


Abstract
Targeted endonucleases including zinc finger nucleases (ZFNs) and clustered regularly interspaced short palindromic repeats (CRISPRs)/Cas9 are increasingly being used for genome editing in higher species. We therefore devised a broadly applicable and versatile method for increasing editing efficiencies by these tools. Briefly, 2A peptide-coupled co-expression of fluorescent protein and nuclease was combined with fluorescence-activated cell sorting (FACS) to allow for efficient isolation of cell populations with increasingly higher nuclease expression levels, which translated into increasingly higher genome editing rates. For ZFNs, this approach, combined with delivery of donors as single-stranded oligodeoxynucleotides and nucleases as messenger ribonucleic acid, enabled high knockin efficiencies in demanding applications, including biallelic codon conversion frequencies reaching 30–70% at high transfection efficiencies and ∼2% at low transfection efficiencies, simultaneous homozygous knockin mutation of two genes with ∼1.5% efficiency as well as generation of cell pools with almost complete codon conversion via three consecutive targeting and FACS events. Observed off-target effects were minimal, and when occurring, our data suggest that they may be counteracted by selecting intermediate nuclease levels where off-target mutagenesis is low, but on-target mutagenesis remains relatively high. The method was also applicable to the CRISPR/Cas9 system, including CRISPR/Cas9 mutant nickase pairs, which exhibit low off-target mutagenesis compared to wild-type Cas9.
INTRODUCTION
Nuclease-based technologies have opened unprecedented possibilities for targeted genome editing in numerous species and cell types previously found challenging for genetic modification. The general principle involves engineering of endonucleases that can create a double-strand break at a desired site in genomic deoxyribonucleic acid (DNA) and vastly stimulate mutagenesis rates at that site. The technology may exploit natural homing endonucleases with specificities redirected towards a desired genomic sequence (1); alternatively, it may exploit non-specific nucleases, such as FokI, that are targeted to a desired genomic location via fusion to protein modules engineered to bind a specific DNA sequence. The latter systems include zinc finger nucleases (ZFNs) (2,3) and transcription activator-like type II effector nucleases (TALENs) (4). ZFNs and TALENs function as heterodimers in which the individual monomers bind offset 9–18-bp target sequences on opposite strands of DNA and subsequently nick their respective strands to produce a double-strand break. Recently, clustered regularly interspaced short palindromic repeat (CRISPR) systems for genome editing have been developed to introduce a double-strand break by the non-specific nuclease Cas9, which is directed to the desired locus by a 20-nt sequence contained within a so-called guide ribonucleic acid (gRNA) through Watson–Crick base pairing with target DNA (5–11). Most recently, pairs of gRNAs that target offset sequences on opposite strands of the target locus have been used in conjunction with nickase mutants of Cas9. This represents an editing system that is analogous to that of ZFNs and TALENs and shows greatly increased specificity as compared to the single CRISPR/Cas9 approach (12–14).
No matter the type of engineered nuclease used, the ultimate goal is to produce a site-specific DNA double-strand break. Such breaks can be resolved via the relatively error-prone non-homologous end joining (NHEJ) pathway, which often inserts or deletes a number of bases at the break. If nucleases are targeted to a coding sequence, a frame shift and functional gene knockout may be the outcome. Alternatively, the DNA break can be repaired by the homology-directed repair (HDR) pathway using the sister chromatid as repair template. However, if an exogenous, homologous DNA template (donor) containing a mutation is co-delivered into cells along with the nucleases, HDR can be exploited to precisely modify a genome in a user-defined manner. Short, homologous single-stranded oligodeoxynucleotides (ssODNs) have also proven highly effective donors (15), exploiting repair mechanisms that are not entirely clear.
The efficiency of nuclease-based generation of genome-edited clones from a targeted cell population is affected by several factors. One critical determinant is nuclease expression levels. Nucleases are most often delivered to cultured cells by transfection of plasmid- or messenger ribonucleic acid (mRNA)-based expression constructs and less frequently via viral or protein delivery (16–19). Regardless of the method, nuclease delivery efficiencies and the resultant expression levels vary greatly between cell types. Even within a given cell population, nuclease expression levels often vary substantially. Consequently, low nuclease expression levels in individual cells and/or nuclease expression in only a small fraction of cells often represent a major barrier to the generation of modified clones from a targeted cell population.
Expression of fluorescent proteins followed by fluorescence-activated cell sorting (FACS) is a powerful method for tracking cells of interest in a mixed population and has also been explored for nuclease genome editing. For instance, a fluorescence-based surrogate target gene reporter was co-transfected along with the nucleases and was used to enrich for cells with high nuclease activity (20). Furthermore, elegantly designed fluorescence-based reporter systems were used to explore repair mechanisms underlying nuclease genome editing, showing that elevated nuclease levels promote NHEJ and HDR and that elevated donor levels increase HDR whilst suppressing NHEJ (21).
Here, we aimed to devise a FACS-based method for obtaining cell populations with desired, high and uniform nuclease expression levels from which genome-edited clones may be derived with increased efficiencies. The method was based on tightly coupled (1:1) co-expression of nuclease and fluorescent protein in the same cell. This was achieved via transfection with constructs in which nuclease and fluorescent protein are encoded in the same transcript and linked by 2A peptide sequence (22), causing expression of nuclease and fluorescent protein as separate entities. Subsequent FACS allowed for easy isolation of cell populations with increasingly higher and uniform nuclease expression level which translated into increasingly higher genome editing frequencies.
We explored this method in several variations that offer specific advantages depending on the application: linking the two nucleases of a pair to distinct fluorescent proteins for standard genome editing, linking nuclease pairs for distinct genes to distinct fluorescent proteins for multiplex editing, and consecutive rounds of fluorescent protein/nuclease delivery and FACS for generation of genome edited cell pools. In the diverse and often challenging applications tested, this method was found to greatly increase genome editing frequencies by ZFNs and CRISPR/Cas9. By combining the method with delivery of ZFNs as mRNA and donors as ssODNs, generation of biallelic knockin clones in large numbers was achieved. Observed ZFN off-target effects associated with the method were minimal. Furthermore, in the event that an off-target effect is encountered, the method may be exploited to balance ZFNs to levels where off-target mutagenesis is low, but on-target mutagenesis remains sufficiently high. For CRISPR/Cas9, off-target effects may be reduced by incorporation of nickase pairs into the method.
MATERIALS AND METHODS
ZFNs and donors
ZFNs were of the CompoZr ZFN format generated by Sigma-Aldrich, except for the RSK4 ZFNs that were engineered by Sangamo Biosciences, Inc. The ZFNs contain the eHiFi (ELD/KKR) FokI domains and function as obligate heterodimers with improved catalytic activity (23). The zinc finger moieties of the dimers were designed to target 30–33-bp recognition sites in total. The sequences of the major ZFNs used here (RSK2, RSK4 and PRMT1) are given in Supplementary Table S1. Plasmid and ssODN donors (Supplementary Table S2) were synthesized by Gene Oracle Inc. and Sigma-Aldrich (Genosys), respectively.
pFluo-2A-ZFN constructs
First, a so-called pFluo-2A-ZFN construct (Figure 1A) was generated to co-express enhanced green-fluorescent protein (EGFP) (24) and ZFN via the 2A peptide. Briefly, the EGFP-2A-FLAG-NLS sequence shown in Supplementary Figure S1 and starting with an EcoRI site and ending with a KpnI site was synthesized (Gene Oracle, Inc.). This sequence was then used to replace the FLAG tag and nuclear localization signal (NLS) sequences excised from the standard CompoZr ZFN expression construct, pZFN (which in this case encoded the left ZFN for a ZFN pair targeting human RSK4), using the same restriction endonuclease sites. The resulting construct was used to generate all other pFluo-2A-ZFN derivatives reported here for co-expression of EGFP, the DsRed2 derivative E2 Crimson (25), Venus (26) or Cerulean (27) together with various ZFNs. First, fluorescent proteins were swapped by polymerase chain reaction (PCR) amplification of the fluorescent protein coding sequence using up- and downstream primers with overhangs containing NsiI and AvrII sites, respectively, followed by cloning of the PCR product into the pEGFP-2A-ZFN construct for RSK4 using these sites. Second, the various fluorescent protein-2A-FLAG-NLS cassettes were cloned into pZFNs for various genes using the EcoRI/KpnI sites, a cloning strategy that works for any CompoZr pZFN construct as long as these sites are absent from the zinc finger moiety of the construct.
Figure 1.
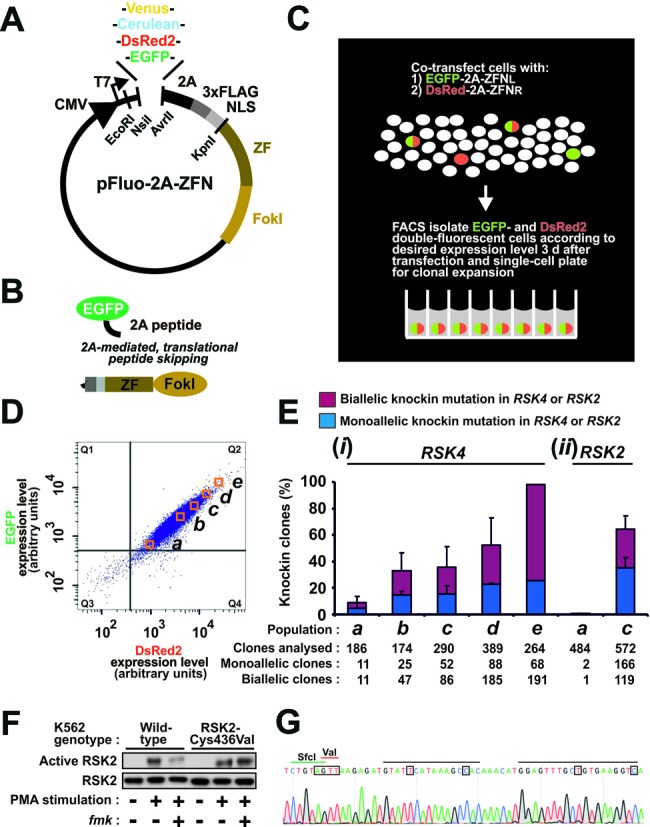
High-efficiency genome editing via 2A-coupled co-expression of ZFNs and fluorescent proteins combined with FACS. (A) Schematic of the pFluo-2A-ZFN construct for 2A-coupled, 1:1 co-expression of ZFN and fluorescent protein. Functional parts indicated are CMV promotor, fluorescent protein (Venus, Cerulean, DsRed2, EGFP), 2A peptide, triple FLAG epitope tag, nuclear localization signal (NLS), zinc finger (ZF) moiety and FokI nuclease moiety. The restriction endonuclease sites NsiI/AvrII and EcoRI/KpnI can be used to swap fluorescent protein and ZFN, respectively. (B) Fluorescent protein and ZFN expressed from Fluo-2A-ZFN transcripts are co-expressed as separate entities due to translational skipping at the 2A sequence. (C) Overview of strategy for high-efficiency genome editing via FACS isolation of cells that co-express fluorescent protein and ZFNs. In this example, left and right ZFNs of a pair (ZFNL and ZFNR) have been coupled to distinct fluorescent proteins. (D) FACS profile of K562 cells 3 days after transfection with mRNA for EGFP-2A-ZFNL and DsRed2–2A-ZFNR targeting the RSK4 locus along with an ssODN knockin donor (RSK4 Cys443Val/BamHI). Cells from populations displaying low to high EGFP/DsRed2 double-fluorescence intensities (a)–(e) were FACS isolated and single-cell seeded for clonal expansion. Cells in the gate Q3 have fluorescence intensities similar to mock-transfected cells. (E) Frequency of monoallelic and biallelic knockin modification at (i) the diploid RSK4 locus in clonal cell lines derived from populations (a)–(e) or at (ii) the diploid RSK2 locus in a similar knockin experiment using reagents for RSK2 and analysing populations (a) and (c) only. Except for population (e) that was isolated only once, the data are means + SD of three independent experiments. (F) Functional codon conversion at the RSK2 locus. Wild-type or clonal K562 cells with biallelic Cys436Val knockin modification of RSK2 derived from (c) in panel (E) were analysed for sensitivity of RSK2 to fmk by immunoblot analysis after exposure of cells to the stimulus PMA in the absence or presence of fmk. The data are representative of those obtained with five clones analysed. (G) Genomic sequence of an RSK2 Cys436Val clone, indicating the ZFN-binding sites (black bars), the Val436 codon (red bar) and the diagnostic SfcI site (green bar). The mutations introduced are boxed. Those occurring in the ZFN target site aimed to prevent re-cutting of the locus once the donor had been knocked in.
pFP-ZFN and pFP-ZFNL-ZFNR constructs
Alternative versions of pFluo-2A-ZFN, called pFP-ZFN, were created. First, pFP-ZFNL and pFP-ZFNR constructs were generated that encode green-fluorescent protein (GFP) (28,29) and red fluorescent protein (RFP) (30), respectively, linked to ZFN via a 2A sequence (Supplementary Figure S7). Fluorescent proteins and ZFNs can be swapped using the NsiI and KpnI/XhoI restriction sites, respectively. To combine both ZFNs of a pair into a single, GFP-coupled construct, the 2A-ZFNR sequence can be excised from a pFP-ZFNR vector using the restriction enzymes BglII and XhoI and then inserted into a pGFP-ZFNL construct digested with the same enzymes. The resulting pFP-ZFNL-ZFNR construct (Figure 3A) contains intervening 2A sequences for co-expression of fluorescent protein, ZFNL and ZFNR as three separate entities.
Figure 3.
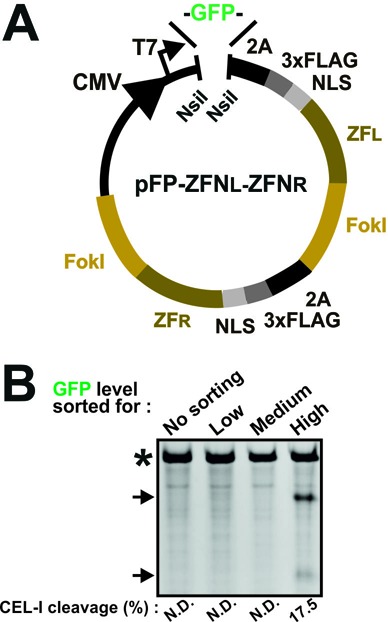
pFP-ZFNL-ZFNR single vector genome editing. (A) Schematic of a pFP-ZFNL-ZFNR vector that co-expresses both ZFNs of a pair along with a fluorescent protein (FP) from the same transcript and as three separate entities. Functional parts indicated are as in Figure 1A. (B) Jurkat cells were transfected with mRNA for a GFP-ZFNL-ZFNR construct targeting the RSK2 locus. Three days after transfection, genomic DNA was isolated from non-sorted cells or from cells FACS isolated for low, medium or high GFP fluorescence intensities and analysed by CEL-I assay. PCR products containing amplicons derived from mutant RSK2 alleles or from wild-type alleles only are indicated by arrows and an asterisk, respectively. The experiment was repeated three times with similar results. N.D.: none detected.
CRISPR/Cas9 constructs
A single vector construct, pCMV-Cas9-GFP, was gene-synthesized to co-expresses gRNA targeting the KRAS or the VEGFA locus under control of the U6 promoter and Cas9–2A-GFP under control of the cytomegalovirus (CMV) promoter (Figure 5A). QuikChange mutagenesis (Stratagene) was used to swap the gRNA in these constructs to gRNAs targeting the EMX1 locus as well as to convert Cas9 into a DNA nickase (Cas9N) by introduction of the D10A point mutation. Sequences of gRNAs and mutagenesis primers are provided in Supplementary Tables S3 and S4, respectively. QuikChange conditions were standard, except that the extension time was 16 min and 18 cycles were performed to obtain robust amplification. Furthermore, 5% dimethyl sulfoxide (DMSO) was included in the reaction mixture. To swap GFP with DsRed2 in these constructs, a MegaQuikChange strategy was employed. Briefly, the DsRed2 sequence was PCR amplified using the following primers: 5′GGAGAATCCTGGCCCACGATCGATGCATGATAGCACTGAGAACG-3′; 5′CCTCTAGACTCGAGTCATCAGTTAACCTGGAACAGGTGGTGGCGGG-3′. The PCR product was purified and 200 ng were used as MegaQuikChange primers using pCMV-Cas9N-GFP as template.
Figure 5.
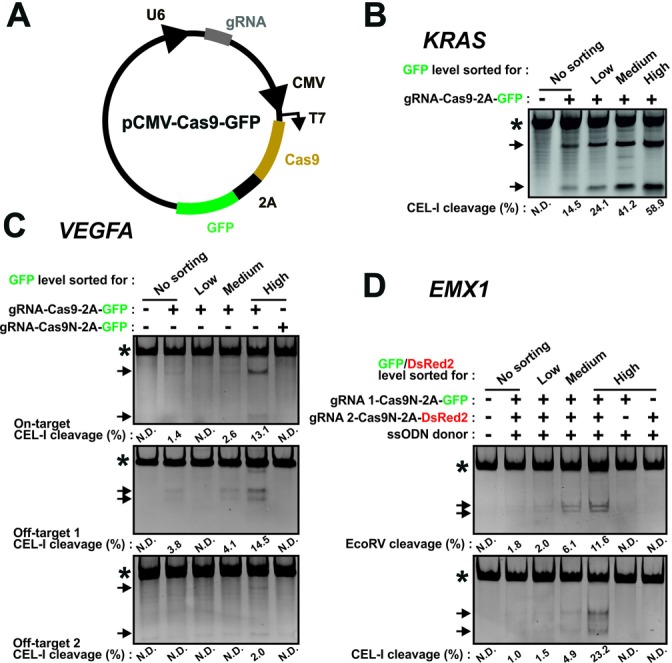
CRISPR/Cas9 genome editing is enhanced using the 2A-coupled fluorescent protein/FACS strategy. (A) Schematic of a construct, pCMV-Cas9-FP, that co-expresses gRNA and Cas6–2A-fluorescent protein. Functional parts indicated are U6 promoter, guide (g)RNA, CMV promoter, Cas9 nuclease, 2A peptide and fluorescent protein (GFP). (B)–(D) K562 cells were transfected (B) with a Cas9-GFP construct targeting the KRAS locus, (C) with a Cas9-GFP construct targeting the VEGFA locus or the same construct containing a nickase version of Cas9 (Cas9N) or (D) with such constructs targeting the EMX1 locus as a nickase pair or as individual nickases coupled to GFP or dsRed2. An ssODN donor (EMX1 gRNA 1/2 EcoRV) for knockin of an EcoRV restriction site in EMX1 was co-transfected. Three days after transfection, genomic DNA was isolated from non-sorted cells or from cells FACS isolated for low, medium or high fluorescence intensities and analysed by RFLP or CEL-I assay, as indicated. In all experiments, repeated three times with similar results, PCR products containing amplicons derived from mutant alleles or from wild-type alleles only are indicated by the arrows and asterisks, respectively. N.D.: none detected.
In vitro synthesis of mRNA
mRNA was in vitro synthesized from pZFN or pFluo-2A-ZFN using the MessageMAX T7 ARCA-Capped Message Transcription Kit (#EPICMMA60710, Epicentre) and the Poly A tailing kit (#EPICPAP5104H, Epicentre) and purified using the MEGAClear kit (Ambion) according to the manufacturers’ instructions.
Cell culture
K562 and HCT116 cells were cultured in Iscove's modified Dulbecco's medium and McCoy's 5A medium, respectively, supplemented with 10% foetal bovine serum. MCF10A cells were maintained in Ham's nutrient mixture F12/Dulbecco's modified Eagle's medium (1:1) supplemented with 5% horse serum, 10-μg/ml insulin, 20-ng/ml epidermal growth factor (EGF), 5-μg/ml hydrocortisone and 100-ng/ml cholera toxin. Jurkat cells were cultured in Roswell Park Memorial Institute-1640 medium supplemented with 10% fetal bovine serum. Chinese hamster ovary (CHO) cells were maintained in EX-CELL CHO CD Fusion serum-free medium (Sigma-Aldrich). All media were supplemented with 1.5-mM L-glutamine (4 mM for CHO cells), 100-U/ml penicillin and 100-μg/ml streptomycin. K562- and Jurkat-conditioned media were prepared by mixing equal amounts of fresh medium with medium collected from either cell type cultured at ∼0.5 × 106 cells per ml for 24 h then sterilized using a 0.2-μm filter (Millipore).
Expression comparison of ZFNs from pFluo-2A-ZFN versus pZFN
For plasmid transfection, HCT116 cells were plated at 1.2 × 105 cells per 3.8 cm2 well. After 24 h, the cells were transfected with 0.4-μg DNA complexed with 2-μl TransIT-LT1 transfection reagent (#MIR2310, Mirus) according to the manufacturer's instructions. For mRNA transfection, HCT116 cells were split and plated at a density leading to ∼70% confluency at the time of transfection 2 days later. Cells were then electroporated using a Nucleofector (Lonza) set to program D-032. 1 × 106 cells were dissolved in 100-μl Nucleofector Solution V (Lonza) with 2.0-μg and 3.3-μg mRNA transcribed from pZFN and pFluo-2A-ZFN, respectively. The difference in mRNA amounts used was due to the size difference of the mRNA constructs (∼1170 and ∼1950 bps, respectively, plus a poly A tail of ∼150 bps) and ensured transfection with roughly equimolar amounts of both mRNA types. Three days post-transfection, cells were lysed in sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (2% SDS, 62-mM Tris-HCl pH 6.8, 10% glycerol, 50-mM dithiothreitol and 0.12% bromophenol blue) and subjected to immunoblotting, as described below.
Functional comparison of ZFNs expressed from pFluo-2A-ZFN versus pZFN
K562 cells were split 2 days prior to electroporation so that the concentration of the cells was ∼0.5 × 106 cells per ml at the time of electroporation. The K562 electroporation was performed in a Nucleofector (Lonza) using program T-016 according to the manufacturer's instructions. Briefly, 1 × 106 cells in 100-μl Nucleofector Solution V (Lonza) were mixed with the following reagents as appropriate for the experiment: for ssODN donor-based genome editing, 3 μl of 100-μM RSK4 Cys443Val/BamHI ssODN donor (equaling ∼6.7 μg) in 10-mM Tris (pH 7.6) was used together with 2-μg ZFN-expressing plasmid (for each ZFN of the pair) or with 2-μg and 3.3-μg ZFN-expressing mRNA (for each ZFN of the pair), when transcribed from pZFN and pFluo-2A-ZFN, respectively. For plasmid donor-based genome editing, 10-μg RSK2 Cys436Val/BamHI plasmid donor was used together with 2-μg ZFN-expressing mRNA when transcribed from pZFN or 3.3-μg ZFN-expressing mRNA when transcribed from pFluo-2A-ZFN. Four hours after nucleofection, cells were shifted to 30°C. Whilst a beneficial effect of this condition termed ‘cold shock’ has so far only been validated for NHEJ editing, we used it also for knockin editing, since it was reported to increase cellular ZFN levels (31). After 3 days, genomic DNA was isolated as described below.
Genome editing using 2A-coupled co-expression of nuclease and fluorescent proteins combined with FACS
K562 cells were electroporated with the various constructs for ZFN or CRISPR/Cas9 2A-linked to fluorescent protein, as described in the section above and in the main text. Cells were then cultured for 72 h with a 30°C cold shock from 4–68 h (ZFNs only). MCF10A and Jurkat cells were electroporated as described for K562 except that Nucleofector solution T/Nucleofector program T-020 and Nucleofector Solution V/Nucleofector program X-001 were used for the two cell lines, respectively. In the comparison of the efficiency of ZFN genome editing using non-linked versus 2A-linked fluorescent protein, the non-linked fluorescent protein was delivered as 250 ng of pGFP plasmid (contained in the Nucleofector kit from Lonza).
Three days after transfection, the cells were subjected to FACS analysis. Briefly, cells (trypsinized if adherent and then diluted with culture medium to inactivate trypsin) were pelleted by centrifugation at 200 g for 4 min and resuspended in normal culture medium to about 0.5–1×106 cells per ml (NB: optimal cell densities will vary depending on cell type and whether bulk sorting or single-cell plating is the goal). Finally, the cells were passed through 50-μm filcons (BD Biosciences) to achieve a single-cell suspension. FACS analysis was performed on an FACS Aria I (BD Biosciences) with excitation and detection of emission as appropriate for the various fluorescent proteins. Fluorescence levels of mock-transfected cells were determined in parallel and considered background level, based on which cell populations with specific fluorescence signals could be assigned (see Supplementary Figure S5 for further details). Fluorescent cell populations were then gated in defined fluorescence intensity intervals (e.g. 50% ± 5% or top 10%) of the total fluorescent cell population and isolated. To generate stably modified clones, cells isolated by FACS according to a desired fluorescence level or cells not subjected to FACS were single-cell sorted into 96-well plates in medium conditioned as appropriate and expanded to clonal cell lines.
Genomic DNA extraction and PCR amplification
For single-cell clones, DNA was isolated when the expanded cell clones grew dense. Briefly, the cells were washed with a 1X phosphate-buffered saline (PBS) solution, lysed in 70-μl lysis buffer [25-mM NaOH, 0.2-mM ethylenediaminetetraacetic acid (EDTA), pH 12], then incubated at 95°C for 30 min. Thereafter, 70-μl neutralization buffer (40-mM Tris-HCL, pH 5) were added. Of this mixture, 1–3 μl were used as template for PCR for genome editing analysis by restriction fragment length polymorphism (RFLP) assay, as described below.
For bulk-sorted cells, genomic DNA was isolated by incubation of the cells for 3 h at 55°C in lysis buffer [20-mM Tris-HCl (pH 8.0), 5-mM EDTA, 400-mM NaCl, 1% SDS, 400-μg/ml proteinase K], followed by phenol-chloroform extraction and isopropanol precipitation of the DNA. Approximately 100 ng of genomic DNA were then used as template for PCR amplification and genome editing analysis by CEL-I or RFLP assays, as described below. PCR was carried out in 12.5-μl reactions using Taq DNA polymerase (Ampliqon). The specific annealing temperatures and additives for the various PCRs are listed in Supplementary Table S5. The following touch-down PCR amplification conditions were used: after an initial 5-min denaturation at 95°C, samples were subjected to 15 cycles consisting of denaturation for 30 s at 95°C, then primer annealing and extension for 70 s at 70°C in the first cycle with a 0.5°C reduction of annealing/extension temperature in each subsequent cycle. This was followed by 23–26 cycles of denaturation for 30 s at 95°C, primer annealing for 30 s at various temperatures (provided in Supplementary Table S5) and extension for 70 s at 72°C. The reactions were terminated by a final elongation step of 5 min at 72°C.
CEL-I assay
CEL-I assays were performed according to the manufacturer's instructions (Transgenomic). Briefly, 10 μl of the PCR products amplified from wild-type and mutant alleles were used to generate heteroduplexes using the following reaction conditions: an initial 10-min denaturation step at 95°C was followed by 1 s at 95°C and a step-wise 2°C reduction in temperature with 1-s incubations down to 85°C, then 1 s at 85°C and a step-wise 0.1°C reduction in temperature with 2-s incubations down to 4°C. Next, the reactions were incubated at 42°C for 30 min with 0.4 μl of SURVEYOR Nuclease S (CEL-I) and SURVEYOR Enhancer S (#706025, Transgenomic). One-tenth of the PCR volume of Stop Solution was added to terminate the reactions. Finally, the digests were resolved on 6% or 8% Tris-borate-EDTA (TBE) acrylamide gels, depending on the anticipated band sizes. Supplementary Table S5 provides the sequences of the primers used, as well as the sizes of diagnostic fragments and PCR products amplified from the targeted alleles.
RFLP assay
PCR reactions were digested by addition of 10 U of the appropriate restriction endonuclease and incubation overnight at 37°C. Digests were resolved on either 6% or 8% TBE acrylamide gels, or on 1–2% 0.5 x Tris-acetate-EDTA (TAE) agarose gels, depending on the anticipated band sizes. When analysing clonal cell lines, mono- and biallelic knockin of diagnostic restriction sites in diploid loci were evidenced by ∼50% and complete digestion of the PCR products, respectively (see Supplementary Figure S4 as an example). For triploid loci, triallelic knockin was evidenced as complete digestion, whereas partial digestion (∼30–60%) was taken as mono- or biallelic knockin. Supplementary Table S5 provides the diagnostic restriction endonucleases, sequences of the primers used, as well as the sizes of diagnostic fragments and PCR products amplified from the targeted alleles.
Densitometric quantization of CEL-I and RFLP assays
Gels with digested PCR products were stained for 10 min with ethidium bromide and imaged with a Quantity One Gel Doc XR imaging system (Bio-Rad). Quantification was performed using ImageJ software (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2012) and based on relative intensities of uncleaved versus cleaved PCR products, and percentage cleavage was determined using the formula: % cleavage = 100 x (1-(1-fraction cleaved) 1/2).
Off-target analysis
To predict candidate off-target sites for the human RSK4 and RSK2 ZFNs, the Homo sapiens genome (GRCh37, hg19) was scanned for sequences highly homologous to their on-target sites. To predict candidate off-target sites for the various CHO ZFNs, the Cricetulus griseus genome version 1.0 was downloaded from NCBI (ID: AFTD01000000) and indexed for use with Bowtie (32) for alignment with the relevant ZFN on-target sites. The Phred score threshold was set to 160, which in our case thus searched for sites with up to five mismatches per monomer binding site. Finally, 100-bps up- and downstream of the candidate sites were extracted for primer design for genomic PCR. For both human and hamster, a 5- or a 6-bp spacer sequence between the ZFN monomer binding sites was allowed. Furthermore, searches were restricted to heterodimeric locations since the ZFNs used contain obligate heterodimeric FokI (ELD/KKR) domains, which have previously been shown to eliminate off-target activity at homodimeric target locations (33).
To perform Sanger sequencing of candidate off-target sites, the appropriate genomic loci were PCR amplified using primers listed in Supplementary Table S5. The resulting PCR products were then purified using carboxyl-coated magnetic beads. Briefly, an equal volume of PCR product and hybridization buffer (2.5-M NaCl, 20% polyethylene glycol (PEG) 8000) supplemented with 10 μl of 10 mg/ml beads pre-washed three times with 0.5-M EDTA (#21353, Pierce) were mixed and incubated for 10 min at room temperature. Then, magnetic beads with bound PCR products were captured using a magnetic stand for 5 min and washed three times with 70% ethanol. The beads were air-dried and re-suspended in 20 μl 10-mM Tris-HCl pH 8.0 buffer. For sequencing (GATC Biotech), 20–80 ng/μl of the purified PCR products were pre-mixed with 25 pmol (in 5 μl) of the appropriate sequencing primers (Supplementary Table S5).
Gene copy number analysis
Genomic DNA was extracted from K562 and Jurkat cells using the FlexiGene DNA kit (Qiagen) and subjected to gene copy number analysis (performed at the Center for Genomic Medicine, Rigshospitalet) using CytoScan HD arrays (Affymetrix). Briefly, 250 ng of genomic DNA were digested and ligated to adapters. Adapter-ligated DNA was amplified, purified, fragmented, labelled with biotin and hybridized to the arrays for 18 h. The Fluidics Station 450 (Affymetrix) and the GeneChip Scanner 3000 7G (Affymetrix) were used to wash, stain and scan the arrays. The generated .CEL files were then analysed in Partek Genomics Suite 6.6. Briefly, data were imported using the recommended default Partek settings: probes were adjusted for probe sequence, settings for no background and quantile normalization were applied and probes were allele-specifically summarized. Gene copy number and single nucleotide polymorphism (SNP) allele ratio were calculated using an unpaired analysis with the baseline distributed by Partek, which is based on 380 CytoScan HD array from the International HapMap project. Copy number status of individual genes was established by examining copy number calls combined with the SNP allele ratios, whilst also taking polyploidy into account.
Stimulation and isolation of RSK2 for functional codon conversion assay
To assay RSK2 activity, wild-type or RSK2-Cys436Val K562 cells plated at 2×106 cells per well were serum-starved for 4 h and then incubated for 1 h with 10-μM fmk. A final 20-min incubation with 100-nM phorbol 12-myristate 13-acetate (PMA) was included, when indicated. The cells were solubilized for 15 min in 1-ml lysis buffer [300-mM NaCl, 50-mM Tris-HCl (pH 7.4), 0.5% Igepal CA-630, 1-mM Na3VO4, 5-mM EDTA, 50-mM NaF, 10-nM calyculin A, 10-μM leupeptin, 5-μM pepstatin and 1-μg/ml aprotinin] on ice and manipulated at <4°C thereafter. Cell extracts were clarified by centrifugation for 5 min at 18 000 g and the supernatants were incubated for 90 min with 2 μg of anti-RSK2 antibody (MCA3429Z, ABC Serotec), with the addition of 20-μl protein G agarose beads (#16–266, Upstate) during the final 30 min. The beads were then precipitated by centrifugation, washed five times with lysis buffer, drained and dissolved in SDS-PAGE sample buffer.
Immunoblot analysis
Immunoprecipitated RSK2 and extracts of cells transfected with ZFN and fluorescent protein expression constructs were subjected to SDS-PAGE and the proteins were then transferred to Hybond-ECL nitrocellulose membranes (GE Healthcare). Membranes were blocked with 2% (w/v) ECL Advance Blocking Agent (GE Healthcare), followed by incubation with primary antibodies against active (Ser386-phosphorylated) RSK2 (#AF1094, R&D Systems), total RSK2 (MCA3429Z, ABC Serotec), FLAG epitope tag (horse radish peroxidase-coupled, #F3165, Sigma), EGFP (#8334, Santa Cruz Biotechnology; this antibody was also used to detect Venus and Cerulean) or DsRed2 antibody (#632496, Clontech), as appropriate. Thereafter, relevant horseradish peroxidase-coupled secondary antibodies and ECL Advance (GE Healthcare) were used for visualization of the primary antibodies.
RESULTS
Generation of a plasmid construct (pFluo-2A-ZFN) for tightly coupled co-expression of fluorescent protein and ZFN as separate entities
We first created the construct pFluo-2A-ZFN that encodes a fluorescent protein linked to a ZFN via a 2A peptide sequence in one single reading frame (Figure 1A and Supplementary Figure S1). The 2A peptide undergoes a translational ribosomal skip of the glycyl-prolyl peptide bond at its C-terminus, which results in the expression of fluorescent protein and ZFN as two separate entities (Figure 1B). Of note, 2A-based approaches are superior with respect to producing 1:1 co-expression levels of proteins of interest, as compared to other co-expression methods (22). We designed pFluo-2A-ZFN such that a single cloning step can swap fluorescent protein (including EGFP, Venus, DsRed2 and Cerulean) by using the sites NsiI/AvrII and ZFN by using the sites EcoRI/KpnI.
pFluo-2A-ZFN can co-express diverse fluorescent proteins and ZFNs at normal expression and activity levels
Next, we determined whether pFluo-2A-ZFN expresses the fluorescent protein and ZFN coding sequences as separate entities and whether ZFN expression and activity levels were comparable to those obtained with the CompoZr pZFN construct from which we derived pFluo-2A-ZFN. HCT116 cells were transfected with pZFN or pFluo-2A-ZFN encoding a variety of fluorescent proteins linked to either monomer of various ZFN pairs, denominated ZFN left (ZFNL) and ZFN right (ZFNR). Two days after transfection, the cells were subjected to immunoblot analysis for the fluorescent protein or the FLAG epitope tag present in the ZFNs. In all instances, pFluo-2A-ZFN expressed the ZFN moieties cleaved from fluorescent protein at levels similar to those expressed from pZFN (Supplementary Figure S2A).
Whereas the above experiments used plasmid-based ZFN delivery, ZFN coding sequences can also be delivered into cells as mRNA synthesized in vitro from vectors that contain the appropriate elements. When delivered as mRNA, Fluo-2A-ZFN constructs also expressed cleaved ZFNs at levels identical to those derived from pZFN (Supplementary Figure S2B). Furthermore, pFluo-2A-ZFN was of comparable efficiency to pZFN in serving as a template for in vitro synthesis of mRNA (Supplementary Figure S2C).
To test whether Fluo-2A-ZFN expresses ZFNs that function effectively, human leukemic K562 cells were transfected with various derivatives of this construct, either as plasmid or mRNA. Along with the ZFN constructs, we co-transfected plasmid or ssODN donor aiming to introduce a specific codon-swap mutation as well as a silent diagnostic restriction endonuclease site in the targeted genes (the sequences of donors are provided in Supplementary Table S2). Three days post-transfection, the cells were lysed and the cell lysates were used as template to PCR amplify genomic sequence spanning the mutation sites. The resulting PCR products were subjected to two different assays that both detect mutagenesis at the target site: either CEL-I assay that detects any alterations at the target site or RFLP assay that specifically detects knockin of the diagnostic restriction site. Both assays revealed that ZFNs derived from either pZFN or pFluo-2A-ZFN produced similar levels of genome editing. This was observed regardless of whether the ZFNs were delivered as plasmid or mRNA or whether the donor was plasmid or ssODN (Supplementary Figure S3). Note that these knockin experiments with plasmid and mRNA ZFN constructs were performed on different occasions and cannot be compared. In side-by-side comparisons, we find that mRNA constructs are often the more efficient for knockin, as we reported previously (15).
Controlled low to high knockin editing frequencies by combining 2A-coupled co-expression of fluorescent proteins and ZFNs with FACS
We next tested whether transfection with Fluo-2A-ZFN constructs followed by FACS for isolation of ZFN transfectants can be used to increase the efficiency of genome editing, as assessed in clonal cell lines. We first focused on knockin editing, which is relatively demanding, and chose to deliver donors as ssODNs and nuclease constructs as mRNA, since these delivery forms offer various advantages over double-strand DNA construct delivery, as detailed in the Discussion section.
First, we transfected K562 cells with mRNA constructs in which each ZFN of a pair was 2A-coupled to a distinct fluorescent protein: EGFP for ZFNL and DsRed2 for ZFNR. The ZFNs targeted the RSK4 locus which was found to be diploid in these cells by CytoScan HD array-based gene copy number analysis (Supplementary Table S6). Along with the ZFNs, we co-transfected an ssODN donor (RSK4 Cys443Val/BamHI) designed to knockin a specific codon-swap mutation and a silent diagnostic restriction site in RSK4. FACS analysis 3 days after transfection revealed that the expression levels of both EGFP and DsRed2 varied ∼100-fold in the cell population (Figure 1D, Q2), illustrating the great lack of uniform expression in cells of a transfected cell pool. Also, a fraction of the fluorescent cells were DsRed2 positive only (Figure 1D, Q4: ∼1.7% of the cells), thus apparently expressing only one ZFN of the pair, which is incompatible with respect to genome editing. Using FACS, we next isolated cell populations that displayed uniform EGFP and DsRed2 double-fluorescence levels ranging from low to high (Figure 1D, a–e), aiming to recapitulate the finding in the model system by Certo et al. that increased nuclease levels result in increased editing frequencies (21). Thereafter, cells from each population were single-cell sorted into 96-well plates and expanded to clonal cell lines. The expanded clones were analysed using RFLP assay to detect knockin of the diagnostic restriction site in a monoallelic or biallelic state at the RSK4 locus (Supplementary Figure S4). Three striking results were obtained [Figure 1E, (i)]. First, the knockin frequencies in the clones increased with the fluorescence level observed in the cells at the time of FACS and single-cell plating. Second, ∼95% of the 264 clones derived from the most highly fluorescent cell population ‘e’ had the diagnostic knockin mutation and, of these, ∼70% had biallelic modification of the targeted locus. Finally, a total of 520 biallelic knockin clonal cell lines were rapidly generated using this high-throughput genome editing method that was streamlined for 96-well plate format and multi-channel pipette handling from FACS to the final step of genotyping of clones by RFLP assay.
In a similar experiment, we targeted the RSK2 locus in K562 cells using mRNA for ZFNL coupled to EGFP and ZFNR coupled to DsRed2. Along with ZFNs, we co-transfected an ssODN donor (RSK2 Cys436Val/SfcI) designed to knockin a codon-swap mutation and a silent diagnostic restriction site in the RSK2 locus, which was found to be diploid in K562 cells by gene copy number analysis (Supplementary Table S6). Three days after transfection, EGFP and DsRed2 double-fluorescent cells were FACS isolated from populations with fluorescence levels similar to ‘a’ and ‘c’ in Figure 1D, single-cell plated and expanded to clonal cell lines. The resulting cell lines were shown to harbour low (∼0.6%) and high (∼64%) stable knockin modification, respectively, of the RSK2 locus [Figure 1E, (ii)]. Of note, biallelic knockin clones constituted ∼29% of those derived from population ‘c’. By contrast, in an earlier study using the same reagents (except for minor differences in the donor), but where we did not implement FACS enrichment, only 2.7% biallelic knockin modification of the RSK2 locus was obtained (15). According to RNA-seq analysis, RSK4 is expressed at >1000 times lower levels compared to RSK2 in K562 cells (our unpublished data), yet the two genes were knockin modified with rather similar efficiencies.
We developed the present high-throughput knockin method with the aim of genetically manipulating the pharmacological sensitivity of many different protein kinases in diverse cell lines. For instance, the Cys436Val codon-swap knockin mutation introduced in RSK2 in Figure 1E aimed at rendering this kinase insensitive to the small-molecule inhibitor fmk. We tested whether such functional codon conversion had occurred in our clones by immunoblot analysis for active RSK2 in cells exposed to the stimulus PMA in the absence or presence of fmk. As shown in Figure 1F, the targeting event generated RSK2-Cys436Val mutant kinase that was expressed and activated similar to wild-type RSK2, but was rendered insensitive to fmk, as predicted. Genomic sequencing of the RSK2-Cys436Val clone revealed homozygous knockin of the desired alterations at the RSK2 locus (Figure 1G).
Challenging knockin applications are facilitated by the use of 2A-coupled co-expression of fluorescent proteins and ZFNs
We next tested the ability of our method to enrich for rare targeting events. First, we knockin-targeted the RSK4 locus by transfecting K562 cells with the same reagents as in Figure 1D. We thereafter diluted the transfected cells with mock-treated cells to obtain a cell pool in which the transfected cells constituted only ∼0.5% of the total cell population. Unsorted cells from this pool were single-cell cloned, whilst another portion of the cell pool was subjected to FACS enrichment for cells displaying EGFP/DsRed2 double-fluorescence followed by single-cell cloning. The non-sorted cell pool yielded no modified clones out of 810 clones analysed (Figure 2A, upper panel). In contrast, the FACS-enriched pool yielded 10 monoallelic (6%) and five biallelic (3%) knockin clones out of the 171 clones analysed, demonstrating that our method can enrich for rare targeting events in this partial model of an inefficient transfection scenario.
Figure 2.
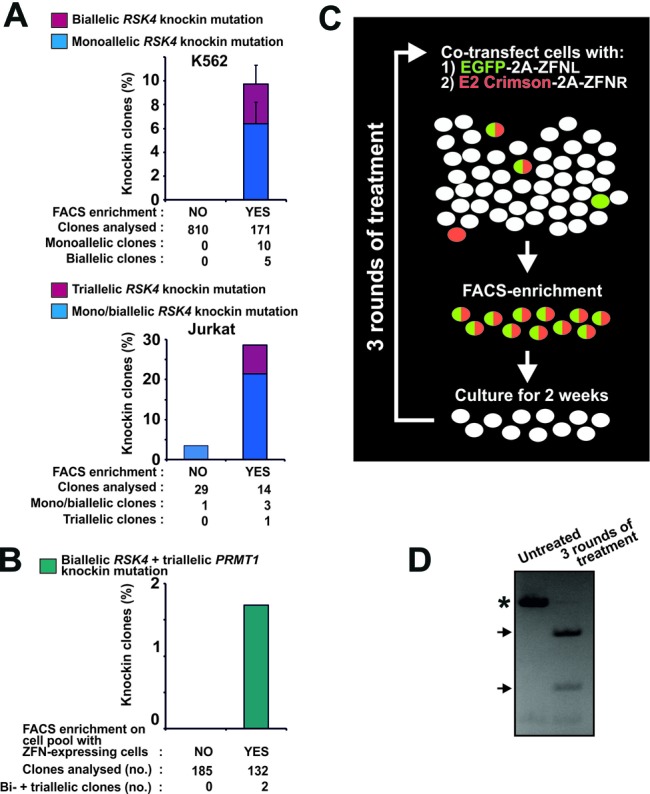
Fluorescent protein-2A-ZFN constructs enable efficient knockin genome editing in demanding applications. (A) K562 or Jurkat cells were transfected with mRNA expressing EGFP-2A-ZFNL and DsRed2–2A-ZFNR for RSK4 along with an ssODN knockin donor (RSK4 Cys443Val/BamHI). For K562, the cells were diluted 3 days post-transfection with mock-transfected cells to produce a pool of ∼0.5% ZFN-expressing cells. Cells of this pool were seeded singly or subjected to FACS isolation of EGFP/DsRed2 double-fluorescent cells (any level) that were thereafter also seeded singly. For Jurkat, non-sorted cells or cells FACS isolated from the 15% most highly EGFP/DsRed2 double-fluorescent cell population were seeded singly 3 days post-transfection. In both experiments, the singly seeded cells were expanded to clonal cell lines and analysed for knockin mutation by RFLP analysis. For K562, data are means + range of two independent experiments. (B) K562 cells were co-transfected with mRNA expressing EGFP-2A-ZFNs (left and right) for RSK4 and DsRed2–2A-ZFNs (left and right) for PRMT1 along with ssODN donors for knockin modification of these loci (RSK4 Cys443Val/BamHI and PRMT1 ScaI). Three days after transfection, non-sorted cells or cells FACS isolated from the 15% most highly EGFP/DsRed2 double-fluorescent cell population were seeded singly, expanded to clonal cell lines and analysed for complete knockin modification of the RSK4 and PRMT1 loci. Data are summed of three independent experiments. (C) Overview of strategy for generation of cell pools with high levels of stable knockin modification. In this case, left and right ZFNs of a ZFN pair (ZFNL and ZFNR) have been coupled to distinct fluorescent proteins. (D) Example of close to 100% knockin in an MCF10A cell pool. Specifically, MCF10A cells were transfected for codon-conversion knockin at the RSK2 locus as described in Figure 1E, (ii). Three days after transfection, the 40% most highly fluorescent cells were FACS isolated and cultured as a pool for 2 weeks. Thereafter, this cell pool was subjected to the same treatment for two more rounds. After the third round, the cell pool was analysed for stable knockin at the RSK2 locus by RFLP analysis with untreated MCF10A cells serving as a control. PCR products derived from wild-type and mutant alleles are indicated by an asterisk and arrows, respectively.
We next tested our method for knockin editing of actual difficult-to-transfect cells such as human leukemic Jurkat T cells. Again, we used the reagents described above to edit the RSK4 locus, which was found to be triploid in Jurkat cells by gene copy number analysis (Supplementary Table S6). In parallel, we transfected K562 cells with the same reagents and also mock-transfected both Jurkat and K562 cells with irrelevant mRNA not encoding fluorescent protein. Three days post-transfection, FACS profiles were established (Supplementary Figure S5). The profiling in mock-transfected cells in Supplementary Figure S5B defined the cell populations (Q3) devoid of detectable specific fluorescence signal from transfected EGFP or DsRed2. Based on these data, cell populations with specific EGFP signal (Q1), specific DsRed2 signal (Q4) or specific signals for both EGFP and DsRed2 could be assigned in the FACS profiles in Supplementary Figure S5A. Comparing the various FACS profiles demonstrated a lower transfection efficiency of Jurkat cells, as compared to K562 cells: thus, a much larger fraction of the Jurkat cell population was present in Q3. Furthermore, the expression levels were also lower in the Jurkat cell population, as evidenced by many fewer medium and highly fluorescent cells, in particular EGFP/DsRed2-double fluorescent cells, as compared to the K562 cell population. Importantly, however, when sorting for the 15% most highly EGFP/DsRed2 double-fluorescent Jurkat cells prior to single-cell plating, we observed an ∼8-fold increase in the isolation of clones with targeted knockin at the RSK4 locus, as compared to non-sorted cells (Figure 2A, lower panel). Moreover, sorting even produced one triallelic knockin clone amongst the 14 clones analysed.
In the challenging Jurkat cells, we also tested the advantage of 2A-coupled co-expression of ZFN and fluorescent protein as compared to non-coupled co-expression, in which ZFN and fluorescent protein are expressed from separate constructs. Thus, Jurkat cells were either transfected with mRNA for EGFP-2A-ZFNL and DsRed2–2A-ZFNR or with mRNA for ZFNL and ZFNR along with a separate plasmid expressing EGFP. Again, the ZFNs targeted the RSK4 locus and the aforementioned ssODN donor Cys443Val/BamHI were co-transfected for knockin in this gene. Prior to sorting, the level of modification in the two cell pools was found to be similar by CEL-I assay and thus the separate EGFP construct did not negatively affect the non-coupled ZFNs (data not shown). Furthermore, FACS analysis showed that the fraction of EGFP fluorescent and EGFP/DsRed2 double-fluorescent cells were similar (∼35%) in the two cell pools (Supplementary Figure S6A). The two cell pools were then sorted for EGFP fluorescence and EGFP/DsRed2 double-fluorescence, respectively, and single cells were plated out for clonal expansion. As shown in Supplementary Figure S6B, 2A-coupled co-expression of ZFN and fluorescent protein increased the knockin editing frequency ∼6-fold as compared to non-coupled co-expression. Thus, 2A-coupled co-expression produced five mono/biallelic and one triallelic knockin clone out of 174 clones, whereas non-coupled co-expression produced only one mono/biallelic knockin clone out of 171 clones. The lower knockin frequencies obtained in this experiment compared to that in Figure 2A are due to the fact that we sorted for cells showing any fluorescence level, as compared to sorting for the top-15% expressers in Figure 2A.
We extended our approach to investigate whether it could enable simultaneous knockin modification of two genes at all alleles in the same cell. K562 cells were co-transfected with mRNA expressing EGFP-linked ZFNs (L and R) for RSK4 as well as DsRed2-linked ZFNs (L and R) for PRMT1 (triploid in these cells—Supplementary Table S6), along with ssODN donors for knockin modification of each locus (RSK4 Cys443Val/BamHI and PRMT1 ScaI). Three days after transfection, cells were subjected to FACS and cells from the 15% most highly EGFP/DsRed2 double-fluorescent population were single-cell cloned. Strikingly, FACS enrichment enabled simultaneous homozygous knockin mutation of the two genes at all five alleles in two clones out of 132, whereas no homozygously modified clones were present amongst 185 clones from unsorted cells (Figure 2B).
Some cell lines and primary cells are inherently recalcitrant to single-cell cloning and so it may be necessary to create pools of cells that are highly enriched for the desired mutation. To this end, we evaluated the utility of several rounds of ZFN transfection and FACS on a cell population to generate pools of cells with high knockin mutation levels (Figure 2C). Human immortalized, but non-transformed, mammary MCF10A cells that are difficult to expand from single cells after FACS were transfected with EGFP-2A-ZFNL and DsRed2–2A-ZFNR mRNA for RSK2 and the aforementioned ssODN donor RSK2 Cys436Val/SfcI. Three days after transfection, the 40% most highly EGFP/DsRed2 double-fluorescent cells were isolated by FACS, followed by 2 weeks in culture. Thereafter, this cell population was subjected to the same treatment two more times. Strikingly, after three rounds of treatment, a pool of MCF10A cells had been obtained with nearly complete targeted codon conversion at the RSK2 locus [diploid in these cells, (34)], as evidenced by the nearly complete digestion in the RFLP assay performed on the cell pool (Figure 2D).
Single vector fluorescent protein-ZFNL-ZFNR methods
To further expand the utility of our approach, we generated variants of the Fluo-2A-ZFN construct, specifically pFP-ZFN constructs that can rapidly be converted into pFP-ZFNL-ZFNR constructs that encode both ZFN monomers of a pair coupled to a single fluorescent protein (Figure 3A). First, pFP-ZFN vectors were generated to encode GFP and RFP 2A-coupled to ZFNL and ZFNR, respectively (Supplementary Figure S7). To combine both ZFNs into a single, GFP-coupled construct, the 2A-ZFNR sequence can be excised from a pRFP-ZFNR vector using the restriction enzymes BglII and XhoI and inserted into a pGFP-ZFNL construct digested with the same enzymes. Intervening 2A sequences ensure that each of the three functional components is expressed as a separate entity. We tested a pGFP-ZFNL-ZFNR construct for NHEJ-based modification in the Jurkat cell line. To this end, Jurkat cells were transfected with mRNA encoding GFP-ZFNL-ZFNR for RSK2 (triploid in these cells—Supplementary Table S6). Three days later, the cells were left unsorted or were FACS isolated for low, medium or high fluorescence intensities roughly corresponding to populations ‘a’, ‘b’ and ‘d’ in Figure 1D). Subsequent CEL-I assay showed no detectable NHEJ modification in the unsorted, low or medium fluorescent cell pools. By contrast, in the highly GFP positive cell pool, ∼18% of the alleles were modified (Figure 3B), demonstrating that FP-ZFNL-ZFNR constructs work effectively.
Off-target effects associated with FACS enrichment for cells with high ZFN levels
We assessed whether enrichment for cells with high ZFN levels was associated with high off-target cutting. First, we analysed a large number of K562 knockin clones established from cell populations with high levels of ZFNs, specifically, the populations ‘d’/‘e’ for RSK4 and ‘c’ for RSK2, as depicted in Figure 1E. We only analysed clones with biallelic (complete) knockin, since these may generally represent clones derived from cells with particularly high levels of ZFN activity and may therefore also be most susceptible to off-target cutting. We queried the human genome for sequences with the highest similarity to the target sites of the RSK4 and RSK2 ZFNs, then analysed top-ranking candidate off-target sites for mutations by Sanger sequencing. Strikingly, no mutations were detected when analysing four top-ranking off-target sites in ∼300 clones, probing a total of 2372 alleles (Figure 4A). These off-target sites had four to seven mismatches relative to the respective on-target sites.
Figure 4.
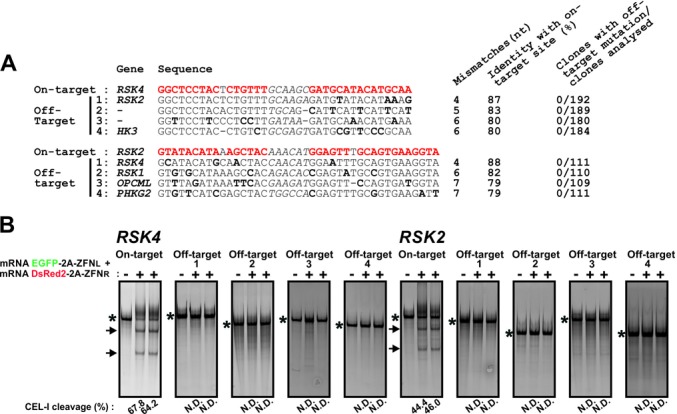
Off-target cutting associated with FACS for high ZFN levels in K562 cells. (A) K562 clones with biallelic knockin at the RSK4 and RSK2 loci established from the populations ‘d’/‘e’ and ‘c’ in Figure 1E, respectively, were analysed for mutations in four top-ranking candidate off-target sites (off-targets 1–4) by Sanger sequencing. The ZFN on-target site is indicated in red and mismatches in off-target sites relative to the target site are boldface. The spacer sequence between the ZFN monomer binding sites is in italics. (B) K562 cells were transfected with EGFP-2A-ZFNL and DsRed2–2A-ZFNR for RSK4 or RSK2 along with ssODN donors for either gene. Three days later, genomic DNA was isolated from the 20% most highly fluorescent cell population and analysed by CEL-1 assay for modification at the target site as well as the four off-target sites shown in panel (A). PCR products containing amplicons derived from mutant alleles or PCR products derived from wild-type alleles only are indicated by the arrows and asterisks, respectively. The experiment was repeated three times with similar results. N.D.: none detected.
The absence of off-target mutagenesis in clones derived from cells with high levels of above ZFNs was confirmed by complementary analyses. Thus, CEL-I assays failed to detect mutations in any of the above sites in pooled genomic DNA isolated from highly double-fluorescent cell populations 3 days after transfection with EGFP-2A-ZFNL and DsRed2–2A-ZFNR for RSK4 or RSK2 along with the aforementioned ssODN donor for either gene (Figure 4B). Neither did CEL-I assays detect any mutations in an additional six top-ranking candidate off-target sites with six and seven mismatches relative to the on-target site of the RSK4 and RSK2 ZFNs, respectively (Supplementary Figure S8).
We further assessed the issue of off-target cutting by analysing a number of CHO cell lines with knockout of enzymes in the protein glycosylation pathway that we generated using our method. The clones had biallelic, NHEJ-derived knockout of either of four genes and were established from the top 2–18% most highly double-fluorescent cell populations after transfection with the relevant EGFP-2A-ZFNL and DsRed2–2A-ZFNR constructs (data not shown). Only one monoallelic off-target mutation was identified by Sanger sequencing of various top-ranking off-target sites for the four ZFNs in a number of clones, with a total of 94 alleles probed (off-target 1 for B4GALT4 ZFNs; Supplementary Figure S9A and C).
CEL-I assay on cell pools 3 days after transfection with B4GALT4 ZFNs confirmed that these ZFNs had a propensity to cut off-target site 1 upon sorting for high expression levels, but with a lower efficiency than the on-target site (Supplementary Figure S9B). We therefore also analysed modification of both these sites in cells FACS enriched for medium ZFN levels to test if it was possible to generate a cell population with an even more favourable on/off-target mutagenesis ratio. Interestingly, in the population with medium ZFN levels, detectable off-target cutting was eliminated, whilst the on-target modification remained high.
CRISPR/Cas9-based genome editing is facilitated using the 2A-coupled fluorescent protein/FACS enrichment strategy
To explore our method in relation to other nucleases used for genome editing, we generated a construct expressing Cas9–2A-GFP under control of the CMV promoter as well as the gRNA sequence for the target gene driven by the U6 promoter (pCMV-Cas9-FP; Figure 5A). K562 cells were transfected with this construct encoding gRNA targeting KRAS. After three days, the cells were left unsorted or were FACS isolated for low, medium or high GFP expression levels and subsequently assayed for NHEJ modification of the KRAS locus (triploid in K562 cells; Supplementary Table S6). In unsorted cells, ∼15% of the alleles were modified, which increased to ∼24%, ∼41% and ∼59% in cells displaying low, medium and high fluorescence levels, respectively (Figure 5B). Qualitatively similar results were obtained with the same construct in which the gRNA targeted the VEGFA locus (Figure 5C, upper panel), which is tetraploid in K562 cells (Supplementary Table S6). Thus, 2A-linked co-expression of fluorescent protein and Cas9 combined with FACS can also be used to effectively increase genome editing frequencies in the CRISPR/Cas9 system.
However with certain gRNAs, the CRISPR/Cas9 system has recently been found to exhibit significant off-target cutting (35–38).These include the particular VEGFA gRNA used here, which was found to target additional genomic sites with relatively high efficiency (36). We analysed two of these sites and found that their modification also increased upon FACS for high Cas9 expression levels, one of them to the same extent as the on-target site (Figure 5C, middle and lower panels).
To increase the utility of our method for CRISPR/Cas9-based editing, we tested whether it functions in conjunction with CRISPR/Cas9 nickase mutant pairs, which exhibit enhanced specificity, partly due to low indel mutagenesis rates by the individual nickases of the pair as compared to CRISPR with wild-type Cas9 (12–14). Accordingly, we demonstrated that the on- as well as off-target NHEJ mutagenesis by the VEGFA CRISPR/Cas9 construct observed in the top-expressing cells by CEL-I assays was non-detectable with the D10A mutant nickase version of Cas9 (Cas9N (9)) (Figure 5C, last lane in all panels). Thus, the use of Cas9N can reduce the level of off-target modification associated with FACS for high-level Cas9 expression.
To incorporate Cas9N into our method, we generated CRISPR/Cas9N pairs, in which one construct contained Cas9N that was 2A-coupled to GFP and the other construct contained Cas9N 2A-coupled to DsRed2. K562 cells were co-transfected with such a pair harbouring gRNA pairs previously used to target the EMX1 locus (12). To further assess our method for knockin editing, we co-transfected an ssODN donor designed to introduce an EcoRV site in the locus. Three days post-transfection, the cells were left unsorted or were FACS isolated for low, medium or high GFP/DsRed2 double-fluorescence levels. As we observed for ZFN pairs, genome modification levels increased with increasing fluorescence levels present in cells transfected with CRISPR/Cas9N pairs (Figure 5D). In the most highly fluorescent cell population, ∼11% of alleles of the EMX1 locus (triploid in K562 cells; Supplementary Table S6) were knockin modified, as evidenced by RFLP assay. Total modification rates were ∼23%, as evidenced by CEL-I assay, indicating NHEJ mutations in addition to the knockin modification. By contrast, no mutagenesis was detectable in top-expressing cells after transfection with only one member of the CRISPR/Cas9N pair (Figure 5D). Qualitatively similar results were obtained with another CRISPR/Cas9N pair, in which one of the gRNAs was swapped for another EMX1 target sequence (Supplementary Figure S10).
DISCUSSION
We have developed a method for genome editing via 2A-coupled co-expression of nuclease and fluorescent protein followed by FACS enrichment of cells with high nuclease levels. In the several and diverse variations tested, the method was found to greatly increase genome editing efficiencies of ZFNs as well as CRISPR/Cas9. In most cases, this occurred in a dose-dependent manner: the higher nuclease levels sorted for, the higher genome editing frequencies achieved, both for NHEJ mutagenesis and for knockin editing.
Our data agree with the finding in the model system of Certo et al. (21) that increased levels of nuclease result in increased levels of mutagenesis. In the present study, we mainly delivered donors as ssODNs and nucleases as mRNA. Whilst high knockin levels may also be achieved with plasmid constructs for donors and nucleases, it is noteworthy that the herein used delivery forms work effectively in our method, since ssODNs and mRNA have several advantages: first, ssODN donors can be obtained fast and inexpensively. Second, nuclease expression from mRNA is independent of issues relating to the efficiency, or lack thereof, of a given plasmid promoter in the targeted cell type. Third, when using ssODN and mRNA delivery, editing can be performed entirely without double-stranded DNA, thus minimizing the risk of random genomic integration of donor and nuclease constructs, which by the nature of our method are introduced in very high numbers when sorting for the most efficiently transfected cells. Finally, we consider it likely that the use of ssODN donors contributes to the high knockin frequencies obtained in several of the present experiments. Accordingly, we previously found that ssODNs are more efficient than plasmids as donor in side-by-side comparisons (15). The reason may be that the small size of ssODN donors (∼100 bp) compared to plasmid donors (4–6 kb) permits inclusion of a greater numerical level of donor constructs in the transfection reaction, which should ultimately increase knockin frequencies. This notion would agree with the study by Certo et al. (21), showing that high levels of donor promotes knockin editing at the expense of NHEJ. Recently, fluorescent proteins have also been 2A-coupled to TALENs followed by FACS for transfected cells (39). Whilst this study did not enrich for specific nuclease levels, it demonstrates that the fluorescent protein-2A strategy also works for TALENs, which may therefore similarly be used in the various enrichment protocols described here.
One advantage of 2A-coupled co-expression of nuclease and fluorescent protein is that FACS enrichment for fluorescent cells guarantees that these cells also took up the nuclease construct. For nucleases acting as a pair, the coupling of each monomer to a distinct fluorescent protein furthermore ensures that double-fluorescent cells took up both monomers of the pair. The benefit of the dual labelling was particularly evident in the Jurkat cells, where a substantial fraction of cells appeared to express only one monomer of the ZFN pair. Whilst both monomers need be present in the cell, a 1:1 expression ratio may not be optimal for all ZFN pairs (40). For such ZFN pairs, different amounts of individual monomer constructs may be transfected, followed by sorting for a population in which the individual monomers are present both at optimal relative levels and at as high levels as possible. The approach of 2A-coupling both ZFNs of a pair to the same fluorescent protein, as in pFP-ZFNL-ZFNR constructs, is another means to ensure that FACS will only enrich for cells that have taken up both ZFNs of a pair. This single vector strategy might be particularly useful for multiplex editing, in which ZFN pairs for several different genes could each be linked to a distinct colour for simultaneous multi-colour FACS enrichment of cells in which several genes may have been modified. Finally, 2A-coupled constructs have the advantage that a ‘normal’ fluorescence signal for a particular cell type can reveal that the co-encoded nuclease construct is of good quality, i.e. not degraded, a control that may be particularly useful with labile RNA nuclease constructs. By contrast, non-coupled co-expression of the fluorescence marker via a separate construct cannot guarantee that fluorescent cells have also taken up the co-transfected nuclease. Such potential discrepancy between fluorescence signal and nuclease uptake may be part of the reason why we observed higher genome editing frequencies with 2A-coupled co-expression of the fluorescent protein and ZFN than with non-coupled co-expression.
In the surrogate gene reporter strategy, nucleases are co-transfected with a reporter that constitutively expresses RFP to mark transfected cells and conditionally expresses GFP upon indel mutagenesis of a nuclease target site placed upstream of the GFP (20), thereby marking cells expressing active nuclease. The authors showed that FACS can thereafter be used to successfully isolate cell populations enriched for active nuclease and NHEJ-based mutation of the chromosomal target gene. Compared to the surrogate reporter strategy, the present method allows for more donor in the transfection reaction, can be performed without the use of double-stranded DNA, has more fluorescence markers available for multi-plexing (since none are used for a reporter construct) and does not require construction of a surrogate reporter target site. Furthermore, since fluorescence signal in the 2A-coupled approach is a more direct measure of nuclease levels, it may be easier to balance nuclease levels for an optimal on/off-target ratio using the present method, as discussed below.
Although ZFNs are rather specific reagents, off-target cleavage can occur and may increase at elevated levels of nuclease (33,41). However, for the most exhaustively characterized ZFNs in this study (RSK4 and RSK2), we found no mutations in top candidate off-target sites in cells enriched for very high ZFN levels, using a depth of analysis that should detect off-target rates occurring with >1% on average. Furthermore, analysis of a number of CHO knockout clones generated using our method also suggested very modest off-target effects. It should be noted, however, that the extent of off-target cutting is inherently dependent on ZFN design. The ZFNs used here had no predictable off-target sites with less than four nucleotide mismatches compared to the on-target site. Furthermore, the target sites were 30–33 bps long in total. It is possible that higher off-target levels will be observed in top-expressing cells if ZFNs with off-target sites containing fewer mismatches and/or having shorter target sites are used. Both of these features promote off-target cutting and were features of several of the ZFNs used in the aforementioned studies on the off-target activity of ZFNs. It should also be noted that we only analysed predictable off-target sites (i.e. homologous off-target sites), and thus did not address potential off-target actions at non-homologous sites, which would require whole-genome sequencing or other unbiased genome-wide strategies (33). Nevertheless, at this point our data suggest that with ZFN designs similar to those used here, off-target effects associated with our method are very modest and that it seems easy to derive clones with no modifications in predictable off-target sites.
Interestingly, in the one case in which off-target mutagenesis occurred at high ZFN levels, it was possible to choose a cell population with medium ZFN levels, which eliminated detectable off-target cutting, whilst maintaining high rates of on-target modification. We speculate that mismatches in off-target sites render their cutting more sensitive to decreasing ZFN concentrations than the on-target-site, a notion supported by ZFN off-target analyses in vitro (41), such that nuclease levels can be obtained where on-target cutting is relatively much higher than the off-target event, as compared to saturating doses of nuclease. With the current knowledge on ZFN action it is not possible to predict a priori an optimal ZFN expression level showing the desired on- and off-target cutting ratio. This must be determined on a case-by-case basis, for example, by performing experiments like the one shown in Supplementary Figure S9B. Such experiments may be able to identify a cell population with a suitable nuclease level, which may subsequently be used to generate modified cell clones or cell pools.
Regarding the CRISPR/Cas9 system, the VEGFA construct used here produced increased cutting at certain off-target sites upon FACS for high nuclease levels. These findings agree with previous studies that some gRNAs, including the present VEGFA gRNA, exhibit significant off-target actions that may increase with increasing amounts of gRNA (35–38). Although we did not test the possibility, in some cases our method might be used to balance CRISPR/Cas9 on- and off-target effects in the same manner we showed for ZFNs. However, this will not be possible for certain gRNAs, as exemplified in the present case with VEGFA, in which the on-target site and off-target site 1 were mutated to the same extent at the various expression levels. It is possible that more careful design and/or new developments in the design of gRNA, such as described in (13), may ameliorate the off-target issue of our method applied with CRISPR/Cas. However, it is noteworthy that our method also increased the genome editing efficiency of CRISPR/Cas9N nickase pairs, a version of the CRISPR/Cas9 system that shows greatly improved specificity (12–14). The individual CRISPR/Cas9Ns of a pair will likely show increased off-target nicking upon FACS enrichment for high Cas9N levels. However, since DNA single-strand breaks are predominantly repaired by the high-fidelity base excision repair pathway (42), off-target nicks will rarely be mutagenic. Accordingly, we detected no indel mutagenic effects of individual CRISPR/Cas9Ns in cells sorted for high-level expression of such constructs. Another reason for the higher specificity of CRISPR/Cas9 nickase pairs is a combined target sequence that is twice as long as that of an individual CRISPR/Cas9 construct. Altogether, nickase pairs seem a particularly safe version of the CRISPR/Cas9 genome editing system for use in our method.
We primarily validated our method by assessing genome editing in stably modified clonal cell lines or stable cell pools. This is opposed to assessing editing in genomic DNA from cell pools 3 days after transfection with editing reagents, as is often performed, but which may not be a reliable indicator of stable modification rates. For instance, in our hands and in the hands of others, modification rates observed 3 days post transfection can be many-fold higher than those eventually observed in stably modified cells (15,20). Altogether, we validated our method in a very large set of clonal cell lines (>4000), of which 426 were monoallelic and 648 were biallelic knockin clones. This also highlights that in some cases our method can serve as a high-throughput strategy for generation of targeted knockin clones. The ability to generate many clones with the same mutation may be a powerful means to minimize the confounding impact of clonal variation on downstream functional analyses, a problem inherent to most studies on clonal cell lines. For example, 25 knockin clones may be generated, pooled and subjected to phenotypic analysis, a strategy that may effectively dilute out non-nuclease-derived phenotypic differences of individual clones in the overall results achieved when studying the clone pool. A similar benefit may be obtained by studying cell pools with close-to-complete genome modification via consecutive rounds of ZFN transfections and FACS on a cell population, as we also found feasible in the present study.
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online.
Acknowledgments
The authors thank Anna Fossum, BRIC, for excellent FACS assistance and Allan Lind-Thomsen, Wilhelm Johanssen Centre for Functional Genome Research for the prediction of CHO ZFN off-target sites. This study is dedicated to Dr Dana Carroll, one of the founding fathers of ZFN genome editing technology.
FUNDING
Danish Cancer Society [R2-A132-02-S2 to M.F., R40-A1936-11-S2 to S.H.H.]; Novo Nordisk Center for Biosustainability [Z.Y.]; University of Copenhagen 2016 Excellence Programme [M.F. and H.H.W.]; Danish Cancer Research Foundation [10114941 to M.F.]; Novo Nordisk Foundation [M.F.]; Danish National Research Foundation [M.F.]; National Institutes of Health [NIH RO1 CA142647 to S.H.H.]. Funding for open access charge: BRIC core funding [M.F.].
Conflict of interest statement. Qiaohua Kang and Gregory D. Davis are present employees and Shondra M. Pruett-Miller is a former employee of Sigma-Aldrich Corporation.
REFERENCES
- 1.Stoddard B.L. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure. 2011;19:7–15. doi: 10.1016/j.str.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188:773–782. doi: 10.1534/genetics.111.131433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Urnov F.D., Rebar E.J., Holmes M.C., Zhang H.S., Gregory P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 4.Bogdanove A.J., Voytas D.F. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 5.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 6.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R., Joung J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen F., Pruett-Miller S.M., Huang Y., Gjoka M., Duda K., Taunton J., Collingwood T.N., Frodin M., Davis G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods. 2011;8:753–755. doi: 10.1038/nmeth.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaj T., Guo J., Kato Y., Sirk S.J., Barbas C.F., III Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat. Methods. 2012;9:805–807. doi: 10.1038/nmeth.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Provasi E., Genovese P., Lombardo A., Magnani Z., Liu P.Q., Reik A., Chu V., Paschon D.E., Zhang L., Kuball J., et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012;18:807–815. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holkers M., Maggio I., Liu J., Janssen J.M., Miselli F., Mussolino C., Recchia A., Cathomen T., Goncalves M.A. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63. doi: 10.1093/nar/gks1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lombardo A., Genovese P., Beausejour C.M., Colleoni S., Lee Y.L., Kim K.A., Ando D., Urnov F.D., Galli C., Gregory P.D., et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 20.Kim H., Um E., Cho S.R., Jung C., Kim J.S. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat. Methods. 2011;8:941–943. doi: 10.1038/nmeth.1733. [DOI] [PubMed] [Google Scholar]
- 21.Certo M.T., Ryu B.Y., Annis J.E., Garibov M., Jarjour J., Rawlings D.J., Scharenberg A.M. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods. 2011;8:671–676. doi: 10.1038/nmeth.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szymczak A.L., Vignali D.A. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert Opin. Biol. Ther. 2005;5:627–638. doi: 10.1517/14712598.5.5.627. [DOI] [PubMed] [Google Scholar]
- 23.Doyon Y., Vo T.D., Mendel M.C., Greenberg S.G., Wang J., Xia D.F., Miller J.C., Urnov F.D., Gregory P.D., Holmes M.C. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat. Methods. 2011;8:74–79. doi: 10.1038/nmeth.1539. [DOI] [PubMed] [Google Scholar]
- 24.Zacharias D.A., Violin J.D., Newton A.C., Tsien R.Y. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 25.Strack R.L., Hein B., Bhattacharyya D., Hell S.W., Keenan R.J., Glick B.S. A rapidly maturing far-red derivative of DsRed-Express2 for whole-cell labeling. Biochemistry. 2009;48:8279–8281. doi: 10.1021/bi900870u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagai T., Ibata K., Park E.S., Kubota M., Mikoshiba K., Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 27.Kremers G.J., Goedhart J., van Munster E.B., Gadella T.W., Jr Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry. 2006;45:6570–6580. doi: 10.1021/bi0516273. [DOI] [PubMed] [Google Scholar]
- 28.Xia N.S., Luo W.X., Zhang J., Xie X.Y., Yang H.J., Li S.W., Chen M., Ng M.H. Bioluminescence of Aequorea macrodactyla, a common jellyfish species in the East China Sea. Mar. Biotechnol. (N.Y.) 2002;4:155–162. doi: 10.1007/s10126-001-0081-7. [DOI] [PubMed] [Google Scholar]
- 29.Subach O.M., Gundorov I.S., Yoshimura M., Subach F.V., Zhang J., Gruenwald D., Souslova E.A., Chudakov D.M., Verkhusha V.V. Conversion of red fluorescent protein into a bright blue probe. Chem. Biol. 2008;15:1116–1124. doi: 10.1016/j.chembiol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merzlyak E.M., Goedhart J., Shcherbo D., Bulina M.E., Shcheglov A.S., Fradkov A.F., Gaintzeva A., Lukyanov K.A., Lukyanov S., Gadella T.W., et al. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods. 2007;4:555–557. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- 31.Doyon Y., Choi V.M., Xia D.F., Vo T.D., Gregory P.D., Holmes M.C. Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat. Methods. 2010;7:459–460. doi: 10.1038/nmeth.1456. [DOI] [PubMed] [Google Scholar]
- 32.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gabriel R., Lombardo A., Arens A., Miller J.C., Genovese P., Kaeppel C., Nowrouzi A., Bartholomae C.C., Wang J., Friedman G. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 34.Marella N.V., Malyavantham K.S., Wang J., Matsui S., Liang P., Berezney R. Cytogenetic and cDNA microarray expression analysis of MCF10 human breast cancer progression cell lines. Cancer Res. 2009;69:5946–5953. doi: 10.1158/0008-5472.CAN-09-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cradick T.J., Fine E.J., Antico C.J., Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding Q., Lee Y.K., Schaefer E.A., Peters D.T., Veres A., Kim K., Kuperwasser N., Motola D.L., Meissner T.B., Hendriks W.T., et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szczepek M., Brondani V., Buchel J., Serrano L., Segal D.J., Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 2007;25:786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 41.Pattanayak V., Ramirez C.L., Joung J.K., Liu D.R. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dianov G.L., Hubscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 2013;41:3483–3490. doi: 10.1093/nar/gkt076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.