [DOI] [PubMed] [Google Scholar]
- 150.Scappaticci F.A., Fehrenbacher L., Cartwright T. Surgical wound healing complications in metastatic colorectal cancer patients treated with bevacizumab. J Surg Oncol. 2005;91:173–180. doi: 10.1002/jso.20301. [DOI] [PubMed] [Google Scholar]
- 151.Gordon M.S., Cunningham D. Managing patients treated with bevacizumab combination therapy. Oncology. 2005;69(Suppl. 3):25–33. doi: 10.1159/000088481. [DOI] [PubMed] [Google Scholar]
- 152.Han E.S., Monk B.J. What is the risk of bowel perforation associated with bevacizumab therapy in ovarian cancer? Gynecol Oncol. 2007;105:3–6. doi: 10.1016/j.ygyno.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 153.Perini R., Wallace J.L., Ma L. Roles of platelets and proteinase-activated receptors in gastric ulcer healing. Dig Dis Sci. 2005;50(Suppl. 1):S12–S15. doi: 10.1007/s10620-005-2801-8. [DOI] [PubMed] [Google Scholar]
- 154.Viglietto G., Maglione D., Rambaldi M. Upregulation of vascular endothelial growth factor (VEGF) and downregulation of placenta growth factor (PlGF) associated with malignancy in human thyroid tumors and cell lines. Oncogene. 1995;11:1569–1579. [PubMed] [Google Scholar]
- 155.Sato K., Terada K., Sugiyama T. Frequent overexpression of vascular endothelial growth factor gene in human renal cell carcinoma. Tohoku J Exp Med. 1994;173:355–360. doi: 10.1620/tjem.173.355. [DOI] [PubMed] [Google Scholar]
- 156.Katoh R., Miyagi E., Kawaoi A. Expression of vascular endothelial growth factor (VEGF) in human thyroid neoplasms. Hum Pathol. 1999;30:891–897. doi: 10.1016/s0046-8177(99)90241-1. [DOI] [PubMed] [Google Scholar]
- 157.Klein M., Picard E., Vignaud J.M. Vascular endothelial growth factor gene and protein: strong expression in thyroiditis and thyroid carcinoma. J Endocrinol. 1999;161:41–49. doi: 10.1677/joe.0.1610041. [DOI] [PubMed] [Google Scholar]
- 158.Fenton C., Patel A., Dinauer C., Robie D.K., Tuttle R.M., Francis G.L. The expression of vascular endothelial growth factor and the type 1 vascular endothelial growth factor receptor correlate with the size of papillary thyroid carcinoma in children and young adults. Thyroid. 2000;10:349–357. doi: 10.1089/thy.2000.10.349. [DOI] [PubMed] [Google Scholar]
- 159.Ramsden J.D. Angiogenesis in the thyroid gland. J Endocrinol. 2000;166:475–480. doi: 10.1677/joe.0.1660475. [DOI] [PubMed] [Google Scholar]
- 160.Nagura S., Katoh R., Miyagi E., Shibuya M., Kawaoi A. Expression of vascular endothelial growth factor (VEGF) and VEGF receptor-1 (Flt-1) in Graves disease possibly correlated with increased vascular density. Hum Pathol. 2001;32:10–17. doi: 10.1053/hupa.2001.21139. [DOI] [PubMed] [Google Scholar]
- 161.Mannavola D., Coco P., Vannucchi G. A novel tyrosine-kinase selective inhibitor, sunitinib, induces transient hypothyroidism by blocking iodine uptake. J Clin Endocrinol Metab. 2007;92:3531–3534. doi: 10.1210/jc.2007-0586. [DOI] [PubMed] [Google Scholar]
- 162.Wong E., Rosen L.S., Mulay M. Sunitinib induces hypothyroidism in advanced cancer patients and may inhibit thyroid peroxidase activity. Thyroid. 2007;17:351–355. doi: 10.1089/thy.2006.0308. [DOI] [PubMed] [Google Scholar]
- 163.Garber J.R., Cobin R.H., Gharib H. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid. 2012;22:1200–1235. doi: 10.1089/thy.2012.0205. [DOI] [PubMed] [Google Scholar]
- 164.Theodossiou C., Skrepnik N., Robert E.G. Propylthiouracil-induced hypothyroidism reduces xenograft tumor growth in athymic nude mice. Cancer. 1999;86:1596–1601. [PubMed] [Google Scholar]
- 165.Goodman A.D., Hoekstra S.J., Marsh P.S. Effects of hypothyroidism on the induction and growth of mammary cancer induced by 7,12-dimethylbenz(a)anthracene in the rat. Cancer Res. 1980;40:2336–2342. [PubMed] [Google Scholar]
- 166.Mishkin S.Y., Pollack R., Yalovsky M.A., Morris H.P., Mishkin S. Inhibition of local and metastatic hepatoma growth and prolongation of survival after induction of hypothyroidism. Cancer Res. 1981;41:3040–3045. [PubMed] [Google Scholar]
- 167.Nelson M., Hercbergs A., Rybicki L., Strome M. Association between development of hypothyroidism and improved survival in patients with head and neck cancer. Arch Otolaryngol Head Neck Surg. 2006;132:1041–1046. doi: 10.1001/archotol.132.10.1041. [DOI] [PubMed] [Google Scholar]
- 168.Hercbergs A.A., Goyal L.K., Suh J.H. Propylthiouracil-induced chemical hypothyroidism with high-dose tamoxifen prolongs survival in recurrent high grade glioma: a phase I/II study. Anticancer Res. 2003;23:617–626. [PubMed] [Google Scholar]
- 169.Trentin A.G., Alvarez-Silva M., Moura Neto V. Thyroid hormone induces cerebellar astrocytes and C6 glioma cells to secrete mitogenic growth factors. Am J Physiol Endocrinol Metab. 2001;281:E1088–94. doi: 10.1152/ajpendo.2001.281.5.E1088. [DOI] [PubMed] [Google Scholar]
- 170.Biondi B., Fazio S., Palmieri E.A. Left ventricular diastolic dysfunction in patients with subclinical hypothyroidism. J Clin Endocrinol Metab. 1999;84:2064–2067. doi: 10.1210/jcem.84.6.5733. [DOI] [PubMed] [Google Scholar]
- 171.Mousa S.A., O’Connor L.J., Bergh J.J., Davis F.B., Scanlan T.S., Davis P.J. The proangiogenic action of thyroid hormone analogue GC-1 is initiated at an integrin. J Cardiovasc Pharmacol. 2005;46:356–360. doi: 10.1097/01.fjc.0000175438.94906.a0. [DOI] [PubMed] [Google Scholar]
- 172.Biondi B., Wartofsky L. Combination treatment with T4 and T3: toward personalized replacement therapy in hypothyroidism? J Clin Endocrinol Metab. 2012;97:2256–2271. doi: 10.1210/jc.2011-3399. [DOI] [PubMed] [Google Scholar]
- 173.Robert C., Soria J.C., Spatz A. Cutaneous side-effects of kinase inhibitors and blocking antibodies. Lancet Oncol. 2005;6:491–500. doi: 10.1016/S1470-2045(05)70243-6. [DOI] [PubMed] [Google Scholar]
- 174.Yang C.H., Lin W.C., Chuang C.K. Hand–foot skin reaction in patients treated with sorafenib: a clinicopathological study of cutaneous manifestations due to multitargeted kinase inhibitor therapy. Br J Dermatol. 2008;158:592–596. doi: 10.1111/j.1365-2133.2007.08357.x. [DOI] [PubMed] [Google Scholar]
- 175.Anderson R., Jatoi A., Robert C., Wood L.S., Keating K.N., Lacouture M.E. Search for evidence-based approaches for the prevention and palliation of hand–foot skin reaction (HFSR) caused by the multikinase inhibitors (MKIs) Oncologist. 2009;14:291–302. doi: 10.1634/theoncologist.2008-0237. [DOI] [PubMed] [Google Scholar]
- 176.Brandt J., Briddell R.A., Srour E.F., Leemhuis T.B., Hoffman R. Role of c-kit ligand in the expansion of human hematopoietic progenitor cells. Blood. 1992;79:634–641. [PubMed] [Google Scholar]
- 177.Gerber H.P., Malik A.K., Solar G.P. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature. 2002;417:954–958. doi: 10.1038/nature00821. [DOI] [PubMed] [Google Scholar]
- 178.Gabrilovich D., Ishida T., Oyama T. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–4166. [PubMed] [Google Scholar]
- 179.Mendel D.B., Laird A.D., Xin X. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- 180.Abrams T.J., Lee L.B., Murray L.J., Pryer N.K., Cherrington J.M. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2:471–478. [PubMed] [Google Scholar]
- 181.Kapiteijn E., Brand A., Kroep J., Gelderblom H. Sunitinib induced hypertension, thrombotic microangiopathy and reversible posterior leukencephalopathy syndrome. Ann Oncol. 2007;18:1745–1747. doi: 10.1093/annonc/mdm454. [DOI] [PubMed] [Google Scholar]
- 182.Trinkaus M., Trudeau M., Callum J. Drug-induced immune thrombocytopenic purpura secondary to sunitinib. Curr Oncol. 2008;15:152–154. doi: 10.3747/co.v15i3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yoo C., Kim J.E., Lee J.L. The efficacy and safety of sunitinib in korean patients with advanced renal cell carcinoma: high incidence of toxicity leads to frequent dose reduction. Jpn J Clin Oncol. 2010;40:980–985. doi: 10.1093/jjco/hyq073. [DOI] [PubMed] [Google Scholar]
- 184.Schmidinger M., Arnold D., Szczylik C., Wagstaff J., Ravaud A. Optimizing the use of sunitinib in metastatic renal cell carcinoma: an update from clinical practice. Cancer Invest. 2010;28:856–864. doi: 10.3109/07357901003631080. [DOI] [PubMed] [Google Scholar]
- 185.Donskov F, et al. Neutropenia and thrombocytopenia during treatment as biomarkers of sunitinib efficacy in patients with metastatic renal cell carcinoma (mRCC). Poster presentation at the European Multidisciplinary Cancer Congress; 2011 [abstr. 1141].
- 186.Puzanov I, et al. Evaluation of hand–foot syndrome (HFS) as a potential biomarker of sunitinib (SU) efficacy in patients (pts) with metastatic renal cell carcinoma (mRCC) and gastrointestinal stromal tumour (GIST). Poster presentation at the European Multidisciplinary Cancer Congress; 2011 [abstr. 1444].
- 187.Davis Meal. Asthenia and fatigue as potential biomarkers of sunitinib efficacy in metastatic renal cell carcinoma. Poster presentation at the European Multidisciplinary Cancer Congress; 2011 [abstr. 1139].
- 188.Kim J.J., Vaziri S.A., Rini B.I. Association of VEGF and VEGFR2 single nucleotide polymorphisms with hypertension and clinical outcome in metastatic clear cell renal cell carcinoma patients treated with sunitinib. Cancer. 2012;118:1946–1954. doi: 10.1002/cncr.26491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Garcia-Donas J., Esteban E., Leandro-Garcia L.J. Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: a multicentre, observational, prospective study. Lancet Oncol. 2011;12:1143–1150. doi: 10.1016/S1470-2045(11)70266-2. [DOI] [PubMed] [Google Scholar]
- 190.Szmit S., Langiewicz P., Zlnierek J. Hypertension as a predictive factor for survival outcomes in patients with metastatic renal cell carcinoma treated with sunitinib after progression on cytokines. Kidney Blood Press Res. 2012;35:18–25. doi: 10.1159/000329933. [DOI] [PubMed] [Google Scholar]
- 191.Sommers Smith S.K., Smith D.M. Beta blockade induces apoptosis in cultured capillary endothelial cells. In Vitro Cell Dev Biol Anim. 2002;38:298–304. doi: 10.1290/1071-2690(2002)038<0298:BBIAIC>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 192.Menezes M.D., McCarter R., Greene E.A., Bauman N.M. Status of propranolol for treatment of infantile hemangioma and description of a randomized clinical trial. Ann Otol Rhinol Laryngol. 2011;120:686–695. doi: 10.1177/000348941112001010. [DOI] [PubMed] [Google Scholar]
- 193.Wasa J., Sugiura H., Kozawa E., Kohyama K., Yamada K., Taguchi O. The tumor suppressive effect of angiotensin II type 1 receptor antagonist in a murine osteosarcoma model. Anticancer Res. 2011;31:123–127. [PubMed] [Google Scholar]
- 194.Gong Q., Davis M., Chipitsyna G., Yeo C.J., Arafat H.A. Blocking angiotensin II Type 1 receptor triggers apoptotic cell death in human pancreatic cancer cells. Pancreas. 2010;39:581–594. doi: 10.1097/MPA.0b013e3181c314cd. [DOI] [PubMed] [Google Scholar]
- 195.Molteni A., Heffelfinger S., Moulder J.E., Uhal B., Castellani W.J. Potential deployment of angiotensin I converting enzyme inhibitors and of angiotensin II type 1 and type 2 receptor blockers in cancer chemotherapy. Anticancer Agents Med Chem. 2006;6:451–460. doi: 10.2174/187152006778226521. [DOI] [PubMed] [Google Scholar]
- 196.Santoni G., Santoni M., Nabissi M. Functional role of T-type calcium channels in tumour growth and progression: prospective in cancer therapy. Br J Pharmacol. 2012;166:1244–1246. doi: 10.1111/j.1476-5381.2012.01908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bono P., Elfving H., Utriainen T. Hypertension and clinical benefit of bevacizumab in the treatment of advanced renal cell carcinoma. Ann Oncol. 2009;20:393–394. doi: 10.1093/annonc/mdn729. [DOI] [PubMed] [Google Scholar]
- 198.Nozawa M, Matsumura N, Yasuda M, Okuda Y, Uemura H. Activity of retreatment with sorafenib for metastatic renal cell carcinoma. In: ASCO Genitourinary Cancers Symposium, February 17–19, Orlando, Florida, 2011 [abstr. 404].
- 199.Rixe O. Diastolic blood pressure (dBP) and pharmacokinetics (PK) as predictors of axitinib efficacy in metastatic renal cell cancer (mRCC) J Clin Oncol. 2009;27(Suppl.):15s. [abstr. 5045] [Google Scholar]
- 200.Feal Bladou. Hypothyroidism and survival during sunitinib therapy in metastatic renal cell carcinoma (mRCC): a prospective observational analysis. J Clin Oncol. 2010;28(Suppl.):13. [abstr. e15013] [Google Scholar]
- 201.Baldazzi V., Tassi R., Lapini A. The impact of sunitinib-induced hypothyroidism on progression-free survival of metastatic renal cancer patients: a prospective single-center study. Urol Oncol. 2012 Sep;30(5):704–710. doi: 10.1016/j.urolonc.2010.07.015. [DOI] [PubMed] [Google Scholar]