


Abstract
Despite declining sequencing costs, few methods are available for cost-effective single-nucleotide polymorphism (SNP), insertion/deletion (INDEL) and copy number variation (CNV) discovery in a single assay. Commercially available methods require a high investment to a specific region and are only cost-effective for large samples. Here, we introduce a novel, flexible approach for multiplexed targeted sequencing and CNV analysis of large genomic regions called multiplexed direct genomic selection (MDiGS). MDiGS combines biotinylated bacterial artificial chromosome (BAC) capture and multiplexed pooled capture for SNP/INDEL and CNV detection of 96 multiplexed samples on a single MiSeq run. MDiGS is advantageous over other methods for CNV detection because pooled sample capture and hybridization to large contiguous BAC baits reduces sample and probe hybridization variability inherent in other methods. We performed MDiGS capture for three chromosomal regions consisting of ∼550 kb of coding and non-coding sequence with DNA from 253 patients with congenital lower limb disorders. PITX1 nonsense and HOXC11 S191F missense mutations were identified that segregate in clubfoot families. Using a novel pooled-capture reference strategy, we identified recurrent chromosome chr17q23.1q23.2 duplications and small HOXC 5′ cluster deletions (51 kb and 12 kb). Given the current interest in coding and non-coding variants in human disease, MDiGS fulfills a niche for comprehensive and low-cost evaluation of CNVs, coding, and non-coding variants across candidate regions of interest.
INTRODUCTION
As candidate genes are identified for human diseases, there is an increasing need to sequence the regions of interest at high depth in larger populations. Although custom capture and amplicon-based sequencing methods are commercially available, they require a high initial investment in probe designs and only become cost-effective when amortized over large sample sizes that are often not available for rare disorders. Additionally, many methods focus on a single type of variant such as SNPs and INDELs and are poorly designed to detect copy number variations (CNVs), or are limited to only the coding region. This presents a paradox for researchers because a significant financial investment must be made for a specific region and type of variation before having the evidence that these variants are relevant for the disorder in question.
Despite declining sequencing costs and the development of novel targeted capture methods, even modestly sized sequencing projects are still cost prohibitive for most investigators. Therefore, we set out to develop a low-cost methodology that combines SNP/INDEL detection with CNV analysis across multiple coding and non-coding regions. The goal was to develop an approach that was scalable for both small and large studies, yet flexible enough to apply to candidate resequencing projects ranging from multiple large genomic regions to large multi-exonic genes that can be especially costly to sequence. To achieve this, we developed a novel approach called multiplexed direct genomic selection (MDiGS), which utilizes a biotinylated bacterial artificial chromosome (BAC) based capture (1) combined with multiplexed pooled capture of up to 96 samples (2) and next-generation sequencing for SNP/INDEL detection and highly accurate CNV analysis. BACs tile the human genome (n = 208 922) with an average size of 147 514 bp (25 001–349 971 bp range). These BACs cover >92% of the human genome, >99.4% of the consensus coding sequence (CCDS), and are readily available for $80 each. Because MDiGS avoids the costly investment required for custom probe designs, MDiGS provides low-cost and comprehensive alternative for candidate screening of large genes and gene clusters, analysis of extensive non-coding regions, analysis of linkage and GWAS loci for small sample sizes, or as a pilot screen before committing to a region of interest with alternative methods.
MDiGS introduces a novel copy number analysis method that provides distinct advantages over currently available capture methods. Major limitations of other targeted capture CNV detection methods are sample-to-sample variability and probe hybridization variability, both of which are minimized in MDiGS. With many non-multiplexed methods, sample variability results from differences in pre- and post-capture library amplification and efficiency of individual hybridization reactions, making it difficult to determine if coverage differences represent true CNVs. The use of small non-contiguous probes as capture baits is also problematic, since individual probes are inherently difficult to quantify precisely and equivalently in batches. Although several analysis methods have been proposed to improve CNV detection in non-contiguous targeted capture data (3–5), MDiGS addresses both of these challenges by performing pre-capture DNA multiplex pooling and hybridization to large contiguous BAC baits. Since all amplification and hybridizations steps are performed as a pool, sample-to-sample variability is reduced allowing for a high signal-to-noise ratio. Because the BAC bait is prepared from a single linear stretch of DNA, region-to-region differences are reduced, the likelihood of identifying reads spanning breakpoints is increased, and CNVs are more accurately detected.
We applied MDiGS to a study of human lower limb disorders in which rare CNVs, SNP/INDELs and non-coding regions had previously been implicated. We captured and sequenced pools of up to 96 indexed samples for 554 kb of genomic sequence across three chromosomal regions associated with lower limb disorders (5′ HOXC cluster (6), PITX1 (7,8) and TBX4 (9)) on a single MiSeq run. Using MDiGS, we found comparable or better coverage of coding and non-coding sequences compared to whole exome sequencing (WES) and whole genome sequencing (WGS), particularly in problematic GC-rich regions (10,11) compared to WGS. Sensitivity and specificity of the method was demonstrated by 100% concordance for all variant and reference genotyping calls for samples sequenced with MDiGS and WES. CNV detection using MDiGS was compared to samples analyzed on the Affymetrix 6.0 platform and comparable sensitivity and specificity for large CNVs (>100 kb) and increased sensitivity for small CNVs (>12 kb) was demonstrated. MDiGS provides a powerful and flexible, low-cost method of candidate screening for SNP/INDELs and CNVs and is widely applicable for resequencing of large genes and multiple genomic regions.
MATERIALS AND METHODS
Patient samples and controls
We identified 253 patients with idiopathic lower limb abnormalities (clubfoot, vertical talus, tibial hemimelia and polydactyly) from the Washington University Musculoskeletal DNA Databank who were recruited from St Louis Children's Hospital and St Louis Shriners Hospital. Patients were excluded from the study if they had additional non-limb congenital anomalies, developmental delay or known underlying etiologies such as arthrogryposis, myelomeningocele or myopathy. DNA was collected from blood and saliva from affected probands and family members after obtaining informed consent. DNA extractions were performed using the manufacturer's protocols using either the DNA Isolation Kit for Mammalian Blood (Roche, Indianapolis, IN, USA) or the Oragene Purifier for saliva (DNA Genotek, Kanata, ON, Canada).
Multiplexed direct genomic selection
Enrichment of large genomic regions was performed on cases and controls using the MDiGS method. MDiGS uses a biotinylated BAC-based capture (1) combined with multiplexed pooled-capture (2) and next-generation sequencing; the protocol is described in detail in Supplementary Methods. Briefly, individual samples were indexed in batches of 96 using 150 ng of genomic DNA. Indexed samples were pooled in batches of 48 prior to BAC capture. BACs were obtained from BACPAC Resources (Oakland, California), purified with NucleoBond BAC 100 (Clonetech), biotinylated with the Nick Translation System (Invitrogen) and used to capture the following coding and non-coding genomic regions: FBN1 (RP11-147E14 and RP11-1144G24 chr15:48676783–48985158), FBN2 (RP11-909P14 and RP11-351A8 chr5:127567172–127906815), PITX1 (RP11-17G15 chr5:134350801–134514499), HOXC cluster (RP11-578A18 chr12:54225979–54410228) and TBX4 (RP11-905P16 chr17:59468421–59674379). Ninety-six multiplexed post-capture samples were sequenced using a single 500 cycle MiSeq Personal Sequencer run (Illumina, San Diego, California).
Exome and whole genome sequencing
Exome enrichment was performed on DNA from controls (n = 121) using the SureSelect Human All Exon 50 MB kits (Agilent Technologies, Santa Clara, California). Ten cases with idiopathic lower limb abnormalities were selected for both exome and MDiGS capture to determine the concordance in variant calling and calculate the median absolute pairwise difference (MAPD) scores for WES and MDiGS. WGS was performed by Illumina on DNA from controls (n = 73) and sequenced to 30× coverage. All WES and WGS samples were sequenced to >95% CCDS at ≥8× coverage.
Next-generation sequencing and variant calling
MDiGS reads were demultiplexed by index, allowing for 1 bp mismatches (index sequence Hamming distance ≥2). Identical pipelines were used for MDiGS, WES and WGS alignments and raw variant calling. Samples were aligned to hg19 human reference sequence (NCBI 37.0) using Novoalign V2.08.02 (Novocraft Technologies, Selangor, Malaysia) using the parameters ‘-H –r none –i 300 100 –a [indexed adapters]’ and output in SAM format. Variant calling was performed using SAMtools v0.1.18 mpileup for all capture regions with ‘-C50 –q 20 –Q20 –Eu’ and piped to bcftools “view –bvcg”. Raw reads were filtered for a read depth of 8 and a minimum of three variant reads using vcfutils ‘varFilter –d 8 –a 3’. To correct for potential index switching during pre- and post-capture amplification, MDiGS heterozygous calls with fewer than 20% of the reads matching the reference or variant allele were changed to the homozygous variant or wild-type genotype, respectively, as described previously (2).
Copy number analysis
Copy number analysis was first performed on MDiGS samples using a custom approach to identify large deletions and duplications spanning the entire capture region. Poor-quality samples with total BAC coverage less than 80% at 8× were excluded from CNV analysis. For large CNV analysis, each individual sample's read counts mapping to each region were first normalized to the other two target regions to correct for sample loading efficiencies. Each normalized test sample and region was individually compared to (Method 1) the normalized read counts of two healthy controls or (Method 2) the average normalized read counts for all other test samples for each corresponding target region from the same capture batch. Samples with large duplications and deletions comprising the entire BAC length, defined as copy number (normalized test:avg normalized reference ratio)/0.5 ≥ 2.5 or ≤ 1.5 respectively, were easily identified using this analysis. For small CNV analysis, breakpoints within the capture region were detected using CNV-seq using global normalization and 1500 bp window size (12).
Copy number analysis was performed individually for each test sample against (Method 1) a known healthy control reference or (Method 2) against 10 randomly selected test samples. Samples with large duplications or deletions identified in the first stage of the analysis were excluded as reference samples for the pooled reference analysis. Log2 ratios for each window were averaged across all pooled reference comparisons for each test sample. CNVs were defined as log2 ± 0.5 and ≥10 kb and were validated by qPCR. One-end anchored (OEA) reads, reads with one mapped and one unmapped read, were used to identify breakpoints. OEA reads with one paired end mapped within 1 kb of predicted breakpoints were identified for all samples and the unmapped paired ends were mapped using UCSC BLAT to identify exact breakpoints.
MAPD scores were calculated as the median absolute log2 difference between the average base coverage for adjacent 500 bp windows or Agilent SureSelect Human All Exon 50 MB target regions within the BAC capture regions. A paired Student's t-test was used to calculate p-values for MDiGS versus exome capture MAPD scores.
RESULTS
Multiplexed direct genomic selection
We first identified BACs on three different chromosomes spanning the entire coding and flanking regulatory sequence (554 kb) of regions previously associated with lower limb development: PITX1, TBX4, and the 5′ HOXC genes and non-coding RNAs (HOXC8-HOXC13, HOTAIR, HOXC-AS5 and MIR196A2). To determine the consistency of BAC capture against different target regions, we also identified BACs spanning two large multi-exonic genes on separate chromosomes that are especially costly to sequence, FBN1 (308 kb, 66 exons) and FBN2 (340 kb, 65 exons) for separate experiments. BACs were individually biotinylated using the Nick Translation System (Invitrogen) and combined at equal molar ratios for capture of multiple target regions (1).
While the initial direct genomic selection method described by Bashiardes et al. required two BAC hybridization steps to sufficiently enrich for targeted regions, we introduced a non-biotin labeled off-target BAC as a simple repeat element blocking agent that allowed us to reduce the hybridization to a single step. Indexed libraries from 150 ng of genomic DNA were pooled in batches of 48 samples at equal concentrations (2) and hybridized with the biotinylated BACs (Figure 1). Enriched, multiplexed libraries were then sequenced in pools of 96 indexed samples on a single MiSeq run.
Figure 1.
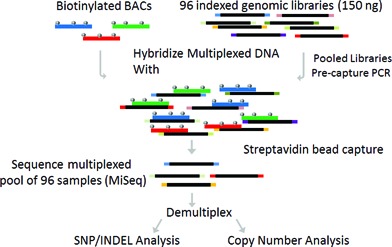
Overview of multiplexed direct genomic selection (MDiGS) methodology. Three BACs were selected for use as baits and biotinylated for targeted capture of 554 kb of coding and non-coding sequence surrounding PITX1, 5′ HOXC genes and TBX4. Genomic input DNA (150 ng) was individually indexed and pooled in batches of 96. Multiplexed DNA pools were hybridized with biotinylated BAC baits and enriched target regions are sequenced on MiSeq Personal Sequencer.
We first evaluated the effects of increasing the number of target regions on target enrichment. We performed MDiGS on pools of 48 samples captured for one, two or three regions and calculated the number of on-target reads for each capture pool (Figure 2A). To determine the technical variability in BAC bait production, we performed each capture in duplicate using separately prepared BAC baits and achieved similar enrichments for each replicate. We observed higher on-target percentage as target regions increased (Figure 2), one region = 25.4% (±2.4), two regions = 39% (±3.5) and three regions = 40% (±2.9), however, on-target enrichment of individual regions decreased as expected as the number of target regions increased. On-target enrichment for each target region was consistent across all samples sequenced on a single MiSeq lane (Figure 2B) and we did not observe a difference in total on-target enrichment for 24 versus 48 sample pre-capture multiplexing (Supplementary Figure S1).
Figure 2.
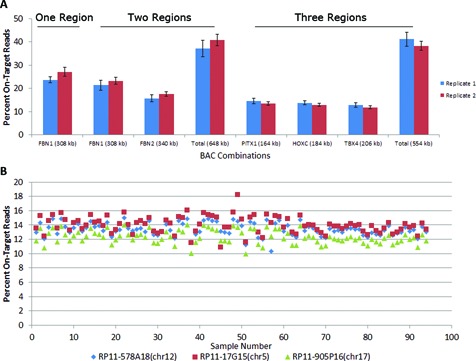
MDiGS Target Enrichment. Pre-capture multiplexed pools of 48 samples were captured for (A) one (FBN1), two (FBN1 and FBN2) or three different chromosome regions (PITX1, HOXC and TBX4). Separately prepared BAC baits were used for each replicate. (B) On-target enrichment of three regions (PITX1, HOXC and TBX4) was consistent for each region and sample (12–14%).
MDiGS coverage compared to WGS and exome capture
MDiGS pooled capture of 96 samples achieved on average 97% (±1.9) at ≥8× coverage across three targeted regions (554 kb) (Supplementary Figure S2), similar to whole-genome sequenced (WGS) samples (n = 73) sequenced to 30× coverage (Figure 3A). MDiGS coverage of coding sequence was comparable to exome captured samples (n = 121), with 90% at 8× coverage, which is lower than the non-coding coverage because of the high GC content within several coding exons of HOXC and PITX1 genes. Coverage was slightly reduced for both exome and MDiGS capture of GC-rich regions (GC > 60%), however, both capture methods performed better than WGS for GC-rich regions (Figure 3B). To determine concordance in variant detection between MDiGS and exome sequencing, 10 samples sequenced using both methods were compared. Overall, 100% concordance was noted for all polymorphic genotypes (n = 120) and all genotypes (n = 161 961) called at positions covered ≥8× by both methods.
Figure 3.
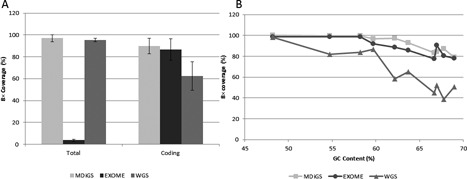
MDiGS coverage of target regions. (A) Total coverage of BAC-targeted coding and non-coding regions were equivalent to whole genome sequencing and coverage of coding regions were equivalent to exome-captured sequencing and superior to whole genome sequencing. (B) Overall coverage of GC-rich regions decreased as GC content increases in all three platforms (MDiGS, exome sequencing and whole genome sequencing). Both BAC- and exome-capture-based methods performed better than whole genome sequencing for high GC-rich genes.
Large copy number analysis
Since most targeted resequencing technologies utilize non-contiguous probes which can introduce region-to-region variability, we compared the median absolute pairwise distance (MAPD) scores of 10 samples sequenced by exome capture and MDiGS. MAPD scores are calculated as the median absolute log2 difference between neighboring probes. We calculated the MAPD scores for all non-contiguous exome target regions within our BAC baits for both MDiGS and exome samples. Since MDiGS utilizes each BAC bait as a large contiguous probe, we also calculated MAPD scores for 500 bp adjacent windows since this is the average size of exome probe targets. MDiGS region-to-region variability was significantly reduced compared to exome samples for non-contiguous exome target regions (p = 1.7 × 10−5) and further reduced using 500 bp adjacent windows (p = 1.1 × 10−8) (Figure 4A).
Figure 4.
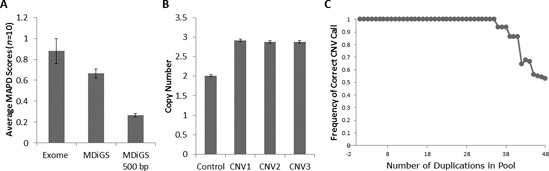
MDiGS copy number analysis. (A) Median absolute pairwise difference (MAPD) scores were calculated as the log2 absolute difference between adjacent target regions for 10 samples enriched by exome capture and MDiGS to compare region-to-region variability of similar target regions. MDiGS MAPD scores were also calculated for 500 bp adjacent windows to demonstrate reduced region-to-region variability achieved by using long contiguous BAC baits. Large duplications spanning an entire BAC can be detected in (B) small sample sizes using Method 1, which compares individual test samples to normal controls or (C) large sample sizes using Method 2. Simulated data demonstrate how individual test samples with large CNVs can be correctly identified within a multiplexed pool of 96 test samples with unknown CNV state provided that less than 36 samples contain similar CNVs.
Most reference-based copy number analysis methods rely on global normalization to correct for sample loading differences (12), which could be particularly problematic for identifying large CNVs spanning an entire BAC captured by MDiGS. To determine if both large and small CNVs could be detected with MDiGS and standard normalization methods, we included cases with known Affymetrix 6.0 microarray validated CNVs spanning an entire capture region as well as cases with CNV breakpoints within a capture region.
To determine if large duplications spanning an entire captured target region (i.e. BAC) could be detected, MDiGS was performed on a pool of 96 DNA samples including three cases with known chromosome 17q23.1q23.2 duplications (9). Data were initially analyzed with CNV-seq (12) that uses a binned coverage comparison of a single test sample compared to a single reference sample using global or chromosomal normalization. Unfortunately, because the chromosome 17q23.1q23.2 duplication represents >37% of the entire captured sequence, the chromosomal and global normalization methods utilized by CNV-seq effectively removed the signal across the capture region and the CNV-seq method therefore failed to detect all three known duplications included in the MDiGS pool.
Therefore, to identify CNVs with MDiGS, we developed two strategies that take advantage of pooled pre-capture and the presence of multiple large chromosomal target regions (BACs). Because pre- and post-capture amplifications, bait hybridization and next-generation sequencing are performed as pooled reactions, MDiGS reduces the technical variability due to library amplification and capture hybridizations that are inherent in individually captured sample comparisons. Thus, MDiGS provides multiple internal capture and sequencing depth controls across each sample and contiguous capture region that can be used to improve CNV detection accuracy. Therefore, two alternative and complementary methods described below were utilized for CNV detection of MDiGS data.
Method 1 compares read counts of individual test samples to negative controls and is ideal for smaller sample sizes. To identify CNVs spanning the entire target region, individual sample total on-target read counts for each BAC capture region were first compared to the on-target read counts for all other capture regions for the same sample: NR1 = R1 / (R2 + R3). Each test sample is then individually compared to the averaged normalized read counts of the two normal controls for the same target regions. Using this method, we individually compared three positive control samples with 17q23.1q23.2 duplications to two normal controls and accurately detected all three patients with duplications (Figure 4B).
Method 2 identifies rare CNVs from a multiplexed pool of MDiGS test samples and is ideal for larger sample sizes. Since comparison to controls is highly dependent on the quality of the control sample data and utilizes valuable sample space within the multiplex pool that otherwise could be used for cases, we sought to determine if rare CNVs could be identified from a pool of test samples with unknown CNV states. We simulated a pooled reference analysis of captured samples and calculated normalized read counts for each test sample region compared to the averaged normalized read counts across all other test samples (n−1) from the same captured batch. Since additional co-captured samples could theoretically contain recurrent CNVs of the same region, increasing replicates of samples containing whole chromosome 17q23.1q23.2 duplications versus normal control samples (validated by Affymetrix genome-wide 6.0 array) were simulated in a single capture analysis. In the simulation, copy number state was correctly called for all 96 samples when less than 36 samples with chromosome 17q23.1q23.2 duplications were included in a multiplexed pool (Figure 4C), suggesting that this method is robust for rare CNV detection.
High-resolution copy number analysis
We next determined whether small CNVs within the target region could be identified and mapped to high resolution using one-end anchored read alignments (OEA), which uses mapped reads within 1 kb of a predicted breakpoint to identify unmapped paired-ends spanning the CNV breakpoints (3,13). To test the sensitivity of small CNV detection, we included two samples with one chromosomal deletion breakpoint within the targeted capture regions at chr12:54165001–54335668 (5′ HOXC) (6) or chr5:134194484–134435123 (PITX1) (7). CNV-seq analysis was performed using global normalization and a 1500 bp window size for each sample compared to 10 randomly selected test samples from the same pool. Log2 differences for each bin were averaged across all reference comparisons to reduce background noise and further refine CNV breakpoints. Only bins with averaged log2 +/− 0.5 were called for CNVs using CNV-seq Perl and R packages (12).
Unlike the chromosome 17q23.1q23.2 duplications that were not detected using CNV-seq because they spanned the entire target region (see above), deletions that were partially contained within the target region were detected for both PITX1 (Figure 5A) and HOXC (Figure 5C) deletions with CNV-seq using global normalization and pooled reference analysis. An OEA read strategy was then used to identify exact breakpoints of CNVs both within and outside the targeted capture regions (3,13). First, we identified all OEAs for which the mapped read was within 1 kb of a predicted breakpoint and UCSC BLAT was used to map the unmapped paired-end spanning the CNV breakpoint. Even though 5′ breakpoints were outside the targeted capture region for both deletions, this analysis resulted in our identification of reads that overlapped both the 5′ and 3′ CNV breakpoints, allowing us to precisely map both CNV breakpoints for each deletion (Figure 5B and D).
Figure 5.
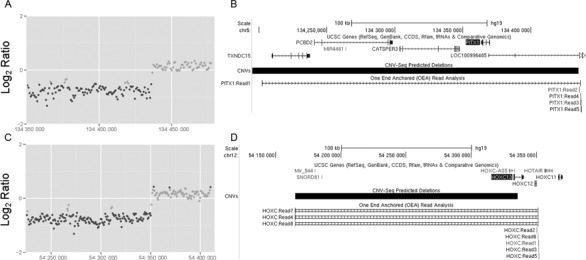
Microdeletions can be detected using MDiGS. (A) Deletions with breakpoints within the MDiGS targeted capture regions of (A) PITX1 and (C) HOXC were detected with CNV-seq analysis. (B and D) Exact locations of both 5′ and 3′ CNV breakpoints were mapped with OEA read analysis and UCSC BLAT.
Novel SNP/INDEL and CNV detection in lower limb malformations
To determine the frequency of SNP/INDELs and small CNV rare variants in PITX1, TBX4 and 5′ HOXC cluster in humans with lower limb disorders, we next performed MDiGS on a cohort of 253 patients with lower limb malformations and 168 controls. We identified and validated 13 rare missense or small INDEL variants (MAF < 0.01) causing protein coding changes in PITX1, HOXC13, HOXC12, HOXC11, HOXC10, HOXC8 and TBX4 and two rare variants in HOTAIR, a non-coding RNA located within the HOXC 5′ gene cluster that is not typically captured by commercial exome capture kits (Supplementary Table S1). Six of these rare variants causing protein coding changes were not previously described in public databases (dbSNP, 1000 Genomes or EVS) or in the control sample sequence with MDiGS. These variants include a novel PITX1 frameshift mutation and a HOXC11 S191F missense mutation that both segregate with clubfoot (data not shown).
To determine whether common variants near PITX1, TBX4 or HOXC genes are associated with lower limb abnormalities, we selected 190 unrelated Caucasian cases and 156 controls for a case/control association analysis using PLINK (14). We excluded rare variants (MAF < 0.05), variants out of Hardy–Weinberg equilibrium (p ≤ 0.001) and variants with missing calls in >10% of cases and controls. None of the common variants were significant after correction for multiple testing.
Because large CNVs involving PITX1, TBX4 and HOXC genes have previously been described in patients with lower limb malformations using Affymetrix 6.0 arrays that have a lower size limit of detection of approximately 100 kb in our prior studies (6), we were interested in determining whether additional smaller CNVs involving these genes could be detected with MDiGS. Therefore, copy number analysis was performed on 253 MDiGS captured samples from patients with clubfoot and other lower limb disorders and 168 controls. To confirm the accuracy of CNV calls, three samples with known chromosome 17q23.1q23.2 duplications were included in this capture. We first performed pooled reference analysis on all cases and controls in order to identify CNVs that span the entire target region. All three samples with known chromosome 17q23.1q23.2 duplications and one additional new case with a chromosome 17q23.1q23.2 duplication were identified (Figure 6A) and no CNVs were predicted in the control samples. The chromosome 17q23.1q23.2 duplication in this clubfoot case was validated by qPCR and segregated with an affected parent (Figure 6B and C).
Figure 6.
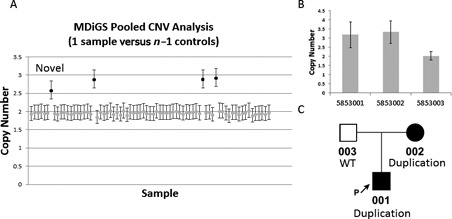
MDiGS identification of recurrent chromosome 17q23.1q23.2 duplications. Recurrent chromosome 17q23.1q23.2 duplications spanning the entire target region were identified using MDiGS method 2, pooled CNV analysis, in (A) three positive controls and one additional clubfoot proband (novel). The novel chromosome 17q23.1q23.2 duplication (in pt 5853001) was (B) validated by qPCR and (C) shown to segregate with the affected parent.
Samples with smaller CNVs were then identified using CNV-seq after excluding cases with large CNVs (detected by pooled CNV analysis) as reference samples. Two novel chromosomal microdeletions were identified in two separate cases on chromosome 12 at the HOXC gene cluster (Figure 7A and B). These include a 51 kb deletion (chr12:54311194–54361982) overlapping the HOXC cluster identified in a patient with vertical talus and a 12.7 kb deletion located 5′ of the HOXC gene cluster identified in a patient with isolated clubfoot (chr12:54303751–54316500). OEA read analysis was used to identify the precise breakpoints as previously described (Figure 7C). This 51 kb microdeletion had not previously been identified with Affymetrix 6.0 microarray analysis (data not shown) nor was it detected by analysis of WES data with CONTRA (15), a copy number analysis tool for targeted resequencing that was specifically designed for whole-exome capture data (Supplementary Figure S3), demonstrating the superiority of MDiGS for detection of small microdeletions.
Figure 7.
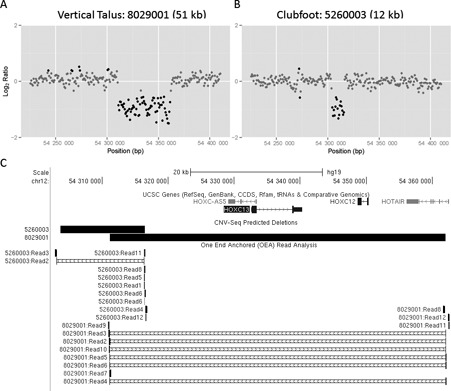
Small internal CNVs detected by MDiGS. (A and B) Two novel microdeletions located within the 5′ HOXC gene cluster were identified in two patients with lower limb malformations, 8029001 (51 kb) and 5260003 (12 kb), using merged CNV-Seq analysis. Breakpoints were identified with (C) one-end anchored (OEA) read analysis.
DISCUSSION
MDiGS provides a flexible, low-cost alternative method for improved CNV detection and SNP/INDEL screening of variable sample sizes. Custom targeted capture with currently available commercial products are suitable for large sample sizes in which the individual sample cost is reduced by spreading the high cost of custom probes or primers over large sample sizes (16–18). However, for rare disorders, large sample sizes are not available, and the cost of gene resequencing per sample becomes prohibitively expensive. Furthermore, custom capture design locks an investigator into a specific experimental design, as the genes and regions to be sequenced need to be selected at the beginning of the experiment, and cannot be modified as samples are captured and sequenced, a process that may take several months to complete. In contrast, the MDiGS method described here utilizes BACs that are readily available at low cost ($80) for nearly all chromosomal regions and genes, making MDiGS ideal for candidate locus screening.
MDiGS capture efficiency is similar to previously described pre-capture multiplexing methods (2,19). Although on-target percentage is reduced compared to post-capture multiplexing (20), enrichment is sufficient to cover >97% of target bases at ≥8× coverage for 554 kb of target sequence in 96 samples on a single MiSeq lane. Using only 150 ng of genomic DNA with our library prep protocol, pre-capture multiplexed DNA libraries can typically be used for up to 10 subsequent captures with different targets, adding flexibility and reducing the cost of sequencing additional target regions by eliminating the need for costly sample preps. BAC-based targeted selection is particularly suitable for resequencing of large genes with multiple exons since other amplicon- and probe-based approaches require individually designed baits for each exon. Also, with new interest in the role of non-coding regions in human disease (21–23), our BAC-based approach comprehensively evaluates non-coding regulatory sequences at significantly lower cost than WGS.
MDiGS is based on direct genomic selection (DiGS), a BAC-based capture method (1) that achieved 52% on-target capture using two BAC hybridization steps (24), although it did not utilize multiplexed patient DNA capture and was not optimized for CNV analysis. Our modifications of their method include use of a non-biotin labeled off-target BAC as a blocking agent to reduce hybridization to a single step. However, the most significant modification of their protocol is our utilization of the BAC-based method to capture multiplexed DNA samples in a pooled capture that optimizes concurrent CNV analysis. In addition to cost advantages, we demonstrate that multiplexed BAC capture of pooled samples is highly accurate for large and small CNV and SNP/INDEL detection. The MDiGS protocol is now designed for 96 multiplexed samples and takes advantage of an Illumina multiplexed library preparation that requires only 150 ng of input genomic DNA (Supplementary Methods). MDiGS can also be used with dual indexing to increase sample numbers or to reduce the potential risk of index switching that would be required for diagnostic sequencing. With MDiGS, a single MiSeq run is sufficient to analyze 96 patient samples for up to 550 kb of sequence across multiple targeted genomic loci. Although we have not identified the upper limits of genomic sequence that can be covered in a single MiSeq run, enriched libraries can be scaled up to HiSeq 2000 lanes for larger number of samples or post-capture libraries can be pooled for increased targeted regions.
A unique strength of MDiGS is the additional high-resolution and accurate CNV detection capabilities obtained from pooled capture in addition to the detection of SNPs and INDELs. Identification of a variety of genetic and genomic abnormalities using a single platform provides more comprehensive candidate gene analysis than is currently available. Using MDiGS, we demonstrate two distinct strategies to reliably detect (i) common CNVs by comparison to healthy reference samples, which is ideal for smaller sample sizes, and (ii) rare CNVs in a multiplexed pool of test samples with unknown CNV state. With MDiGS, smaller CNVs with one or both breakpoints within the capture region are identifiable with available CNV detection software such as CNV-seq. We also demonstrated that using large contiguous BAC baits provides lower region-to-region variability and higher sensitivity CNV detection compared to non-contiguous probes used for exome targeted capture. Furthermore, OEA reads can be used to map exact breakpoints with higher resolution than microarray-based CNV detection methods.
Our application of the MDiGS method to candidate gene screening of 253 patients with lower limb malformations identified novel single nucleotide polymorphisms and novel copy number variants in PITX1, TBX4 and HOXC genes, confirming the importance of both CNVs and point mutations in the etiology of these genetically heterogeneous disorders. Furthermore, our method allowed us to identify additional mutations in the non-coding regulatory sequences of all three genes that can now be studied for their role in hindlimb development. While there are a variety of methods available for targeted sequencing and CNV analysis, MDiGS offers a valuable cost-effective alternative for comprehensive screening of genomic loci.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank the patients and their families for their role in this work. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for providing Illumina MiSeq Personal Sequencer support.
FUNDING
Health Eunice Kennedy Shriver National Institute of Child Health and Human Development [R03HD068649NIH]; Shriners Hospital for Children; Children's Discovery Institute of Washington University; St. Louis Children's Hospital; NCI Cancer Center Support Grant [P30 CA91842 to the Siteman Cancer Center]; National Center for Research Resources (NCRR), National Institutes of Health (NIH) [ICTS/CTSA UL1RR024992]; NIH Roadmap for Medical Research. Source of open access funding: Shriners Hospital for Children.
This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bashiardes S., Veile R., Helms C., Mardis E.R., Bowcock A.M., Lovett M. Direct genomic selection. Nat. Methods. 2005;2:63–69. doi: 10.1038/nmeth0105-63. [DOI] [PubMed] [Google Scholar]
- 2.Ramos E., Levinson B.T., Chasnoff S., Hughes A., Young A.L., Thornton K., Li A., Vallania F.L., Province M., Druley T.E. Population-based rare variant detection via pooled exome or custom hybridization capture with or without individual indexing. BMC Genomics. 2012;13:683. doi: 10.1186/1471-2164-13-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nord A.S., Lee M., King M.C., Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics. 2011;12:184. doi: 10.1186/1471-2164-12-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W., Carvalho B., Miller N.D., Pevsner J., Chakravarti A., Irizarry R.A. Estimating genome-wide copy number using allele-specific mixture models. J. Comput. Biol. 2008;15:857–866. doi: 10.1089/cmb.2007.0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao M., Wang Q., Wang Q., Jia P., Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14(Suppl. 11):S1. doi: 10.1186/1471-2105-14-S11-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarado D.M., Buchan J.G., Frick S.L., Herzenberg J.E., Dobbs M.B., Gurnett C.A. Copy number analysis of 413 isolated talipes equinovarus patients suggests role for transcriptional regulators of early limb development. Eur. J. Hum. Genet. 2013;21:373–380. doi: 10.1038/ejhg.2012.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvarado D.M., McCall K., Aferol H., Silva M.J., Garbow J.R., Spees W.M., Patel T., Siegel M., Dobbs M.B., Gurnett C.A. Pitx1 haploinsufficiency causes clubfoot in humans and a clubfoot-like phenotype in mice. 2011;20:3943–3952. doi: 10.1093/hmg/ddr313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gurnett C.A., Alaee F., Kruse L.M., Desruisseau D.M., Hecht J.T., Wise C.A., Bowcock A.M., Dobbs M.B. Asymmetric lower-limb malformations in individuals with homeobox PITX1 gene mutation. Am. J. Hum. Genet. 2008;83:616–622. doi: 10.1016/j.ajhg.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alvarado D.M., Aferol H., McCall K., Huang J.B., Techy M., Buchan J., Cady J., Gonzales P.R., Dobbs M.B., Gurnett C.A. Familial isolated clubfoot is associated with recurrent chromosome 17q23.1q23.2 microduplications containing TBX4. Am. J. Hum. Genet. 2010;87:154–160. doi: 10.1016/j.ajhg.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tewhey R., Nakano M., Wang X., Pabon-Pena C., Novak B., Giuffre A., Lin E., Happe S., Roberts D.N., LeProust E.M., et al. Enrichment of sequencing targets from the human genome by solution hybridization. Genome Biol. 2009;10:R116. doi: 10.1186/gb-2009-10-10-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirby A., Gnirke A., Jaffe D.B., Baresova V., Pochet N., Blumenstiel B., Ye C., Aird D., Stevens C., Robinson J.T., et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat. Genet. 2013;45:299–303. doi: 10.1038/ng.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie C., Tammi M.T. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics. 2009;10:80. doi: 10.1186/1471-2105-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F., et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J., Lupat R., Amarasinghe K.C., Thompson E.R., Doyle M.A., Ryland G.L., Tothill R.W., Halgamuge S.K., Campbell I.G., Gorringe K.L. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012;28:1307–1313. doi: 10.1093/bioinformatics/bts146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodges E., Xuan Z., Balija V., Kramer M., Molla M.N., Smith S.W., Middle C.M., Rodesch M.J., Albert T.J., Hannon G.J., et al. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 17.Porreca G.J., Zhang K., Li J.B., Xie B., Austin D., Vassallo S.L., LeProust E.M., Peck B.J., Emig C.J., Dahl F., et al. Multiplex amplification of large sets of human exons. Nat. Methods. 2007;4:931–936. doi: 10.1038/nmeth1110. [DOI] [PubMed] [Google Scholar]
- 18.Gnirke A., Melnikov A., Maguire J., Rogov P., LeProust E.M., Brockman W., Fennell T., Giannoukos G., Fisher S., Russ C., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shearer A.E., Hildebrand M.S., Ravi H., Joshi S., Guiffre A.C., Novak B., Happe S., LeProust E.M., Smith R.J. Pre-capture multiplexing improves efficiency and cost-effectiveness of targeted genomic enrichment. BMC Genomics. 2012;13:618. doi: 10.1186/1471-2164-13-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodi K., Perera A.G., Adams P.S., Bintzler D., Dewar K., Grove D.S., Kieleczawa J., Lyons R.H., Neubert T.A., Noll A.C., et al. Comparison of commercially available target enrichment methods for next-generation sequencing. J. Biomol. Tech. 2013;24:73–86. doi: 10.7171/jbt.13-2402-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeong Y., Leskow F.C., El-Jaick K., Roessler E., Muenke M., Yocum A., Dubourg C., Li X., Geng X., Oliver G., et al. Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 2008;40:1348–1353. doi: 10.1038/ng.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benko S., Fantes J.A., Amiel J., Kleinjan D.J., Thomas S., Ramsay J., Jamshidi N., Essafi A., Heaney S., Gordon C.T., et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat. Genet. 2009;41:359–364. doi: 10.1038/ng.329. [DOI] [PubMed] [Google Scholar]
- 23.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yigit E., Zhang Q., Xi L., Grilley D., Widom J., Wang J.P., Rao A., Pipkin M.E. High-resolution nucleosome mapping of targeted regions using BAC-based enrichment. Nucleic Acids Res. 2013;41:e87. doi: 10.1093/nar/gkt081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.