


Abstract
Instead of a classical single-stranded deoxyribonuleic acid (DNA)-binding protein (SSB), some hyperthermophilic crenarchaea harbor a non-canonical SSB termed ThermoDBP. Two related but poorly characterized groups of proteins, which share the ThermoDBP N-terminal DNA-binding domain, have a broader phylogenetic distribution and co-exist with ThermoDBPs and/or other SSBs. We have investigated the nucleic acid binding properties and crystal structures of representatives of these groups of ThermoDBP-related proteins (ThermoDBP-RPs) 1 and 2. ThermoDBP-RP 1 and 2 oligomerize by different mechanisms and only ThermoDBP-RP2 exhibits strong single-stranded DNA affinity in vitro. A crystal structure of ThermoDBP-RP2 in complex with DNA reveals how the NTD common to ThermoDBPs and ThermoDBP-RPs can contact the nucleic acid in a manner that allows a symmetric homotetrameric protein complex to bind single-stranded DNA molecules asymmetrically. While single-stranded DNA wraps around the surface or binds along channels of previously investigated SSBs, it traverses an internal, intersubunit tunnel system of a ThermoDBP-RP2 tetramer. Our results indicate that some archaea have acquired special SSBs for genome maintenance in particularly challenging environments.
INTRODUCTION
Deoxyribonucleic acid (DNA) replication, repair, recombination and telomere maintenance require the transient unwinding of duplex DNA (1,2). To maintain and protect DNA in the single-stranded (ss) state, all organisms harbor essential single-stranded DNA-binding proteins (SSBs) that bind ssDNA with high affinity and low sequence specificity. Classical SSBs harbor one of four distinct DNA-binding domains: the oligonucleotide/oligosaccharide-binding (OB) fold; the K homology (KH) domain; the ribonucleic acid (RNA) recognition motif (RRM) or the whirly domain (3). The sequences of the proteins are poorly conserved in either group (4) and different SSBs adopt a variety of oligomeric states (5), thereby bringing several ssDNA-binding domains together (6).
Recently, a group of hyperthermophilic crenarchaea has been found to lack a classical SSB and instead to contain a distinct SSB, termed ThermoDBP (7). The crystal structure of the ssDNA-binding N-terminal domain (NTD) of Thermoproteus tenax (tte) ThermoDBP, comprising a four-stranded β-sheet packed against four α-helices (7), differs markedly from the classical SSB DNA-binding domains, and tteThermoDBP was predicted to dimerize in a parallel fashion via a C-terminal leucine zipper (7). However, the exact mode of oligomerization and the mechanism of DNA binding of ThermoDBPs so far remained elusive.
ThermoDBPs share sequence similarity with domain of unknown function (DUF) 2258 proteins, which have a broader phylogenetic distribution in archaea (Thermoproteales, Sulfolobales, Desulfurococcales, Thermococci and Archaeoglobi). DUF2258 proteins can be divided into two groups that we term ThermoDBP-related proteins (ThermoDBP-RPs) 1 and 2. Both groups contain a ThermoDBP-like NTD but differ from ThermoDBPs and from each other in their C-terminal regions (Supplementary Figure S1). The ThermoDBP-like NTD suggests that ThermoDBP-RPs might also be SSBs. However, tteThermoDBP-RP1 was not recovered from cell lysates via a biotinylated oligodeoxynucleotide used for isolation of tteThermoDBP (7). Furthermore, Sulfolobus solfataricus (sso) ThermoDBP-RP1 was reported to associate with box C/D small (s) RNAs and with the 30S ribosomal subunit (8), but the protein was not detected in a subsequent proteomic characterization of sso ribosomes (9). We therefore set out to further characterize the structures and functions of ThermoDBP-RPs.
Our analyses show that ThermoDBP-RP2s are unconventional SSBs that bind ssDNA in an asymmetric fashion at an internal, intersubunit tunnel system of a symmetric protein tetramer. The apo-structure of ThermoDBP-RP1 suggests that DNA binding by this group of proteins may be auto-inhibited, explaining their weak nucleic acid affinities observed in vitro. Structural comparisons reveal that the eukaryotic DNA polymerase eta has acquired a domain that closely resembles the N-terminal ssDNA-binding domain of ThermoDBPs and ThermoDBP-RPs but employs a different molecular surface for binding to double-stranded (ds) DNA.
MATERIALS AND METHODS
Protein production
Coding sequences of Pyrococcus furiosus (pfu) ThermoDBP-RP1 and Aeropyrum pernix (ape) ThermoDBP-RP2 were polymerase chain reaction (PCR)-amplified from P. furious and A. pernix genomic DNA. The PCR fragments encoding full-length ThermoDBP-RP genes were cloned into pET-M11 (EMBL, Heidelberg) via NcoI/BamHI restriction sites in frame with an N-terminal His6-tag followed by a Tobacco Etch Virus (TEV) protease cleavage site. For expression, cells were grown in auto-inducing medium (10) at 37°C to an OD600 of 0.8, transferred to 18°C and harvested after 60 h. Cell pellets were resuspended in 20 mM HEPES-NaOH, pH 7.5, 500 mM NaCl and 5 mM β-mercaptoethanol supplemented with 10 μg/ml DNaseI, 3 mM MgCl2 and 100 μg/ml lysozyme. Cells were lyzed by four passes through a microfluidizer at 80 kPsi. The cell lysates were clarified by centrifugation. For purification, clarified lysates were loaded onto Ni2+-NTA resin equilibrated with lysis buffer. Beads were washed with one bed volume of 20 mM HEPES-NaOH, pH 7.5, 2 M LiCl and 5 mM β-mercaptoethanol followed by washing with 5 bed volumes of 20 mM HEPES-NaOH, pH 7.5, 500 mM NaCl, 20 mM imidazole and 5 mM β-mercaptoethanol. Proteins were eluted with 20 mM HEPES-NaOH, pH 7.5, 200 mM NaCl, 5 mM β-mercaptoethanol and 300 mM imidazole. Eluates were mixed with TEV protease (mass ratio protease/protein 1:50) and dialyzed overnight at 4°C against 20 mM HEPES–NaOH, pH 7.5, 100 mM NaCl and 2 mM β-mercaptoethanol. The dialyzed samples were again loaded on Ni2+-NTA resin equilibrated with dialysis buffer to remove the TEV protease and uncleaved protein. The flow-throughs were further purified by ion exchange chromatography. Samples were loaded on a MonoQ column (GE Healthcare) equilibrated with dialysis buffer. Proteins were eluted with a linear gradient from 100 mM to 500 mM NaCl over 50 column volumes. Fractions containing the proteins of interest were loaded on Superdex 75 (pfuThermoDBP-RP1) or Superdex 200 (apeThermoDBP-RP2) size-exclusion chromatography columns (GE Healthcare) pre-equilibrated with 10 mM Tris–HCl, pH 7.6, 50 mM NaCl and 1 mM DTT. Fractions containing pure protein were pooled and concentrated to 20 mg/ml using Amicon concentrators (Millipore). To prepare selenomethionine (SeMet)-labeled proteins, E. coli BL21(DE3) cells were grown in SeMet auto-inducing medium (10). The other steps for expression and protein purification were the same as described above.
Electrophoretic gel mobility shift assays
Box C/D small RNAs (sRNAs) were synthesized by in vitro transcription using T7 RNA polymerase and PCR amplified templates from P. furious genomic DNA. Transcription products were purified on a 6% urea-TBE denaturing polyacrylamide gel followed by phenol-chloroform extraction and ethanol precipitation. All other oligonucleotides (Supplementary Table S1) were chemically synthesized (Dharmacon and MWG/Operon). To prepare DNA or RNA duplexes, complementary strands were mixed in equimolar ratios, heated to 80°C and slowly cooled (0.2°C/min) in a thermocycler to 20°C. The samples were then separated on agarose gels and duplex bands were extracted and ethanol precipitated. For electrophoretic gel mobility shift assays, oligonucleotides were 5′-end labeled with γ-[32P]-ATP. Labeled oligonucleotides were mixed with recombinant proteins in 20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 2 mM DTT and incubated at 20°C or 80°C for 10 min. To compete RNA binding, 0.5 g/l E. coli transferRNAs (tRNAs) were added to the reactions. Samples were then fractionated on a 6% (60:1) polyacrylamide gel and visualized using a PhosphorImager (Typhoon 8600, GE Healthcare). Binding of apeThermoDBP-RP2 to single-stranded and circular M13 phage DNA was tested on a 1% agarose gel. Increasing concentrations of the protein were mixed with 100 ng DNA and incubated at 4°C for 1 h. DNA was visualized by ethidium bromide staining.
Ribosome binding assays
Ribosomes and ribosomal subunits from Thermococcus kodakarensis were prepared as described (9). In a total reaction volume of 150 μl, 50 pmoles of 30S, 50S or 70S T. kodakarensis ribosomal subunits or ribosomes were incubated with 400 pmoles of His6-tagged pfuThermoDBP-RP1 or apeThermoDBP-RP2 in a buffer containing 10 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 1 mM DTT, 45 mM NH4Cl and the varying concentrations of NaCl. The mixture was incubated for 20 min at 37°C and was then placed on 90 μl of a 20% sucrose cushion. The cushion was centrifuged for 30 min at 78 000 rpm in a TLA 100 rotor. After centrifugation the supernatant was removed and the pellet was resuspended. The input sample before centrifugation, the supernatant and the pellet were analyzed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis followed by Western blotting. The membranes were stained by amido black to check the protein content in each lane and were then treated by anti-His6 antibody for detection of ThermoDBP-RPs.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed at 25°C on an iTC200 microcalorimeter equipped with a 300-μl syringe (Microcal, GE Healthcare). DNA oligonucleotides and apeThermoDBP-RP2 were dialysed against the same buffer overnight. apeThermoDBP-RP2 was titrated at 10 μM concentration in a cell volume of 250 μl with 50–100 μM of different DNA oligonucleotides in 16 injections of 2.5 μl volumes with 5-min intervals. The released heat was obtained by integrating the calorimetric output curves. Binding parameters were calculated using the Origin5 software using the 'One Set of Sites' curve fitting model provided by the software.
Analytical size-exclusion chromatography
Analytical size-exclusion chromatography was conducted on Superdex 75 (pfuThermoDBP-RP1) or Superdex 200 (apeThermoDBP-RP2) PC 3.2/30 analytical size-exclusion columns (GE Healthcare) in 20 mM HEPES-NaOH, pH 7.5, 100 mM NaCl. Exclusion volumes (Vo) were estimated using Blue Dextran (approximate molecular mass 2 MDa). Bovine γ-globulin (158 kDa), rabbit lactate dehydrogenase (140 kDa), chicken conalbumin (75 kDa), bovine serum albumin (67 kDa), chicken ovalbumin (45 kDa) and horse myoglobin (17 kDa) were used as molecular mass standards. Linear regression of plots of log (molecular mass) versus the ratios of the observed elution and exclusion volumes (Ve/Vo) allowed calculation of the apparent molecular masses of pfuThermoDBP-RP1 and apeThermoDBP-RP2 based on the observed elution volumes of the proteins.
Crystallization of ThermoDBP-RPs and of an apeThermoDBP-RP2-dT10 complex
Crystallization of pfuThermoDBP-RP1 was carried out at 20°C using the sitting drop vapor diffusion method. pfuThermoDBP-RP1 crystallized in two different crystal forms. Form 1 crystals of SeMet-labeled pfuThermoDBP-RP1 were obtained by mixing 1 μl of SeMet-labeled protein solution with 1 μl of reservoir solution containing 50 mM sodium cacodylate, pH 6.4, 2.3 M (NH4)2SO4 and 10 mM MgSO4. Prior to flash-cooling in liquid nitrogen, form 1 crystals of pfuThermoDBP-RP1 were soaked in reservoir buffer supplemented with 25% (v/v) glycerol. Form 2 crystals of SeMet-labeled pfuThermoDBP-RP1 were obtained by mixing 0.5 μl of protein solution with 1 μl of reservoir solution containing 0.1 M Tris–HCl, pH 8.5, 0.2 M Li2SO4 and 40% (v/v) PEG400. These crystals were flash-cooled in a 100 K cryogenic stream in their mother liquor. Crystals of apeThermoDBP-RP2 were produced by mixing equal volumes of SeMet-labeled apeThermoDBP-RP2 with a reservoir solution containing 0.1 M imidazole, pH 8.0, 0.4 M NaH2PO4, 1.6 M K2HPO4, 0.2 M NaCl and 0.25 M glycine. Crystals were cryo-protected by addition of 25% (v/v) glycerol to the mother liquor.
For co-crystallization with ssDNA, apeThermoDBP-RP2 (17 mg/ml) was mixed with a 10-fold molar excess of dT10 in 10 mM Tris–HCl, pH 7.6, 50 mM NaCl and 1 mM DTT. The mixture was incubated for 5 min at 80°C. Crystallization experiments were performed in a sitting drop format at 18°C. Crystals appeared after three days over a reservoir solution composed of 200 mM NaOAc, pH 4.3, 1.3 M NaH2PO4 and 0.7 M K2HPO4. For cryo-protection, crystals were transferred to mother liquor supplemented with 15% (v/v) glycerol and subsequently flash-cooled in liquid nitrogen.
Crystallographic procedures
Diffraction data were collected at beamline BL14.2 of the BESSY II storage ring (Berlin, Germany; Supplementary Table S2). Data were processed with the XDS package (11). The structure of pfuThermoDBP-RP1 form 1 crystals was solved by the multiple-wavelength anomalous dispersion method using the program SHARP/autoSHARP (12). The structure of the second crystal form of pfuThermoDBP-RP1 was solved by molecular replacement using the structure coordinates of the crystal form 1 as a search model with the program MOLREP (13). The structure of apeThermoDBP-RP2 was solved via the single-wavelength anomalous dispersion method using the program HKL2MAP (14,15). Finally, the crystal structure of an apeThermoDBP-RP2-dT10 complex was solved by molecular replacement using the structure coordinates of an apeThermoDBP-RP2 tetramer as a search model with the program PHASER (16). Model building was done manually with COOT (17). For all the structures, refinement was done with phenix.refine (18,19) or Refmac5 (20) including TLS refinement (21). Intermediate and final structures were evaluated with MOLPROBITY (22). All structure figures were drawn using PYMOL (23). Electrostatic surfaces were calculated with APBS (24).
RESULTS
Nucleic acid binding by ThermoDBP-RPs
To investigate the possible cellular functions of ThermoDBP-RPs, we first tested the reported interactions of the proteins with box C/D sRNAs and ribosomes or ribosomal subunits in vitro. In electrophoretic mobility shift assays (EMSAs), pfuThermoDBP-RP1 and apeThermoDBP-RP2 bound to box C/D sRNAs but the interactions were efficiently competed by the addition of unlabeled Escherichia coli tRNAs (Supplementary Figure S2), suggesting that the proteins do not recognize sequence or structural features of box C/D sRNAs with high specificity. At low salt concentrations (20–120 mM NaCl), the proteins were pelleted by 30S and 50S ribosomal subunits and by 70S ribosomes (Supplementary Figure S3A; left and middle panels; lanes 3, 6 and 9). Increasing the salt concentration to 200 mM abrogated binding to the subunits and ribosomes, suggesting that these interactions are only marginally stable (Supplementary Figure S3B; left and middle panels; lanes 3, 6 and 9).
While these results do not rule out the possibility that ThermoDBP-RP proteins interact with box C/D sRNAs or ribosomes in vivo, they prompted us to investigate if the proteins interact also with other nucleic acids. Indeed, EMSAs (Figure 1A and B) and isothermal titration calorimetry (ITC; Supplementary Figure S4) showed that apeThermoDBP-RP2 bound to a 21-mer ssDNA of mixed sequence or to 21-mer homo-pyrimidine ssDNAs with apparent Kd values of about 50–100 nM but very weakly or not at all to homo-purine ssDNAs, DNA duplexes, ssRNAs or an RNA duplex of the same length. A longer (45-mer) ssDNA was gradually shifted in EMSAs (Figure 1C), indicating a stepwise binding of more than one apeThermoDBP-RP2 molecule or complex. Under the same experimental conditions, pfuThermoDBP-RP1 did not bind to any of the 21-mer nucleic acids (Figure 1A and B) but weakly bound to a 45-mer ssDNA (about 50% shift at 50–100 μM protein concentration; Figure 1C). Increasing the temperature or time of incubation had no effect on any of these interactions. These data suggest that at least ThermoDBP-RP2 proteins can act as SSBs.
Figure 1.
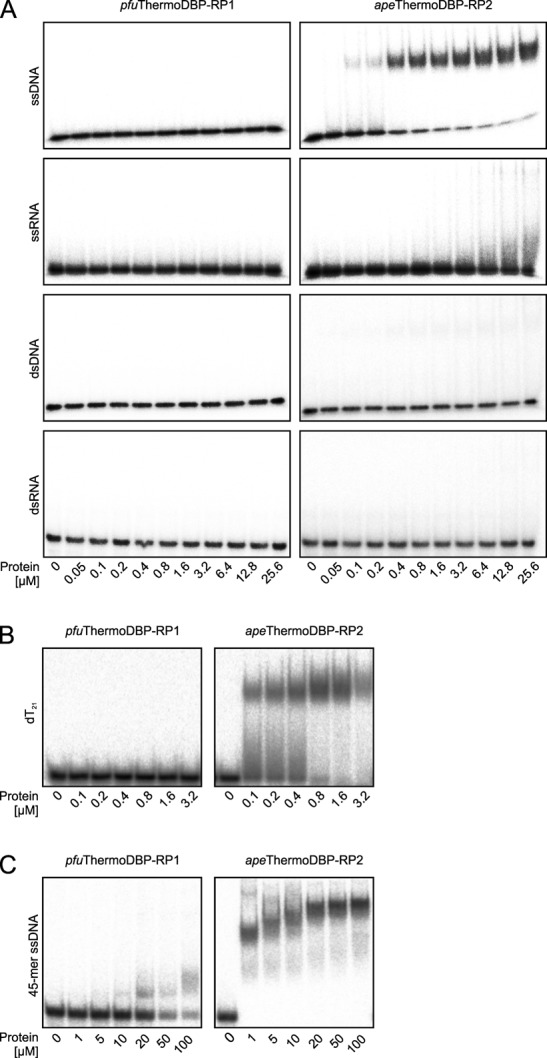
Nucleic acid binding properties of ThermoDBP-RP proteins. (A) Electrophoretic gel mobility shift assays testing the nucleic acid binding capabilities and preferences of pfuThermoDBP-RP1 and apeThermoDBP-RP2. ssDNA–d(ACTGCTAGAGATTTTCCACAT); ssRNA–r(ACTGCTAGAGATTTTCCACAT); dsDNA and dsRNA additionally contained the corresponding complementary strands. (B) Binding of pfuThermoDBP-RP1 and apeThermoDBP-RP2 to a 21-mer homo-pyrimidine (dT21) DNA. (C) Binding of pfuThermoDBP-RP1 and apeThermoDBP-RP2 to a 45-mer ssDNA–d(CTTGCTAGGACGGATCCCTCGAGGTTTTTTTTTTTTTTTTTTTT).
Crystal structures and modes of oligomerization of pfuThermoDBP-RP1 and apeThermoDBP-RP2
We next determined the crystal structures of pfuThermoDBP-RP1 and apeThermoDBP-RP2 at 2.43 and 2.05 Å resolution, respectively (Figure 1A, B and Supplementary Table S2). As expected from sequence analysis (Supplementary Figure S1), pfuThermoDBP-RP1 contains an NTD similar to the tteThermoDBP NTD (Figure 2C; root-mean-square deviation (r.m.s.d.) of 2.0 Å for 81 pairs of Cα atoms). Compared to the pfuThermoDBP-RP1 NTD, the tteThermoDBP NTD is expanded by two additional N-terminal β-strands and an extra helix between pfuThermoDBP-RP1 helices α1 and α2 (Figure 2C and Supplementary Figure S1). The C-terminal part of pfuThermoDBP-RP1 forms an amphipathic α-helix. The C-terminal α3-helices of four pfuThermoDBP-RP1 molecules associate as an antiparallel, four-helix bundle, positioning two pairs of NTDs at the opposite ends of a central four-helix rod (Figure 2A). About 14 000 Å2 of solvent accessible surface area is buried upon tetramerization, and an interface analysis (25) suggested that the observed tetramer is stable in solution. In agreement with this analysis, pfuThermoDBP-RP1 eluted from an analytical size-exclusion column consistent with the size of a tetramer (apparent molecular mass 65.5 kDa; calculated tetramer molecular mass 68.9 kDa).
Figure 2.
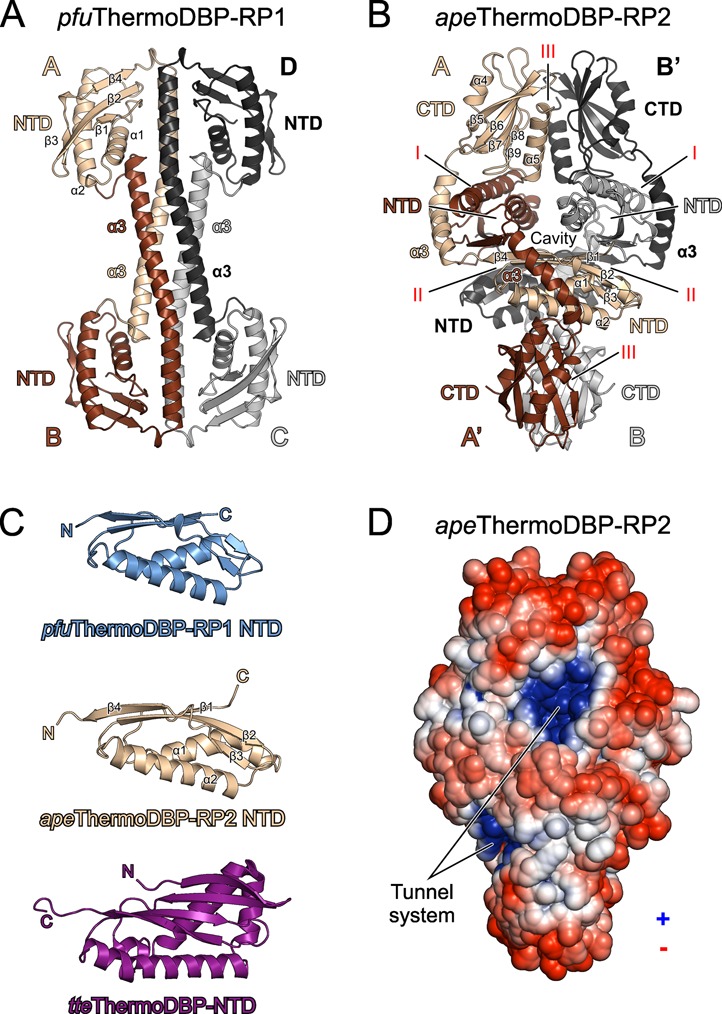
Structures and oligomerization of ThermoDBP-RP proteins. (A) Ribbon plot of the pfuThermoDBP-RP1 tetramer (subunits A–D). Subunits A–D are colored in beige, brown, black and gray (or shaded differently). The NTD and the C-terminal α3 helices forming an antiparallel coiled-coil are labeled. (B) Ribbon plot of the apeThermoDBP-RP2 tetramer. Monomers are colored/shaded as in A. Subunits A/A′ and B/B′ form the tight dimers that associate into tetramers. NTDs, connecting helices α3 and CTDs are labeled. Roman numerals (I–III) denote the different interfaces through which the tetramer forms. (C) Comparison of the NTDs of pfuThermoDBP-RP1 (top), apeThermoDBP-RP2 (center) and tteThermoDBP (bottom). Secondary structure elements are labeled. (D)Electrostatic surface representation of an apeThermoDBP-RP2 tetramer (electrostatic coloring/shading scaled in the range of ±5 kT/e). Entries/exits of an intersubunit tunnel system, which is lined with electropositive surface potential, are indicated.
apeThermoDBP-RP2 contains two globular domains, an NTD and a C-terminal domain (CTD) that are connected by a bent, α-helical linker (helix α3; Figure 2B). The NTD is again similar to those of tteThermoDBP and of pfuThermoDBP-RP1 (r.m.s.d. of 2.1 Å and 0.8 Å, respectively, for 87 Cα atoms; Figure 2C). The apeThermoDBP-RP2 CTD is composed of a five-stranded, anti-parallel β-sheet sandwiched between two α-helices (Figure 2B). apeThermoDBP-RP2 also crystallized as a tetramer, which was arranged as a dimer of tightly intertwined dimers (Figure 2B). Analytical gel filtration analyses indicated that apeThermoDBP-RP2 also forms tetramers in solution (apparent molecular mass 111.3 kDa; calculated tetramer molecular mass 105.4 kDa). Within a tight dimer (subunits A/A′ and B/B′; Figure 2B), each molecule cradles the NTD of the partner molecule between its own CTD and the connecting helix α3 (interface I; Figure 2B), thereby burying ∼3300 Å2 of solvent accessible surface area. Each molecule of a tight dimer interacts via its own NTD with the NTD of one molecule of a neighboring tight dimer (burying ∼1300 Å2 of solvent accessible surface area; interface II; Figure 2B), and via its own CTD with the CTD of the other molecule of the neighboring tight dimer (burying ∼1600 Å2 of solvent accessible surface area; interface III; Figure 2B). Only interface I within a tight dimer is predicted to be stable in isolation, while isolated interfaces II and III (between tight dimers) are predicted to be unstable (25). Consistently, interface I is largely hydrophobic (0.24 salt bridges/100 Å2) while interfaces II and III are more hydrophilic (1.5 and 0.38 salt bridges/100 Å2, respectively). Based the interface analyses, we expect that pfuThermoDBP-RP1 forms permanent tetramers but that interfaces II and III in apeThermoDBP-RP2 may intermittently break either partly (forming open tetramers) or entirely (forming isolated tight dimers).
Crystal structure of a ThermoDBP-RP2-DNA complex
The apeThermoDBP-RP2 tetramer in the crystal exhibits largely electronegative outer surfaces and an interior tunnel system that is lined with electropositive surface potential (Figure 2D), possibly suitable for ssDNA binding. To reveal how apeThermoDBP-RP2 binds ssDNA, we determined its crystal structure in complex with a decameric oligo-dT DNA (dT10) at 2.9 Å resolution (Figure 3A, B and Supplementary Table S2). The crystals contained two crystallographically independent apeThermoDBP-RP2 tetramers per asymmetric unit, each associated with two dT10 molecules (Figure 3A). We will focus our description on one of these virtually identical complexes with better defined electron density.
Figure 3.

Binding of ssDNA by apeThermoDBP-RP2. (A) Ribbon plot of the apeThermoDBP-RP2-dT10 complex with the DNAs in cartoon representation (DNA1: green/dark; DNA2: yellow/light). Monomers are labeled and colored/shaded as in Figure 2B. (B) Close-up view of two neighboring NTDs of the apeThermoDBP-RP2 (from subunits A and B′; ribbons) in contact with two dT10 molecules (DNA1 and DNA2 with nucleotides dT1–dT10 and dT5′–dT11′, respectively; ball-and-stick; coloring/shading as in A). The two DNA strands run across the α1 helices of the respective NTDs. The rotation symbol in this and the following panels indicates the orientation relative to (A). (C) Views on the DNA-binding surfaces of the two tight dimers (A/A′ left; B/B′ right) with the associated DNA1 and DNA2. Relative orientations of the two tight dimers with respect to the tetramer are indicated by the rotation symbols. Two surface representations are shown for each tight dimer. Representations on the left show coloring/shading by molecules (as in Figure 2B). Black and gray patches on tight dimer A/A′ indicate contact residues to subunits B′ and B, respectively, of their neighboring tight dimer (belonging to interfaces II or III as labeled); beige and brown/light and dark patches on tight dimer B/B′ indicate contact residues to subunits A and A′, respectively, of their neighboring tight dimer. Representations on the right show surfaces of the tight dimers colored/shaded by electrostatic surface potential (as in Figure 2A). DNA1 is proposed to outline part of the path that an elongated ssDNA molecule could take through the tetramer. This molecule could either traverse the tetramer diagonally (green/dark dashed extensions; path 1), or turn back along the path that DNA2 is bound in opposite orientation (yellow/light dashed extensions; path 2). Both putative paths are lined with electropositive surface potential. (D) Electrophoretic gel mobility shift assay testing the binding of increasing amounts of apeThermoDBP-RP2 to oligo-dT ssDNAs of increasing length. Interpretation of bands is given on the right. dT15, dT20, dT25 and dT30 show only one shifted band, a second, slower migrating band appears with dT30 at higher apeThermoDBP-RP2 concentrations, indicating that a ssDNA molecule of 35 nucleotides can accommodate two apeThermoDBP-RP2 tetramers. (E) Electrophoretic gel mobility shift assay testing the binding of apeThermoDBP-RP2 to circular M13 phage ssDNA on a 1% agarose gel. DNA was visualized by ethidium bromide staining. Upon addition of increasing concentrations of the protein (indicated below the gel), the DNA migrates progressively slower, indicating that increasing numbers of apeThermoDBP-RP2 molecules bind to the circularly closed DNA. (F) Model for binding of long ssDNAs to an apeThermoDBP-RP2 tetramer along path1 (top) and path 2 (bottom). Left panels: Views on the ssDNAs running across the apeThermoDBP-RP2 A/A′ tight dimer with the other dimers shown as a semi-transparent outline. Right panels: Same views with the B/B′ tight dimer as solid surfaces.
All 10 nucleotides of the first DNA strand (DNA1) could be traced, whereas only seven nucleotides of the second DNA strand (DNA2) were visible in the electron density. As shown in Figure 3A–C, the two DNA molecules meander in the 5′-to-3′ direction from the bottom of the complex into the interior of the apeThermoDBP-RP2 tetramer, where they are cradled between the juxtaposed NTDs of the four subunits. The central portions of both DNA molecules are bound in the same orientation across equivalent surfaces of two non-contacting NTDs of two tight dimers; i.e. DNA1 lies across the NTD of the A subunit (NTDA), while DNA2 binds across NTDB′ (Figure 3B). Superimposing the subunit B′-DNA2 sub-complex on the subunit A-DNA1 sub-complex revealed that nucleotides 5–9 of DNA1 and the first five nucleotides visible in DNA2 are bound in a similar fashion across NTDA and NTDB′, respectively (Figure 3B). We attributed the same numbers to those nucleotides of the two DNA molecules that occupy equivalent positions on NTDA and NTDB′ and engage in similar contacts, irrespective of the total length of the oligos (ten nucleotides). Thus, visible nucleotides of DNA1 were numbered dT1–dT10 while visible nucleotides of DNA2 were numbered dT5′–dT11′, with nucleotides dT5–dT9 of DNA1 and nucleotides dT5′–dT9′ of DNA2 being bound in an equivalent fashion on NTDA and NTDB′, respectively (Figure 3B and Supplementary Figure S6). The two DNAs meet in the center of the apeThermoDBP-RP2 tetramer, with the 3′-terminal nucleobase (dT10) of DNA1 stacking on the fifth nucleobase (dT9′) of DNA2 (Figure 3B and Supplementary Figure S6). The 3′-terminal two nucleotides of DNA2 (dT10′ and dT11′) are deflected from the center of the tetramer towards the upper part of the complex (Figure 3B and C).
We observed contacts of the protein subunits to the sugar-phosphate backbone as well as to the nucleobases of the DNA molecules (Supplementary Table S3; Figure 3B and Supplementary Figure S6). In addition to the interactions of the central portions of DNA1 and DNA2 with NTDA and NTDB′, respectively, they both engage in additional contacts to all other protein subunits of the apeThermoDBP-RP2 tetramer. These interactions again involve primarily residues from the NTDs of the other subunits (Supplementary Table S3 and Supplementary Figure S6). Thus, the NTD of one protein subunit provides the main binding platform for one ssDNA molecule and NTDs from the other subunits engage in different contacts to this DNA molecule and thereby guide it asymmetrically through the symmetric protein tetramer.
In DNA1, dT1 and dT2 stack on each other and on N211 of helix α6 from CTDB (the only CTD-DNA interaction seen in the entire structure). The side chain of dT3 is rotated outwards, introducing a first kink in the DNA backbone. The kinked backbone is stabilized by contacts to K63 and L64, originating from the loop following helix α2 of NTDB′. The following dT4, dT5 and dT6 again form a continuous stack that is capped by F23 from helix α1 of NTDA. This helix is wedged between nucleotides dT6 and dT7, introducing a second kink in the nucleic acid. dT8 stacks on dT7 and the two nucleotides are sandwiched between the central portions of the NTDA and NTDB α1 helices. NTDB helix α1 imposes a third kink and guides the 3′-terminal two-nucleotide stack (dT9–dT10) towards the center of the apeThermoDBP-RP2 tetramer, where the backbone of dT10 is held between K17 and R20 from helix α1 of NTDA′. The conformation and path through the apeThermoDBP-RP2 tetramer of the second DNA strand is similar except for its last two nucleotides, dT10′ and dT11′ (see above). While the protein imposes a multiply kinked structure on the DNA ligands, the apeThermoDBP-RP2 tetramer itself did not undergo any significant conformational changes upon DNA binding (r.m.s.d. ∼0.9 Å for 880 Cα-pairs between DNA-free and DNA-bound forms).
Molecular basis for the nucleic acid-binding preferences of apeThermoDBP-RP2
The kinks in the DNA backbone are incompatible with continuous Watson-Crick base pairing in a DNA duplex and the size of the central tunnel system in an apeThermoDBP-RP2 tetramer could not accommodate duplex DNA (or RNA), rendering apeThermoDBP-RP2 specific for single-stranded nucleic acids. Purines could be accommodated at most positions but a purine at positions 9/9′ would clash with the α1 helices of NTDB/NTDA′, respectively, explaining why apeThermoDBP-RP2 can bind ssDNA of mixed sequence but fails to bind homo-purine sequences (Supplementary Figure S4). Alternatively or additionally, homo-purine sequences may be excluded because of their tendency to form higher-order structures, such as dG-quadruplexes (26) or poly-dA parallel helices (27), which could not be accommodated in the restricted internal tunnel system of the apeThermoDBP-RP2 tetramer. Although the protein interacts with several nucleic acid bases, none of these contacts would be expected to further restrict the nucleic acid sequence that can be bound. Furthermore, a 2′-hydroxyl group on nucleotides 2/2′ would clash with the base of nucleotides 4/4′, a 2′-hydroxyl group on nucleotides 4/4′ would sterically interfere with the ribose of nucleotides 5/5′ and a 2′-hydroxyl on nucleotides 6/6′ would clash with the backbone of Arg20 of NTDA, explaining the preference of apeThermoDBP-RP2 for ssDNA over ssRNA.
Extent of an apeThermoDBP-RP2 binding site and engagement of ssDNA lacking free termini
The precise stoichiometry and ssDNA binding mode seen in the present crystal structure may be a consequence of the short DNA oligomers and high DNA concentrations used during the crystallization experiment. To estimate the number of nucleotides occupied by one apeThermoDBP-RP2 tetramer on ssDNA in solution, we conducted EMSA experiments with increasing length oligo-dT ssDNAs (Figure 3D). Up to a length of 30 nucleotides only a single shifted band was detected, indicating binding of one apeThermoDBP-RP2 tetramer (Figure 3D). However, starting with a length of 35 nucleotides, a second, slower migrating band appeared at higher apeThermoDBP-RP2 concentrations (Figure 3D), indicative of two apeThermoDBP-RP2 tetramers being accommodated on a dT35 ssDNA. These results suggest that one apeThermoDBP-RP2 tetramer occupies a stretch of 17 to 18 nucleotides on a ssDNA target, very similar to the combined number of nucleotides from DNA1 and DNA2 that we see bound to an apeThermoDBP-RP2 tetramer in the crystal structure (17 nucleotides).
Physiologically, apeThermoDBP-RP2 may engage extended ssDNA stretches that lack free ends. While we also obtained crystals of apeThermoDBP-RP2 with longer DNA oligomers, they diffracted poorly (about 5 Å resolution) and the electron densities for the DNA ligand(s) were poorly defined. To test whether apeThermoDBP-RP2 is capable of binding ssDNA molecules that lack free termini, we performed EMSAs with single-stranded circular M13 phage DNA (Figure 3E). Upon addition of increasing concentrations of apeThermoDBP-RP2, the ssDNA was progressively retarded on the gels, indicative of the protein occupying multiple binding sites in a stepwise manner. Due to the large size of the M13 DNA (7249 nucleotides) and the limited number of nucleotides required to accommodate one apeThermoDBP-RP2 tetramer, it is likely that at each increment in protein concentration multiple additional apeThermoDBP-RP2 tetramers bind.
DNA-binding may be auto-inhibited in the pfuThermoDBP-RP1 tetramer
Residues of apeThermoDBP-RP2 that contact the DNA molecules in the present structure are conserved in pfuThermoDBP-RP1 (Supplementary Figure S1). To investigate why the latter protein nevertheless binds poorly to ssDNA (Figure 1B and C), we superimposed the NTDA domain of apeThermoDBP-RP2 in complex with the DNA1 molecule on an NTD of the pfuThermoDBP-RP1 tetramer (Supplementary Figure S5A, left panel). The superposition revealed that in pfuThermoDBP-RP1, the putative DNA-binding surface on the NTD is occluded by the C-terminal tail of another pfuThermoDBP-RP1 subunit (Supplementary Figure S5A; close-up in left panel). Residues 128–139 of subunit B clash with a DNA molecule modeled on the NTD of subunit A. Thus, the present crystal structure of pfuThermoDBP-RP1 may represent an auto-inhibitory conformation with respect to ssDNA binding. We attempted to test whether the NTD of pfuThermoDBP-RP1 alone interacts more strongly with nucleic acids but failed to produce the protein in soluble form. The weak affinity for longer DNA oligonucleotides seen with pfuThermoDBP-RP1 (Figure 1C) may indicate that the protein can bind ssDNA only in special contexts, where the presumed auto-inhibition is relieved, for example by other interacting proteins.
DISCUSSION
ThermoDBP-RP2 proteins can act as non-canonical SSBs
We have demonstrated that the NTD, conserved among ThermoDBPs and ThermoDBP-RPs, is a versatile ssDNA-binding domain in ThermoDBP-RP2 proteins in vitro. ssDNA can come to lie on various sites and in different orientations on multiple copies of this domain in an oligomeric protein. While these results show that ThermoDBP-RP2 proteins can function as SSBs in vitro, a similar activity in vivo remains to be demonstrated.
The fold and mode of ssDNA binding of ThermoDBP-RP2 differ markedly from previously characterized SSBs (3). Canonical SSBs typically bind ssDNA or ssRNA on their outer surfaces with the bases pointing towards the protein and the backbone solvent exposed. In the case of bacteriophage T4 gene 32 protein, ssDNA binds along a deep channel formed between two subdomains (28). In contrast, a ThermoDBP-RP2 tetramer binds ssDNA inside an internal, intersubunit tunnel system. This unprecedented binding mode may be particularly useful to protect ssDNA in challenging environments such as high temperatures.
Molecular mechanism of ssDNA engagement
Our crystal structure of an apeThermoDBP-RP2-DNA complex shows directly that the protein can bind short or nicked ssDNAs. Such a function could be required to intermittently hold on to DNA ends during recombination or repair events. It is even imaginable that such an activity could be used to detect linear, foreign DNA molecules as part of a simple immune system, but there is no direct evidence supporting this latter idea.
On the other hand, we could show that apeThermoDBP-RP2 can bind efficiently to a circular ssDNA molecule that lacks free termini, suggesting that it could also substitute for canonical SSBs in general ssDNA-binding functions. We suggest that the short oligomers bound in our co-crystal structure also mark the possible paths that a single, long ssDNA molecule would take through the tetramer. The DNA1 molecule most likely outlines a major part of the binding site, as it represents that largest coherent stretch of DNA that could be traced in the electron density. The 5′-end of that molecule emerges from the internal tunnel system and lies on the surface of the apeThermoDBP-RP2 tetramer. Additional nucleotides on this side of the DNA most likely simply extend into the solvent. Additional nucleotides at 3′-end would come to lie in positions that in our crystal structure are occupied by the DNA2 molecule. Topologically, the 3′-end of DNA1 could be directly connected to the 3′-terminal two nucleotides (dT10′ and dT11′) of the DNA2 molecule. Thus, these terminal two nucleotides of DNA2 point out a possible exit path for an extended ssDNA molecule between the CTDs of subunits A and B and the NTDs of subunits A’ and B (path 1; green dashed line or tube in Figure 3C and F). Alternatively, an extended DNA1 molecule could bind along the path outlined by nucleotides dT9′–dT5′ of the DNA2 molecule but in opposite direction (path 2; yellow dashed line or tube in Figure 3C and F). Although in this scenario it would traverse the same surfaces of NTDA and NTDB′ in opposite orientation as seen for DNA2 in the present structure, such a binding mode is not entirely unlikely given the versatile contacts that are seen between different NTDs and portions of the bound DNA molecules in our crystal structure. It is also supported by the quasi-continuous stacking interaction observed between the 3′-terminal nucleotide of DNA1 (dT10) and an internal nucleotide (dT9′) of DNA2. Both putative paths for extended ssDNA molecules through the apeThermoDBP-RP2 tetramer are lined with positive electrostatic surface potential on the protein (Figure 3C). However, due to ensuing steric hindrance, it would be impossible to bind two extended ssDNA molecules to an apeThermoDBP-RP2 tetramer at the same time (e.g. along both of the two putative paths through the internal tunnel system).
Another immediate question is how an apeThermoDBP-RP2 tetramer can engage a ssDNA stretch that lacks free 3′-ends and thus could not be threaded end-on into the tunnel system of the tetramer. Given the predicted lability of interfaces II and III of the apeThermoDBP-RP2 tetramer, we envision that the protein complex can open up along these interfaces to engage extended ssDNA molecules at its center. The complex would not have to completely dissociate into tight dimers; opening of one interface III at the top or bottom of the tetramer would suffice to allow accommodation of an extended ssDNA molecule at the internal tunnel system.
Proteins bearing ThermoDBP-RP2-like NTDs
Although the ThermoDBP-RP1 NTD closely resembles the NTD of ThermoDBP-RP2 proteins, the physiological function of ThermoDBP-RP1 proteins remains unclear. Our in vitro binding studies do not support, but also do not ultimately rule out, a specific ribosome or box C/D sRNA association of these proteins, as had been reported previously (8). Weak DNA binding and the observation that the equivalent of the main ssDNA-binding surface of the ThermoDBP-RP2 NTD is obstructed in the tetrameric organization of pfuThermoDBP-RP1 suggest that the proteins may require special activatory mechanisms to engage ssDNA or other nucleic acids. Such activation (or relief of auto-inhibition) may be provided by other interacting proteins that force the pfuThermoDBP-RP1 NTDs into a different relative arrangement with respect to the central four-helix bundle.
Gel filtration and sequence analyses suggested that ThermoDBPs form parallel dimers via a C-terminal leucine zipper motif (7). If these putative, parallel ThermoDBP dimers resemble a parallel dimer of our present pfuThermoDBP-RP1 structure (e.g. subunits A and D; Figure 2A and Supplementary Figure S5A), the main ThermoDBP-RP2-like ssDNA binding surface of the ThermoDBP NTDs would remain unobstructed (Supplementary Figure S5A; right panel). However, most of the ssDNA-contacting residues of the apeThermoDBP-RP2 NTDs are poorly conserved in ThermoDBPs (Supplementary Figure S1), suggesting that ssDNA could also be bound via a different surface in the latter proteins. The NTDs in the presumed parallel ThermoDBP dimers also would not cluster as seen in the apeThermoDBP-RP2 tetramer and must bind ssDNA non-cooperatively or cooperate in a different fashion in ssDNA binding than observed in apeThermoDBP-RP2. Irrespective of the exact mode of ssDNA binding in ThermoDBPs, the observation of strong ssDNA binding by full-length tteThermoDBP (7) indicates that ThermoDBPs are not auto-inhibited.
A search of the Protein Data Bank with the Dali server (29) using the NTD of apeThermoDBP-RP2 revealed its similarity to a C-terminal dsDNA-binding domain in the eukaryotic DNA polymerase eta (r.m.s.d. of 1.75 Å for 97 Cα atoms; Supplementary Figure S5B). Unlike the binding of the apeThermoDBP-RP2 NTDs to ssDNA, the DNA polymerase eta dsDNA-binding domain employs its β-sheet surface to engage dsDNA along its major groove. Together, our analyses show that the ThermoDBP-RP2-like NTD provides a structural scaffold that can be adapted in diverse ways for ssDNA binding (as in the cases of ThermoDBPs and ThermoDBP-RP2s) or for dsDNA binding (as in the case of the C-terminal domain of DNA polymerase eta).
ACCESSION NUMBERS
PDB IDs: 4PSL, 4PSM, 4PSN and 4PSO.
SUPPLEMENTARY DATA
Supplementary Data is available at NAR Online, including [1–5].
Acknowledgments
We thank C. Alings for help in crystallization experiments. We accessed beamlines of the BESSY II (Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung II) storage ring (Berlin, Germany) via the Joint Berlin MX-Laboratory sponsored by the Helmholtz Zentrum Berlin für Materialien und Energie, the Freie Universität Berlin, the Humboldt-Universität zu Berlin, the Max-Delbrück Centrum, and the Leibniz-Institut für Molekulare Pharmakologie. Funding for open access charge: Freie Universität Berlin Kaiserswerther.
Conflict of interest statement. None declared.
Footnotes
These authors contributed equally to this work.
Present addresses:
Homa Ghalei, Department of Cancer Biology, The Scripps Research Institute, Jupiter, FL 33458, USA.
Holger von Moeller, moloX GmbH, Takustr. 6, 14195 Berlin, Germany.
REFERENCES
- 1.Pestryakov P.E., Lavrik O.I. Mechanisms of single-stranded DNA-binding protein functioning in cellular DNA metabolism. Biochemistry (Mosc) 2008;73:1388–1404. doi: 10.1134/s0006297908130026. [DOI] [PubMed] [Google Scholar]
- 2.Shereda R.D., Kozlov A.G., Lohman T.M., Cox M.M., Keck J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008;43:289–318. doi: 10.1080/10409230802341296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickey T.H., Altschuler S.E., Wuttke D.S. Single-stranded DNA-binding proteins: multiple domains for multiple functions. Structure. 2013;21:1074–1084. doi: 10.1016/j.str.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sancar A., Williams K.R., Chase J.W., Rupp W.D. Sequences of the ssb gene and protein. Proc. Natl. Acad. Sci. U.S.A. 1981;78:4274–4278. doi: 10.1073/pnas.78.7.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Theobald D.L., Mitton-Fry R.M., Wuttke D.S. Nucleic acid recognition by OB-fold proteins. Annu. Rev. Biophys. Biomol. Struct. 2003;32:115–133. doi: 10.1146/annurev.biophys.32.110601.142506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohman T.M., Ferrari M.E. Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and cooperativities. Annu. Rev. Biochem. 1994;63:527–570. doi: 10.1146/annurev.bi.63.070194.002523. [DOI] [PubMed] [Google Scholar]
- 7.Paytubi S., McMahon S.A., Graham S., Liu H., Botting C.H., Makarova K.S., Koonin E.V., Naismith J.H., White M.F. Displacement of the canonical single-stranded DNA-binding protein in the Thermoproteales. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E398–E405. doi: 10.1073/pnas.1113277108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciammaruconi A., Gorini S., Londei P. A bifunctional archaeal protein that is a component of 30S ribosomal subunits and interacts with C/D box small RNAs. Archaea. 2008;2:151–158. doi: 10.1155/2008/472786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marquez V., Frohlich T., Armache J.P., Sohmen D., Donhofer A., Mikolajka A., Berninghausen O., Thomm M., Beckmann R., Arnold G.J., et al. Proteomic characterization of archaeal ribosomes reveals the presence of novel archaeal-specific ribosomal proteins. J. Mol. Biol. 2011;405:1215–1232. doi: 10.1016/j.jmb.2010.11.055. [DOI] [PubMed] [Google Scholar]
- 10.Studier F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 11.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vonrhein C., Blanc E., Roversi P., Bricogne G. Automated structure solution with autoSHARP. Methods Mol. Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 13.Vagin A., Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 14.Sheldrick G.M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 2010;66:479–485. doi: 10.1107/S0907444909038360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pape T., Schneider T.R. HKL2MAP: a graphical user interface for macromolecular phasing with SXELX programs. J. Appl. Cryst. 2004;37:843–844. [Google Scholar]
- 16.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murshudov G.N., Skubak P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winn M.D., Isupov M.N., Murshudov G.N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 22.Chen V.B., Arendall W.B., III, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeLano W.L. San Carlos, CA: DeLano Scientific; 2002. The PyMOLmolecular graphics system. [Google Scholar]
- 24.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Gellert M., Lipsett M.N., Davies D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. U.S.A. 1962;48:2013–2018. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saenger W., Riecke J., Suck D. A structural model for the polyadenylic acid single helix. J. Mol. Biol. 1975;93:529–534. doi: 10.1016/0022-2836(75)90244-2. [DOI] [PubMed] [Google Scholar]
- 28.Shamoo Y., Friedman A.M., Parsons M.R., Konigsberg W.H., Steitz T.A. Crystal structure of a replication fork single-stranded DNA binding protein (T4 gp32) complexed to DNA. Nature. 1995;376:362–366. doi: 10.1038/376362a0. [DOI] [PubMed] [Google Scholar]
- 29.Holm L., Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W 549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.