


Abstract
Although several studies have suggested that the functions of heterochromatin regulators may be regulated by post-translational modifications during cell cycle progression, regulation of the histone methyltransferase Suv39H1 is not fully understood. Here, we demonstrate a direct link between Suv39H1 phosphorylation and cell cycle progression. We show that CDK2 phosphorylates Suv39H1 at Ser391 and these phosphorylation levels oscillate during the cell cycle, peaking at S phase and maintained during S-G2-M phase. The CDK2-mediated phosphorylation of Suv39H1 at Ser391 results in preferential dissociation from chromatin. Furthermore, phosphorylation-mediated dissociation of Suv39H1 from chromatin causes an enhanced occupancy of JMJD2A histone demethylase on heterochromatin and alterations in inactive histone marks. Overexpression of phospho-mimic Suv39H1 induces early replication of heterochromatin, suggesting the importance of Suv39H1 phosphorylation in the replication of heterochromatin. Moreover, overexpression of phospho-defective Suv39H1 caused altered replication timing of heterochromatin and increases sensitivity to replication stress. Collectively, our data suggest that phosphorylation-mediated modulation of Suv39H1-chromatin association may be an initial step in heterochromatin replication.
INTRODUCTION
Heterochromatin is a highly condensed and darkly stained fraction of the chromatin, even during interphase. Both facultative and constitutive heterochromatins influence gene expression, although constitutive heterochromatin such as centromeres and telomeres also plays a critical role in maintenance of genome integrity. The basic structures and regulation mechanisms of heterochromatin are highly conserved from yeasts to humans (1,2). During the last decade, a number of chromatin regulators that are involved in the control of heterochromatin assembly have been identified (3–5). These include an RNA interference pathway that is essential for nucleation of histone methylation in fission yeast (6) and non-coding RNAs produced from pericentromeric repeats that are involved in heterochromatin assembly in mammals (7). The suppressor of variegation 3-9 (Su(var)3-9) family of proteins specifically methylates histone H3 at lysine-9 (Lys-9) prior to recruitment of heterochromatin protein 1 (HP1), a key effector molecule of heterochromatin (8,9). Furthermore, Lys-20 methylation of histone H4 by Suv420H1/H2 and DNA methylation by DNA methyltransferases are well established as additional stages in the completion of heterochromatin assembly (1).
Although these accumulating lines of evidence help us understand how heterochromatin is established and maintained (1,4,5,9–12), the signaling pathways that control key heterochromatic regulators during cell cycle progression are not fully understood. Intriguingly, Ezh2, a member of the polycomb protein family, is phosphorylated by cyclin dependent kinase 1 (CDK1) and CDK2 (13–15). These phosphorylation events are important for its binding to polycomb repressive complex 2 (PRC2) recruiters such as HOTAIR and XIST non-coding RNAs or the other PRC2 components SUZ12 and EED, thereby ensuring the transmission of histone H3 Lys-27 tri-methylations through cell division cycles (13–15). In addition, HP1 is a target for post-translational modifications such as phosphorylation and sumoylation (16–19). For example, N-terminal phosphorylation of HP1α promotes its chromatin binding in mammals, and casein kinase 2-mediated phosphorylation of Swi6/HP1, a fission yeast homolog, modulates the interaction between Swi6/HP1 and other regulators (20,21). In our previous study, we demonstrated that the fission yeast Swi6/HP1 protein is sumoylated by Hus5/Ubc9, leading to completion of heterochromatin assembly via modulation of its chromatin binding ability (18). This link between heterochromatin and sumoylation was further supported by elegant studies in a mammalian model system (19,22). Sirtuin 1 (SIRT1) mediated deacetylation of Suv39H1 plays a positive role in its enzymatic activity and protein level (23). This modulation may prevent Mouse Double Minute 2 (MDM2)-mediated polyubiquitination and degradation, thereby contributing to genome protection in response to stress signals (24). Furthermore, Suv39H1 is phosphorylated in a cell-cycle-dependent manner during mitosis and G1-S transition (25,26) although the functional relevance of such modifications remains to be elucidated.
In this study, we show that CDK2 phosphorylates Suv39H1 at Ser-391 residues during G1-S transition in vivo and in vitro. We found that a phospho-mimic mutant of Suv39H1 preferentially dissociated from heterochromatic regions compared with its phospho-defective counterpart, leading to a significant reduction in histone H3 Lys9 tri-methylation, a hallmark of inactive heterochromatin. Importantly, the phosphorylation-mediated dissociation of Suv39H1 from chromatin is accompanied by enhanced preoccupancy of histone demethylase JMJD2A at heterochromatic loci. Moreover, defective phosphorylation of Suv39H1 affected replication timing of heterochromatin and caused increased sensitivity to replication stress such as hydroxyurea (HU) treatment. Our data suggest that phosphorylation-mediated Suv39H1 dissociation from heterochromatin is linked to control of heterochromatin replication during cell cycle progression. Taken together, our results indicate that the CDK2-dependent phosphorylation pathway plays a direct role in modulating key heterochromatin regulators such as Suv39H1.
MATERIALS AND METHODS
Cell culture, transfection and cell synchronization
Cell lines were grown in Dulbecco's modified Eagle's medium with 4 mM L-glutamine and 10% fetal bovine serum (FBS) adjusted to contain 1.5 g/l sodium bicarbonate and 4.5 g/l glucose (all from GE Healthcare Life Sciences) at 37°C in a humidified atmosphere of 5% CO2. We used 293T and HeLa cells from American Type Culture Collection (ATCC) in this study. All transfections were performed using Effectene reagent (QIAGEN) according to the manufacturer's instructions. Cell synchronization was performed as previously mentioned elsewhere (15,27,28).
For production of lentivirus and retrovirus, 293FT cells were transfected with 3 μg pLKO.1 empty vector or shRNA vector targeting Suv39h1/h2 or CDK1/2, 2.25 μg of envelope vector pMD.G (Addgene plasmid 12259; kindly provided by Dr Didier Trono), 6.75 μg of ps.PAX (Addgene plasmid 12260; kindly provided by Dr Didier Trono) and transfected with 5 μg pMSCV empty vector or Flag-Suv39h1 cloned into pMSCV, 4.5 μg of gag-pol, 0.5 μg of VSV-G, respectively. Virus particles were harvested 24 and 48 h after transfection and filtered through a 0.45 μM filter. Cells were infected with lentivirus or retrovirus for at least 6 h in the presence of 6 μg/ml polybrene and were then allowed to recover for 24–48 h.
HeLa cells were synchronized by 1 mM HU (Sigma-Aldrich) for 24 h. The cells were then harvested (G1/S stage) or released into normal growth media and harvested 5 h after release, which represent mid-S phase. Mitotic cells were harvested by treating with nocodazole (100 nM) (Sigma-Aldrich) for 24 h. HeLa cells were synchronized by serum starvation for 48 h and stimulated with fresh medium containing 10% FBS. Cells were harvested at indicated time points. In addition, HeLa cells were synchronized at the G2/M transition of the cell cycle by the thymidine-nocodazole block method. In brief, cells in the exponential growth phase were exposed to 2 mM thymidine for 24 h, released into thymidine-free medium for 9 h, and then re-exposed to 100 ng/ml nocodazole for 12 h. The M phase-arrested cells were released by transfer of cells to fresh nocodazole-free medium.
Antibodies and chemicals
Antibodies that recognize histone modification(s), including H3K9me1 (ab9045), H3K9me2 (ab1220), H3K9me3 (ab8898), H3K4me1 (ab8895), H3K4me2 (ab32356) and H3 (ab1791), were purchased from Abcam Limited (Cambridge, UK). H3K9me3 (07-442) and H3K9Ac (06-942) were purchased from Upstate Biologicals, Inc. (Lake Placid, NY).
Other antibodies' information is indicated as below. Anti-Flag (F3165, F7425) and anti-BrdU (B2531) are from Sigma-Aldrich (St Louis, Missouri). Anti-Myc (ab9106) is from Abcam. Anti-GAPDH (abc-2003) is from Abclon lnc. (Seoul, Korea). Anti-cyclin B1 (sc-245) is from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho-cdc2Y15 (#9111) is from Cell Signaling Technology, Inc. (Danvers, MA). Antibody against phospho-Suv39h1S391 was raised in rabbit using a synthetic peptide, amino acids 387–397 protein (C-GLPGSPKKRVR) (AbFrontier, Seoul, Korea). The CDK1 inhibitor IVRO-3306 (217699) and CDK2 inhibitor II (219445) were purchased from Merck Millipore (Darmstadt, Germany), dissolved in dimethyl sulfoxide (DMSO).
Colony formation assay
Colony formation assay was performed as in the previous report (29). For colony formation assay, cells were seeded at 1 × 103 cells per well in 60 mm dishes in triplicate and maintained for 14 days. Cells were infected with lentivirus vector carrying 3×Flag-Suv39h1 wild-type, phospho-mutant, or empty vector alone. Cells were fixed with fixation solution (methanol:acetic acid, 7:1) for 10 min and stained with 0.02% crystal violet for 20 min at room temperature. The colonies were photographed and counted.
Chromatin immunoprecipitation assay
Cells were seeded in five 15 cm dishes 24 h prior to the experiment and transfected with plasmids or infected with retrovirus as appropriate. HeLa cells were starved in serum-free media for 48 h followed by stimulation with complete serum media for an additional 16 h (27). Chromatin immunoprecipitation (ChIP) experiments were performed using an EZ-ChIP kit (Millipore) according to the manufacturer's instructions. The cells were cross-linked for 10 min by addition of formaldehyde to a final concentration of 1%. The cross-linking was stopped by addition of glycine, followed by cell lysis and sonication. Cross-linked chromatin was incubated with antibodies overnight at 4°C. Reversal of cross-linking was carried out at 65°C for 2 h and the purified DNA was analyzed by quantitative polymerase chain reaction (PCR) using primer sets for target heterochromatic loci (Supplementary Table S1).
Co-immunoprecipitation assay
Cultured cells were washed and lysed with lysis buffer (20 mM Tris-HCl, pH 8, 137 mM NaCl, 10% glycerol, 1% NP-40, 2 mM EDTA) containing protease inhibitors (Roche, USA). Five hundred micrograms of cell lysate proteins were immunoprecipitated with Flag M2 agarose (Sigma) overnight at 4°C, washed five times with lysis buffer, eluted by boiling for 10 min in SDS-sample buffer, separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore), and processed for immunoblotting.
RNA isolation and quantitative real-time PCR
Total cellular RNA was extracted using the RNeasy Mini kit (Qiagen, USA) according the manufacturer's instructions and quantitated using a nanodrop spectrophotometer (Thermo Scientific, Waltman, MA). cDNA was synthesized using the RevertAidTM M-MuLV Reverse Transcriptase (MBI Fermentas Inc., Hanover, MD) with oligo dT primers. Expression levels were analyzed by quantitative real-time PCR (qRT-PCR) with SYBR Premix Ex Taq II (Takara, Japan) and an ABI7000 sequence detector (Applied Biosystems, USA) using primer sets for target genes described in Supplementary Table S1 according to the manufacturers’ protocols. All samples were normalized by comparison to GAPDH transcript.
Bromodeoxyuridine immunostaining and image acquisition
Bromodeoxyuridine (BrdU) immunostaining and image acquisition were largely carried out as previously described (30). NIH3T3 cells were grown in 12-well plates for 24 h prior to infection with retrovirus. Twenty-four hours after infection, cells were treated with 10 μg/ml aphidicolin for 16–18 h. The following day the cells were treated with 10 μM BrdU (CalBiochem, USA) for 1 h before harvesting each time point. Cells were fixed with 2% formaldehyde and ice-cold MtOH, incubated with 50 mM glycine to stop cross-linking, denatured and permeabilized by 0.1% Triton X-100 in 4 N HCl, and blocked with 10% FBS. The fixed cells were incubated with anti-BrdU antibodies. A Carl Zeiss confocal microscope equipped with a ×63 objective lens was used for image acquisition.
Recombinant proteins and in vitro kinase assay
Purification of Maltose-Binding Protein (MBP)-fused Suv39H1 recombinant proteins and in vitro kinase assay were carried out as previously described in manufacturer's manual and elsewhere (New England Biolabs, Beverly, MA; (14,15)). Full-length and mutant Suv39h1 cDNA was subcloned into pMAL-HV vector. Codon plus competent cells and Escherichia coli system were used for production of recombinant proteins. The MBP-tagged Suv39h1 protein was purified using amylose resin (New England Biolabs, Beverly, MA) and maltose elution as mentioned in manufacturer's manual. In vitro kinase reactions were performed by incubating CDK2/cyclin A2 or CDK2/cyclin E1 (Sigma-Aldrich) with purified bacterially expressed Suv39h1 proteins in a buffer containing 50 mM Tris-HCl, 10 mM MgCl2, 0.1 mM EDTA, 2 mM DTT, 0.01% Brij 35 and 0.1 mM NaF for 1 h at 30°C. One microliter of 10 μCi gamma-ATP (Perkin-Elmer Corp., Norwalk, CT) was used (14,15). The reactions were terminated with SDS sample buffer, boiled and resolved by SDS-PAGE gel. Phosphorylation was detected by autoradiogram.
shRNA knockdown
Lentiviral expression was used for shRNAs against Suv39H1/H2 and luciferase. The pLKO.1-TRC control vector (Addgene plasmid 10879; kindly provided by Dr David Root) was used for the subcloning of shRNA sequences (31). The protocol on how to use the pLKO.1 vector is available elsewhere (http://www.addgene.org/plko). The following oligonucleotides were annealed and cloned into a pLKO shRNA expression vector (Supplementary Table S1).
Flow Cytometric Analysis (FACS)
The fluorescence-activated cell sorting (FACS) analysis was performed as previously mentioned elsewhere (32). The brief protocols are as follows. Cells were seeded in a 10 cm dish. Cells were treated with 10 μM BrdU (CalBiochem, USA) for 1 h prior to harvesting. Cells were harvested after trypsinization, fixed in 70% ethanol, denatured by 2 N HCl/Triton X-100 for 30 min at room temperature, neutralized by 0.1 M Na2B4O7·10H2O, pH 8.5, stained with anti-BrdU FITC in 0.5% Tween 20/BSA/PBS for 30 min at room temperature followed by staining with propidium iodide solution at final concentration of 5 μg/ml, and subjected to FACS analysis.
RESULTS
CDK2 phosphorylates Suv39H1 at Ser-391
Previous reports demonstrated phosphorylation of Suv39H1 during cell cycle progression (25,26), and proteomic analysis data and computation prediction programs indicate that Suv39H1 can be phosphorylated at Ser-391 in mouse and human cells (http://www.phosphosite.org; http://www.phosida.com; (33)). In addition, the consensus phosphorylation motif at Ser-391 for CDK1 or CDK2 is completely conserved between mouse and human (Figure 1A). Previous reports clearly documented that CDK2 is activated by the cyclin E and cyclin A subunits and plays important roles in cell cycle control, primarily in G1 and S phases (34,35).
Figure 1.
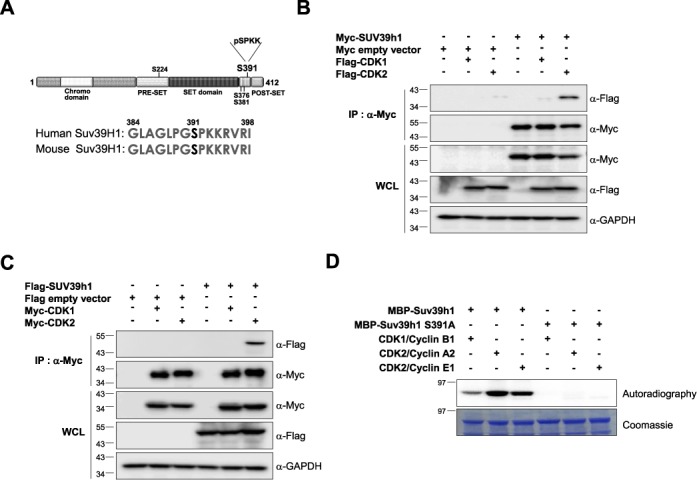
Suv39H1 can form complex with CDK2 but not with CDK1 in vivo. (A) Potential phosphorylation sites of human Suv39H1. Putative phosphorylation sites are shown in black and the phosphorylation site with the CDK1 or CDK2 consensus motif (S391 residue) is shown in red. (B and C) Suv39H1 interacts with CDK2 but not with CDK1 in vivo. Total cell lysates from 293T cells co-transfected with Myc-Suv39H1 and Flag-CDK1 or Flag-CDK2 (in panel (B)) or Flag-Suv39H1 and Myc-CDK1 or Myc-CDK2 (in panel (C)) were co-immunoprecipitated (IP) using anti-Myc agarose beads. The IP fractions were analyzed by western blotting using anti-Myc or anti-Flag antibody. The expression of each gene in whole cell lysate (WCL) was confirmed by western blot analysis with anti-Flag or anti-Myc antibodies. (D) In vitro kinase assay with purified MBP-fused Suv39H1-WT or MBP-Suv39H1-S391A mutant protein. Purified MBP-Suv39H1-WT and MBP-Suv39H1-S391A mutant proteins were incubated with gamma-32P-ATP and CDK2/cyclin complexes. The phosphorylation was visualized by exposure to X-ray film. The data showed that MBP-Suv39H1-WT was phosphorylated by both CDK2/cyclin A2 and CDK2/cyclin E1 complexes but not MBP-Suv39H1-S391A mutant.
To identify which kinase(s) is responsible for the phosphorylation of Suv39H1, we investigated the physical association between Suv39H1 and CDK1 or CDK2 by co-immunoprecipitation assay. The data revealed that Suv39H1 could form a complex with CDK2 but not with CDK1, implying CDK2-dependent phosphorylation of Suv39H1 (Figure 1B and C). To further confirm if CDK2 can phosphorylate Suv39H1 at Ser-391 residue, we carried out in vitro kinase assay using MBP-fused Suv39H1-WT, and Suv39H1-S391A mutant purified from bacterial cells. Our data showed that both CDK2-cyclin A2 and CDK2-cyclin E1 complexes could phosphorylate wild-type Suv39H1 but not Suv39H1-S391A mutant (Figure 1D). The phosphorylation levels were higher in CDK2-cyclin containing reaction than in CDK1-cyclin B1 sample (Figure 1D), suggesting that CDK2-cyclin complexes are the major enzymes for Suv39H1 phosphorylation.
To confirm if the Ser-391 residue is the phosphorylation site of Suv39H1, we generated a phospho-specific antibody against Ser-391 phosphopeptide (pSer-391). We mutated the Ser-391 residue of Flag-tagged Suv39H1 to alanine (S391A; phospho-defective mutant) and to glutamate (S391E; phospho-mimic mutant). We also made two corresponding point mutants for the Ser-224 residue (S224A and S224E), which were used as negative controls. Flag-tagged Suv39H1 and its point mutant derivatives were introduced into 293T cells and the cell extracts were subjected to immunoprecipitation using anti-Flag agarose beads. Analysis of the immunoprecipitates by western blotting with anti-pSer-391 antibody showed that wild-type Suv39H1 (Suv39H1-WT), Suv39H1-S224A and Suv39H1-S224E are phosphorylated normally whereas Ser-391 phosphorylation was abolished in Suv39H1-S391A and Suv39H1-S391E mutant proteins and phosphatase-treated Suv39H1-WT (Supplementary Figure S1). The Ser-391 phosphorylation was further confirmed in the mouse fibroblast NIH3T3 cell lines (Supplementary Figure S2). These results suggest that the Ser-391 residue represents an in vivo phosphorylation site in Suv39H1 in both mouse and human cells. Moreover, we confirmed CDK2-dependent phosphorylation of Suv39H1 using shRNA-based knockdown, CDK1/2-specific inhibitors, and dominant negative mutants of CDK1 or CDK2 (36–39). Treatment of CDK2 inhibitor resulted in decreased phosphorylation of Suv39H1 at Ser-391 compared to that of CDK1 inhibitor (Figure 2A). Consistent with this result, the phosphorylation status of Suv39H1 was reduced in shRNA-based CDK2-knockdown cells but not in CDK1-knockdown cells (Figure 2B). The knockdown of CDK1 and CDK2 was confirmed by qRT-PCR (Supplementary Figure S3). In addition, phosphorylation of Suv39H1 at Ser-391 was barely detectable with ectopic expression of dominant negative kinase-dead mutant CDK2 (CDK2 D145N) compared with expression of wild-type CDK2, whereas two forms of dominant negative CDK1 caused little change in the phosphorylation status of Suv39H1 at Ser-391 (Figure 2C). These data confirm that Suv39H1 is predominantly phosphorylated at Ser-391 in vivo by CDK2 but not by CDK1. Together, the results suggest that the S391 residue is bona fide phosphorylation site for Suv39H1 by CDK2.
Figure 2.

CDK2 phosphorylates Suv39H1 at Ser-391 in vivo. (A) Suv39H1 phosphorylation at Ser-391 was decreased by treatment of CDK2 inhibitor but not by CDK1 inhibitor. The 293T cells stably expressing Flag-Suv39H1 were treated with CDK1 or CDK2 inhibitor for 20 h and 12 h, respectively. Total cell lysates were immunoprecipitated using anti-Flag M2 and analyzed by western blotting using ant anti-phosphoSuv39H1 (pS391). (B) The shRNA-based CDK2 knockdown resulted in decrease of Suv39H1 phosphorylation at Ser-391 residue whereas the phosphorylation was little changed by CDK1 knockdown. The 293T cells stably expressing Flag-empty vector (negative control) or Flag-Suv39H1 were infected with lentivirus expressing CDK1 shRNA or CDK2 shRNA. Cell lysates were immunoprecipitated using anti-Flag M2 and then analyzed by western blotting using ant anti-phosphoSuv39H1 (pS391). (C) Phosphorylation of Suv39H1 at Ser-391 was decreased by overexpression of a dominant negative mutant of CDK2 but not CDK1. The 293T cells were co-transfected with Flag-Suv39H1-WT and Myc-CDK2 or its dominant negative mutant Myc-CDK2 D145N, or with Myc-CDK1 or its dominant mutant derivatives (CDK1 K33R or CDK1 D146N). Total lysates were immunoprecipitated using anti-Flag M2 agarose beads and analyzed by western blotting using anti-pSer-391 antibody. (D) Ser-391 of Suv39H1 is phosphorylated predominantly during S phase and maintained during S-G2-M phases. To profile the cell-cycle-dependent phosphorylation of Suv39H1, the HeLa cell lines stably expressing Flag-tagged Suv39H1-WT were treated by nocodazole and release. Total cell lysates of cells released from cell-cycle blockage were subjected to immunoprecipitation using anti-Flag M2 agarose beads and analyzed by western blotting using anti-pSer-391 antibody.
Phosphorylation of Suv39H1 at Ser-391 during cell cycle progression
Since CDK2 activity is restricted to the G1-S transition phase, we examined the levels of phospho-Suv39h1 Ser-391 at different stages of the cell cycle. Cells stably expressing Flag-tagged Suv39H1-WT were synchronized in M phase with thymidine-nocodazole block and released from the block and collected at different time points indicated. The phosphorylation levels of Flag-Suv39H1 were examined as described in Figure 2A. The G1/S and G2/M phases of each sample were confirmed by the expression of inhibitory phospho-Cdc2 Y15 (lowest levels at G1 and M phases) and cyclin B1 (expressing through G2/M). The results revealed that the phosphorylation levels of Flag-tagged Suv39H1 were detected at M phase, barely detectable at M-G1 transition, subsequently increased from G1/S phase, and the level was still maintained during S-G2-M phases (Figure 2D). The cell cycle profile was confirmed by FACS analysis (Supplementary Figure S4). These data can be explained by the reports that demonstrate the mitosis-specific increase in protein stability of SirT1, acting as a stabilizer of Suv39H1 protein (24,40). In addition, our phosphorylation profile is consistent with the previous quantitative mass data showing its phosphorylation status of endogenous Suv39H1 protein (33). These data suggest that Suv39H1 phosphorylation may occur from late G1 onward and its levels are maintained during S-G2-M phases in which CDK2 is biologically active.
Phosphorylation of Suv39H1 at Ser-391 influences its binding to heterochromatin, occupancy of histone demethylases, and histone methylation patterns at heterochromatin
To assess the biological significance of Suv39H1 phosphorylation during S phase, we first tested whether phosphorylation affects its enzymatic activity using an in vitro histone methyltransferase assay. As previously described (8), we used MBP-fused Suv39H1-WT, Suv39H1-S391A and Suv39H1-S391E mutants purified from E. coli cells. Phosphorylation-defective and mimic mutants of Suv39H1 have histone H3 Lys9 di- and tri-methylation levels comparable to those of wild-type Suv39H1, suggesting no direct linkage between Suv39H1 phosphorylation and enzymatic activity (Supplementary Figure S5). Next, we examined whether phosphorylation of Suv39H1 affects its interaction with CDK2 using a co-immunoprecipitation assay as described in Figure 1B and C. The results demonstrated that the point mutants of Suv39H1 have comparable ability to form a complex with CDK2 to wild-type Suv39H1 protein, suggesting that phosphorylation is not involved in the interaction with its partner protein (Supplementary Figure S6).
Since Suv39H1 is one of the proteins that show dynamic interaction with heterochromatin regions and some inactive promoters of silenced genes (25,41–43), we next assessed whether phosphorylation of Suv39H1 affects its binding capacity to heterochromatin regions using a ChIP assay. Under asynchronous growth conditions, phospho-mimic Suv39H1 (Suv39H1-S391E) had decreased binding affinity to heterochromatin regions relative to wild-type or phospho-defective Suv39H1 (S391A) proteins in both transiently and stably expressing cells (Figure 3A and B, Supplementary Figure S7), suggesting that phospho-mimic Suv39H1 protein preferentially dissociates from heterochromatin regions. The protein expression of each gene in both transiently and stably expressing 293T cells were confirmed by western blot analysis (Supplementary Figure S8). Next, we investigated whether phosphorylation-mediated Suv39H1 dissociation from heterochromatin causes alterations in histone H3 Lys9 tri-methylation patterns, a hallmark of heterochromatin, by performing ChIP assays using antibody specific for tri-methylated H3 protein. As expected, overexpression of phospho-mimic Suv39H1 resulted in a significant decrease in histone H3 Lys9 tri-methylation levels relative to those of wild-type and phospho-defective Suv39H1 proteins in asynchronous growth conditions (Figure 3C). The effects of phospho-mimic and phospho-defective Suv39H1 expression were confirmed under genetic backgrounds lacking endogenous Suv39H1 and Suv39H2. The double knockdown mutant cells were characterized by analysis of histone modification patterns and mRNA levels (Supplementary Figure S9). We transfected constructs of Flag-tagged Suv39H1-WT, Suv39H1-S391A and Suv39H1-S391E into Suv39H1/Suv39H2 double knockdown cells. The ChIP data for these rescued samples were consistent with previous data obtained in this study (Supplementary Figure S10). Together, these observations suggest that phosphorylation of Suv39H1 by CDK2 may affect histone H3 Lys9 methylation patterns at heterochromatic regions via modulation of its binding to heterochromatin.
Figure 3.
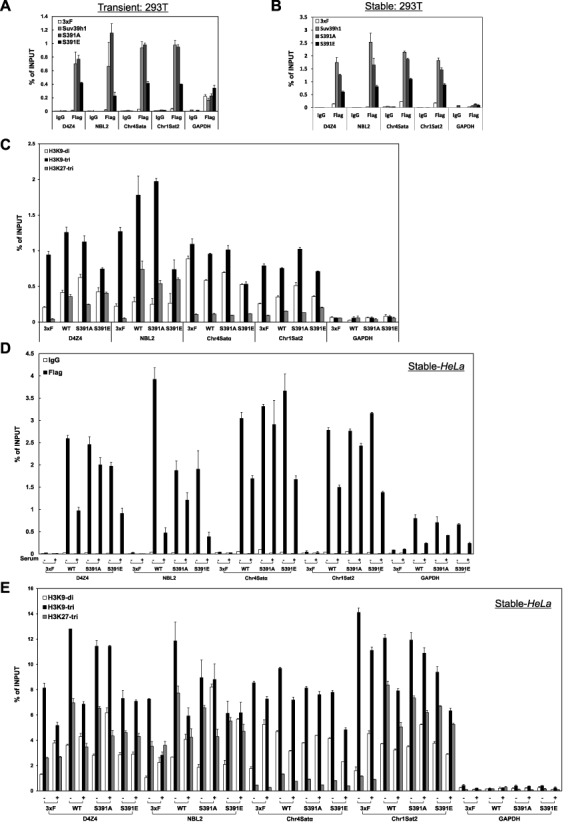
Ser391 phosphorylation of Suv39H1 influences its binding to heterochromatin and histone methylation patterns, a hallmark of inactive chromatin. (A and B) Phospho-mimic Suv39H1 preferentially dissociates from all heterochromatic loci tested compared with wild-type Suv39H1 or phospho-defective Suv39H1. Asynchronously growing 293T cells transiently transfected with Flag empty vector, wild-type Suv39H1, phosphorylation-defective mutant (Suv39H1-S391A), or phosphorylation-mimic mutant (Suv39H1-S391E) were analyzed by ChIP assays using anti-Flag M2 agarose beads. qPCR data are presented as means ± s.d., and are representative of 3–4 independent experiments. Transient: 293T, transiently transfected plasmids. (B) ChIP analysis as in (A) but using cells stably expressing Flag empty vector, Suv39H1-WT, Suv39H1-S391A or Suv39H1-S391E. qPCR data are presented as means ± s.d., and are representative of 3–4 independent experiments. Stable: 293T, stably integrated plasmids. (C) Phosphorylation-mimic form of Suv39H1 at Ser391 residue causes decreased levels of histone H3 Lys9 tri-methylation (H3K9me3), a hallmark of heterochromatin. ChIP assay was performed using the same sample as in (B) and anti-histone H3K9me3 antibody. In addition, histone H3 Lys27 tri-methylation (H3K27me3) levels are displayed as negative control because it is catalyzed by Ezh2 but not by Suv39H1/H2. qPCR data are presented as means ± s.d., and are representative of 3 independent experiments. Stable: 293T, stably integrated plasmids. (D) Phosphorylation-mediated Suv39H1 dissociation from heterochromatin preferentially occurs at the G1-S border. ChIP analysis using anti-Flag M2 agarose beads was performed on four different heterochromatic loci in HeLa cells synchronized in G1 or G1-S phases by the serum-starvation and release method (27). qPCR data are presented as means ± s.d., and are representative of 3 independent experiments. Stable: 293T, stably integrated plasmids. (E) Levels of H3K9me3 are significantly decreased at heterochromatic loci upon overexpression of phospho-mimic mutant Suv39H1-S391E compared with phospho-defective mutant Suv39H1-S391A. qPCR data are presented as means ± s.d. for representative experiment. Stable: 293T, stably integrated plasmids.
Since our data pointed to a role of Suv39H1 phosphorylation in S phase events, we examined whether phosphorylation of Suv39H1 affects its occupancy on heterochromatin regions and histone H3 Lys9 methylation patterns as a hallmark of inactive chromatin during the G1-S transition. G1-S phase samples of HeLa cells stably expressing Flag-tagged Suv39H1-WT, Suv39H1-S391A and Suv39H1-S391E were prepared by serum-starvation and release. The protein expressions from each construct were confirmed by western blot analysis (Supplementary Figure S8). Cells that were serum-starved for 48 h were designated G1-arrested cells, while cells released for 16 h after adding serum were designated G1-S transition phase cells as described elsewhere (27). We performed ChIP assays using these G1-arrested and G1-S transition cells as previously described in Figure 3A and C. The results showed that overexpression of Suv39H1-WT and phospho-mimic Suv39H1-S391E significantly decreased their heterochromatin occupancy in G1-S transition cells compared with G1-arrested cells whereas overexpression of phospho-defective Suv39H1-S391A caused little change in heterochromatin occupancy (Figure 3D). In terms of histone H3 Lys9 tri-methylation, overexpression of Suv39H1-S391E mutant resulted in different types of effects for heterochromatins such as satellite repeats (Chr4 Satα and Chr1 Sat2) or non-satellite repeats (D4Z4 and NBL2). The H3 Lys9 tri-methylation levels at D4Z4 and NBL2 were little changed in either G1 phase or G1-S transition cells but the levels at both G1 phase and G1-S transition in Suv39H1-S391E overexpressing cells were lower than in Suv39H1-S391A overexpressing cells (Figure 3E). In addition, overexpression of Suv39H1-S391E causes a modest reduction of H3 Lys9 tri-methylation levels at satellite repeat-containing heterochromatic loci in G1-S transition cells relative to in G1-arrested cells (Figure 3E) whereas the histone methylation levels in Suv39H1-S391A overexpressing cells were higher than in Suv39H1-S391E overexpressing cells. The reasons to demonstrate these differential effects among different heterochromatins remain to be investigated in future studies. Thus, these data raise the possibility that phosphorylation-mediated modulation of Suv39H1-chromatin association and decreased levels of heterochromatin-specific histone marks may contribute to proper replication of heterochromatin during S phase progression.
The previous report demonstrated that occupancy of JMJD2A histone demethylase during S phase at Chr1 Sat2 was increased in JMJD2A-overexpressing cells, suggesting the role of antagonism between JMJD2A and HP1γ in regulation of DNA replication (28). Based on this mechanism, we hypothesized that phosphorylation-mediated dissociation of Suv39H1 from chromatin may serve as an initial step for JMJD2A occupancy on heterochromatin. To verify this hypothesis, we examined if overexpression of Suv39H1-S391A or Suv39H1-S391E mutants influences the occupancy of JMJD2A on heterochromatin in HU-treated G1/S-arrested cells or during S phase progression. Cells stably expressing Flag-tagged Suv39H1-S391A or Suv39H1-S391E were transfected with Myc-tagged JMJD2A or LSD1 and synchronized in G1/S phase with HU treatment and released from the block and collected at different time points indicated. The heterochromatin occupancy of histone demethylases was determined by using ChIP assay with anti-Myc antibody. The protein expression of each gene and cell cycle profile were confirmed by western blot and FACS, respectively (Supplementary Figure S11). While overexpression of phospho-defective Suv39H1-S391A abolished JMJD2A and LSD1 preoccupancy at heterochromatic loci in G1/S-arrested cells, overexpression of phospho-mimic Suv39H1-S391E resulted in highly enhanced preoccupancy of histone demethylases (Figure 4). Therefore, the data presented here may suggest that unlocking chromatin-bound Suv39H1 via its CDK2-mediated phosphorylation during S phase progression is needed for heterochromatin association of histone demethylases that erase inactive histone marks for proper replication timing of heterochromatin.
Figure 4.

Overexpression of phospho-mimic Suv39H1-S391E mutant proteins results in enhanced preoccupancy of JMJD2A at heterochromatic loci in G1-S border compared to overexpression of phospho-defective Suv39H1-S391A. Cells stably expressing Flag-tagged Suv39H1-S391A or Suv39H1-S391E were transfected with empty Myc vector (control), Myc-tagged Jmjd2A or LSD1 and synchronized in G1/S phase with HU treatment and released from the block and collected at different time points indicated. ChIP assay using anti-Myc antibody revealed that in Suv39H1-S391E overexpressing cells, JMJD2A was highly enriched at heterochromatic loci just after HU release compared to Suv39H1-S391A overexpressing cells. Heterochromatin occupancies of JMJD2A or LSD1 in Suv39H1-S391A or S391E overexpressing cells are presented as a relative ratio of percent immunoprecipitates (IP) of Myc-tagged JMJD2A or Myc-tagged LSD1 to percent IP of empty Myc control. qPCR data are presented as means ± s.d. for representative experiment.
Phosphorylation of Suv39H1 at Ser-391 affects replication timing of heterochromatin
Several chromatin factors including HP1 are known to be involved in the control of heterochromatin replication in yeasts and mammals (30,44–52). In particular, histone demethylation events mediated by JMJD2A are involved in S phase progression via control of replication timing at heterochromatic regions suggesting a functional role of histone H3 Lys-9 methylation in heterochromatin replication (28). Since our data suggest a potential role of phosphorylation of Suv39H1 in G1-S transition, we hypothesized that phosphorylation of Suv39H1 regulates the timing of heterochromatin replication by acting as an initial event in replication. To test this hypothesis, we examined S phase progression using FACS analysis and visualized nuclear morphology in mouse fibroblast NIH3T3 cells as described elsewhere (47,50,53) because the amino-acid sequences of the phosphorylation motif at Ser-391 are completely conserved between mouse and human, and several proteomic data and our data in this study (Supplementary Figure S2) support Ser-391 phosphorylation of Suv39H1 in both mouse and human cells. We established NIH3T3 cells stably expressing empty vector, Flag-tagged Suv39H1-WT, Suv39H1-S391A and Suv39H1-S391E, and the protein expression in each stable cell was analyzed by western blot (Supplementary Figure S8). Our FACS data demonstrate that the late S phase proportion in Suv39H1-S391E overexpressing cells was about 2.3-fold higher than in Suv39H1-S391A overexpressing cells at 3 h and 4 h after release from the block, suggesting the early accumulation of late S phase cells by Suv39H1-S391E overexpression and the delay of S phase by Suv39H1-S391A overexpression (Supplementary Figure S12). Thus, the data presented suggest that phosphorylation of Suv39H1 is involved in regulation of late S phase progression. Since mouse NIH3T3 cells show discrete pericentric heterochromatin foci, we can monitor the replication timing of heterochromatin by observation of BrdU-stained heterochromatic foci in mid-S phase (50). When empty vector or wild-type Suv39H1 was ectopically expressed, clear BrdU-stained heterochromatin foci were largely found in mid-S phase and early time points of late S phase (Figures 5A and B). In contrast, ectopic expression of phospho-mimic Suv39H1 caused early appearance of BrdU-stained heterochromatic foci whereas the replication of heterochromatin was delayed upon expression of phospho-defective Suv39H1 (Figures 5A and B). These results suggest that phosphorylation of Suv39H1 plays a role in the control of heterochromatin replication.
Figure 5.
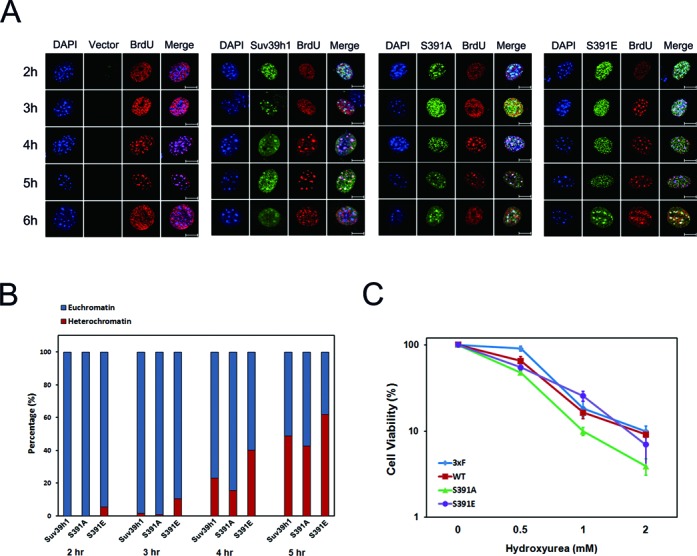
Ser-391 phosphorylation of Suv39H1 affects replication timing of heterochromatin. (A) Immunostaining assay of NIH3T3 cells stably expressing Flag-Suv39H1-WT, Flag-Suv39H1-S391A or Flag-Suv39H1-S391E, treated by aphidicolin blockage and released as described in Supplementary Figure S12. BrdU incorporation and Flag-tagged Suv39H1 were immunostained using antibodies against either BrdU or Flag-tag, respectively. Immunostained images were acquired by using confocal microscope and the representative images are presented here. Green colored images mean expression of Flag-tagged Suv39H1and red-colored images indicate DNA incorporated by BrdU. Replication of heterochromatin was judged by co-localization of BrdU-stained heterochromatin foci (red color) and DAPI-stained DNA foci (blue color) as previously described elsewhere (19,20,50). Scale bar presents 10 μm. (B) Quantitative analysis of the proportion of BrdU-stained heterochromatin foci at the time intervals indicated in (A). The percentages of cells positive for BrdU-stained heterochromatin foci (red bar) or BrdU-stained euchromatin (blue bar) were scored as indicated. Total cell numbers counted in each sample are presented as follows: n = 600 for WT at 2 h, n = 535 for S391A at 2 h, n = 630 for S391E at 2 h, n = 635 for WT at 3 h, n = 645 for S391A at 3 h, n = 718 for S391E at 3 h, n = 296 for WT at 4 h, n = 385 for S391A at 4 h, n = 481 for S391E at 4 h, n = 256 for WT at 5 h, n = 461 for S391A at 5 h, and n = 466 for S391E at 5 h. (C) Phosphorylation state of Suv39H1 at Ser-391 affects sensitivity to the replication inhibitor HU. Compared with control 293T cells expressing empty vector, overexpression of phospho-defective Suv39H1-S391A results in increased sensitivity to HU treatment whereas overexpression of wild-type and Suv39H1-S391E results in HU sensitivity comparable to that of control cells. These experiments were conducted at least three independent times.
Defective phosphorylation of Suv39h1 results in enhanced sensitivity to replication stress
Defects in fission yeast Clr4/Suv39H1, nematode JMJD2 and many other DNA replication factors cause cellular sensitivity to replication stress such as treatment with HU (28,54). Alteration in the replication timing of heterochromatin due to depletion of human JMJD2A histone demethylase also results in HU sensitivity (28). Since our data suggest that overexpression of phospho-mimic Suv39H1-S319E or phospho-defective Suv39H1-S391A could alter the replication timing of heterochromatin (Figures 5A and B, Supplementary Figure S12), we examined whether overexpression of phospho-defective or phospho-mimic Suv39H1 affects HU sensitivity. Overexpression of phospho-defective Suv39H1-S391A resulted in increased HU sensitivity compared with phospho-mimic Suv39H1 and wild-type protein while the standard growth was little changed among cells overexpressing three different forms of Suv39H1 protein (Figure 5C and Supplementary Figure S13). The results suggest that overexpression of Suv39H1-S391A mimics the increased HU sensitivity of JMJD2A-depleted cells (28). In other words, the increased HU sensitivity of Suv39H1-S391A overexpressing cells may be explained by the altered replication timing of heterochromatin as previously observed in JMJD2A-depleted cells (28). Thus, our data suggest that alteration of heterochromatin replication by overexpression of phospho-defective Suv39H1 gives rise to the phenotypic outcome of cellular sensitivity to replication stress.
DISCUSSION
In this study, we identified CDK2-mediated phosphorylation of Suv39H1 as a key step in the replication of heterochromatin during cell cycle progression in mammals (model in Supplementary Figure S14). The data presented demonstrate that CDK2 phosphorylates Suv39H1 at Ser-391 during cell cycle progression and that this phosphorylation is required for heterochromatin replication. Two previous reports from different research groups suggest that Suv39H1 can be phosphorylated at both G1-S phase and mitosis (25,26). Although the link between phosphorylation of Suv39h1 at Ser-391 and centromere function during mitosis is not yet fully understood, mitotic phosphorylation of Suv39H1 occurs at multiple unidentified sites and such phosphorylation and H3 Lys-9 methylation patterns on centromeres may be correlated with novel roles in active centromere function such as chromosome segregation (25,42).
Our data showed that S391E phospho-mimic mutant with low binding affinity to heterochromatin still occupied on chromatin in the absence of serum (G0/G1 phase) comparable to S391A phospho-defective mutant (Figure 3D). How to explain the behavior of Suv39H1-S391E protein? A previous report showed that 20–40% of Suv39H1 protein is stably bound within heterochromatin loci, suggesting the structural role in heterochromatic regions (43). In addition, they showed that Suv39H1-ΔSET domain mutants are more dynamic than the full-length protein. These mutants are very dynamic but can still bind to heterochromatin. Based on these findings, we suggest that Suv39H1 may become more dynamic on its phosphorylation but still can bind to chromatin until input of repelling forces such as recruitment of histone demethylase JMJD2A on heterochromatin during S phase progression.
In our present study, it was suggested that the S391A phospho-defective mutant might be always associated with chromatin regardless of the cell cycle stages. Despite the immobile behavior of Suv39H1-S391A protein, heterochromatin occupancy of histone demethylase JMJD2A increased in S391A-overexpressing cells after 1 h release as compared to 0 h release from HU-arrest, implying the presence of unknown mechanism for repelling chromatin-bound Suv39H1-S391A with lower dynamics as shown in Figure 4. How to explain the interplay between Suv39H1-S391A and JMJD2A? A recent report has revealed that the protein level of JMJD2A was increased at G1/S transition. Importantly, in JMJD2A-overexpressing cells, heterochromatin occupancy of JMJD2A has already increased about 2-fold at G1/S transition (i.e. after 0 h release from HU-arrest). The occupancy further increased to 3-fold after 1 h release and then went down to basal level after 2.5 h release from HU-arrest. These data suggest that removal of histone inactive marks by JMJD2A for heterochromatin replication requires the G1/S transition-specific increase of steady-state level of JMJD2A protein and accelerated recruitment of JMJD2A to heterochromatin. Intriguingly, the binding profile of JMJD2A in the previous report (28) is very similar to that in the present study. Thus, the data raise a possibility that overexpressed JMJD2A is competent to overcome the blockade such as highly condensed configuration of heterochromatin via function of immobile Suv39H1-S391A. Furthermore, an unknown factor may be involved in this process. The X factor may become active after 1 h release as compared to 0 h and then helps JMJD2A be competent to repel Suv39H1-S391A from heterochromatin.
A number of chromatin regulators have been identified as key players in the control of heterochromatin replication as mentioned in the previous section of this study. In particular, a histone H3 Lys-9 demethylation event catalyzed by JMJD2A is a key step in the replication of heterochromatin (28). Overexpression of wild-type JMJD2A or phospho-mimic Suv39H1 has similar effects in terms of replication timing at heterochromatic loci: both cause early replication of heterochromatin ((28); Figure 5).
Our data show that phosphorylation-mediated Suv39H1 dissociation is accompanied by decreased levels of histone H3 Lys-9 methylation at heterochromatin. However, since Suv39H1 is a histone methyltransferase responsible for the creation of inactive histone marks, we speculate that the demethylation associated with phosphorylation-mediated Suv39H1 dissociation may be a passive mode. Thus, we propose that phosphorylation-mediated Suv39H1 dissociation may be an initial step that allows active histone demethylation events by histone demethylase(s) such as JMJD2A thereby leading to proper replication of heterochromatin. Our data presented in this study support this hypothesis since overexpression of Suv39H1-S391E has increased JMJD2A preoccupancy on heterochromatin as shown in Figure 4. This raises the possibility that overexpression of phospho-mimic Suv39H1 may allow histone demethylase(s) to be recruited on heterochromatin and erase inactive histone marks, leading to control of proper replication timing of heterochromatin.
Recent studies support the idea that post-translational modification(s) of chromatin regulators provides the cell other levels of regulation modes for modulation of chromatin dynamics. Among them, phosphorylation(s) of chromatin proteins plays a key role in regulation of chromatin modulation. For examples, CDK1-dependent phosphorylation of mammalian Ezh2 is important for its binding to HOTAIR non-coding RNAs or the other PRC2 components SUZ12 and EED (13–15), and, in addition, CK2-mediated phosphorylation of Swi6/HP1 is required for the interaction between itself and other regulators in fission yeast (21). Another elegant study showed that ‘methyl-phos switching’ by histone H3S10 phosphorylation uniformly disrupts HP1-chromatin (the chromodomain-H3K9me3) interaction, leading to dissociation of HP1 from mitotic chromosome (55), implying the competition of H3S10 phosphorylation with H3K9 methylation for regulation of HP1-chromatin binding during mitosis. On the contrary, our data in this study suggest that CDK2-mediated phosphorylation of Suv39H1 regulates its binding to chromatin during S phase progression. This regulation mode is very similar to those examples: for instances, phosphorylation of H4K20me1 demethylase PHF8 and mammalian HP1, and sumoylation of Swi6/HP1 (18,20,56). However, the phosphorylation of Suv39H1 and PHF8 mediates their preferential dissociation from chromatin while phosphorylation or sumoylation of Swi6/HP1 promotes its chromatin binding. Thus, all these data suggest that post-translational modifications such as phosphorylation are important for regulation of chromatin dynamics via control of target protein–chromatin binding and interaction between chromatin regulators.
Collectively, our findings provide direct evidence that phosphorylation-mediated Suv39H1 dissociation from chromatin plays a role in the replication of heterochromatin during S phase progression. Importantly, data presented here show that CDK2 phosphorylates Suv39H1 at Ser-391 residue resulting in its preferential dissociation from heterochromatin and accompanied by JMJD2A occupancy on heterochromatin and alterations in inactive histone marks. Overexpression of phospho-mimic Suv39H1 causes early replication of heterochromatin whereas overexpression of phospho-defective Suv39H1 results in enhanced sensitivity to replication stress possibly due to the altered replication timing of heterochromatin. Thus, our data suggest that phosphorylation-mediated modulation of Suv39H1-chromatin association may be an initial step in proper replication of heterochromatin.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors are grateful to Drs Didier Trono and David Root for kindly providing plasmids for lentiviral shRNA knockdown. They also thank In Kwon Chung, Kwang-Min Choe and Sang-Mo Han for their encouragement and Jang's Lab members for their helpful suggestions.
FUNDING
National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) [2011-0030049, in part]; National R&D Program for Cancer Control, Ministry for Health, Welfare and Family Affairs, Republic of Korea [0920260]; Basic Science Research Program through the NRF funded by the Ministry of Education, Science and Technology [2010-0010639]; NRF Grant funded by the Korean Government [2009-0075197]; Mid-career Researcher Program through NRF grant funded by the MEST [2009-0083772, in part]; Yonsei University Research Fund [2013-12-0087, in part, to S.H.P]; Brain Korea21Plus (BK21+) Program (S.H.P., S.E.Y. and Y.K.J.). Source of open access funding: NRF grant funded by the Korea government (MSIP) [2011-0030049].
Conflict of interest statement. None declared.
REFERENCES
- 1.Allis C.D., Jenuwein T., Reinberg D. Overview and concepts. In: Allis CD, Jenuwein T, Reinberg D, editors. Epigenetics. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 23–61. [Google Scholar]
- 2.Northrop E.L., Wong L.H. Centromeres and telomeres. In: Craig JM, Wong NC, editors. Epigenetics: A Reference Manual. Norfolk, UK: Caister Academic Press; 2011. pp. 99–132. [Google Scholar]
- 3.Vasiljeva L., Kim M., Terzi N., Soares L.M., Buratowski S. Transcription termination and RNA degradation contribute to silencing of RNA polymerase II transcription within heterochromatin. Mol. Cell. 2008;29:313–323. doi: 10.1016/j.molcel.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 4.Elgin S.C.R., Reuter G. Position-effect variegation, heterochromatin formation, and gene silencing in drosophila. In: Allis CD, Jenuwein T, Reinberg D, editors. Epigenetics. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 81–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F., Kan H., Castranova V. Methylation of lysine 9 of histone H3: role of heterochromatin modulation and tumorigenesis. In: Tollefsbol T, editor. Handbook of Epigenetics: The New Molecular and Medical Genetics. San Diego: Academic Press; 2011. pp. 149–157. [Google Scholar]
- 6.Martienssen R., Moazed D. RNAi and heterochromatin assembly. In: Allis CD, Jenuwein T, Reinberg D, editors. Epigenetics. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 151–166. [Google Scholar]
- 7.Eymery A., Callanan M., Vourch C. The secret message of heterochromatin: new insights into the mechanisms and function of centromeric and pericentric repeat sequence transcription. Int. J. Dev. Biol. 2009;53:259–268. doi: 10.1387/ijdb.082673ae. [DOI] [PubMed] [Google Scholar]
- 8.Rea S., Eisenhaber F., O’Carroll D., Strahl B.D., Sun Z-W., Schmid M., Opravil S., Mechtler K., Ponting C.P., Allis C.D., et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 9.Kouzarides T., Berger S.L. Chromatin modifications and their mechanism of action. In: Allis CD, Jenuwein T, Reinberg D, editors. Epigenetics. New York: Cold Spring Harbor Laboratory Press; 2007. pp. 191–209. [Google Scholar]
- 10.Johnson A., Li G., Sikorski T.W., Buratowski S., Woodcock C.L., Moazed D. Reconstitution of heterochromatin-dependent transcriptional gene silencing. Mol. Cell. 2009;35:769–781. doi: 10.1016/j.molcel.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon S.H., Workman J.L. The changing paces of HP1: from heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription. Bioessays. 2011;33:280–289. doi: 10.1002/bies.201000138. [DOI] [PubMed] [Google Scholar]
- 12.Kim J.H., Workman J.L. Histone acetylation in heterochromatin assembly. Genes Dev. 2010;24:738–740. doi: 10.1101/gad.1922110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng X., Chen S., Huang H. Phosphorylation of EZH2 by CDK1 and CDK2. Cell Cycle. 2011;10:579–583. doi: 10.4161/cc.10.4.14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaneko S., Li G., Son J., Xu C-F., Margueron R., Neubert T.A., Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell-cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–2620. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei Y., Chen Y.H., Li L., Lang J., Yeh S., Shi B., Yang C., Yang J., Lin C., Lai C., et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat. Cell Biol. 2011;13:87–94. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lomberk G., Bensi D., Ferandez-Zapico M.E., Urrutia R. Evidence for the existence of an HP1-mediated subcode within the histone code. Nat. Cell Biol. 2006;8:407–415. doi: 10.1038/ncb1383. [DOI] [PubMed] [Google Scholar]
- 17.Zhao T., Heyduk T., Eissenberg J.C. Phosphorylation site mutations in heterochromatin protein 1 (HP1) reduce or eliminate silencing activity. J. Biol. Chem. 2001;276:9512–9518. doi: 10.1074/jbc.M010098200. [DOI] [PubMed] [Google Scholar]
- 18.Shin J.A., Choi E.S., Kim H.S., Ho J.C.Y., Watts F.Z., Park S.D., Jang Y.K. SUMO modification is involved in the maintenance of heterochromatin stability in fission yeast. Mol. Cell. 2005;19:817–828. doi: 10.1016/j.molcel.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 19.Maison C., Bailly D., Roche D., de Oca R.M., Probst A.V., Vassias I., Dingli F., Lombard B., Loew D., Quivy J.P., Almouzni G. SUMOylation promotes de novo targeting of HP1α to pericentric heterochromatin. Nat. Genet. 2011;43:220–227. doi: 10.1038/ng.765. [DOI] [PubMed] [Google Scholar]
- 20.Hiragami-Hamada K., Shinmyozu K., Hamada D., Tatsu Y., Uegaki K., Fujiwara S., Nakayama J. N-terminal phosphorylation of HP1α promotes its chromatin binding. Mol. Cell. Biol. 2011;31:1186–1200. doi: 10.1128/MCB.01012-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada A., Dohke K., Sadaie M., Shinmyozu K., Nakayama J-I., Urano T., Murakami Y. Phosphorylation of Swi6/HP1 regulates transcriptional gene silencing at heterochromatin. Genes Dev. 2009;23:18–23. doi: 10.1101/gad.1708009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maison C., Romeo K., Bailly D., Dubarry M., Quivy J.P., Almouzni G. The SUMO protease SENP7 is a critical component to ensure HP1 enrichment at pericentric heterochromatin. Nat. Struct. Mol. Biol. 2012;19:458–460. doi: 10.1038/nsmb.2244. [DOI] [PubMed] [Google Scholar]
- 23.Vaquero A., Scher M., Erdjument-Bromage H., Tempst P., Serrano L., Reinberg D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature. 2007;450:440–444. doi: 10.1038/nature06268. [DOI] [PubMed] [Google Scholar]
- 24.Bosch-Presegue L., Raurell-Vila H., Marazuela-Duque A., Kane-Goldsmith N., Valle A., Oliver J., Serrano L., Vaquero A. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensure genome protection. Mol. Cell. 2011;42:210–223. doi: 10.1016/j.molcel.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 25.Agaard L., Schmid M., Warburton P., Jenuwein T. Mitotic phosphorylation of SUV39H1, a novel component of active centromere, coincide with transient accumulation at mammalian centromeres. J. Cell Sci. 2000;113:817–829. doi: 10.1242/jcs.113.5.817. [DOI] [PubMed] [Google Scholar]
- 26.Firestein R., Cui X., Huie P., Cleary M.L. Set domain-dependent regulation of transcriptional silencing and growth control by SUV39H1, a mammalian ortholog of Drosophila Su(var)3–9. Mol. Cell. Biol. 2000;20:4900–4909. doi: 10.1128/mcb.20.13.4900-4909.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L., Lin C., Liu W., Zhang J., Ohgi K.A., Grinstein J.D., Dorrestein P.C., Rosenfeld M.G. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell. 2011;147:773–788. doi: 10.1016/j.cell.2011.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Black J.C., Allen A., Rechem C.V., Forbes E., Longworth M., Tschop K., Rinehart C., Quiton J., Walsh R., Smallwood A., et al. Conserved antagonism between JMJD2A/KDM4A and HP1γ during cell cycle progression. Mol. Cell. 2010;40:736–748. doi: 10.1016/j.molcel.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Yu S.E., Jang Y.K. The histone demethylase LSD1 is required for estrogen-dependent S100A7 gene expression in human breast cancer cells. Biochem. Biophys. Res. Commun. 2012;427:336–342. doi: 10.1016/j.bbrc.2012.09.057. [DOI] [PubMed] [Google Scholar]
- 30.Bozhenok L., Wade P.A., Varga-Weisz P. WSTF-ISWI chromatin remodeling complex targets heterochromatin replication foci. EMBO J. 2002;21:2231–2241. doi: 10.1093/emboj/21.9.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moffat J., Grueneberg D.A., Yang X., Kim S.Y., Kloepfer A.M., Hinkle G., Piqani B., Eisenhaure T.M., Luo B., Grenier J.K., et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 32.Harper J.V. Synchronization of cell populations in G1/S and G2/M phases of the cell cycle. In: Humphrey T, Brooks G, editors. Cell Cycle Control: Mechanisms and Protocols. Methods in Mol. Biol. Vol. 296. Totowa, NJ: Humana Press Inc; 2005. pp. 157–166. [DOI] [PubMed] [Google Scholar]
- 33.Olsen J.V., Vermeulen M., Santamaria A., Kumar C., Miller M.L., Jensen L.J., Gnad F., Cox J., Jensen T.S., Nigg E.A., et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010;3 doi: 10.1126/scisignal.2000475. ra3. [DOI] [PubMed] [Google Scholar]
- 34.Harper J.W., Adams P.D. Cyclin-dependent kinases. Chem. Rev. 2001;101:2511–2526. doi: 10.1021/cr0001030. [DOI] [PubMed] [Google Scholar]
- 35.Morgan D.O. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 36.Meraldi P., Nigg E. A. Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J. Cell Sci. 2001;114:3749–3757. doi: 10.1242/jcs.114.20.3749. [DOI] [PubMed] [Google Scholar]
- 37.Hong K.U., Kim H-J., Kim H-S., Seong Y-S., Hong K-M., Bae C-B., Park J. Cdk1-cyclin B1-mediated phosphorylation of tumor-associated microtubule-associated protein/cytoskeleton-associated protein 2 in mitosis. J. Biol. Chem. 2009;284:16501–16512. doi: 10.1074/jbc.M900257200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dinarina A., Ruiz E.J., O'Loghlen A., Mouron S., Perez L., Nebreda A.R. Negative regulation of cell cycle progression by RINGO/Speedy E. Biochem. J. 2008;410:535–542. doi: 10.1042/BJ20071453. [DOI] [PubMed] [Google Scholar]
- 39.van den Heuvel S., Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262:2050–2054. doi: 10.1126/science.8266103. [DOI] [PubMed] [Google Scholar]
- 40.Fatoba S.T., Okorokov A.L. Human SIRT1 associates with mitotic chromatin and contributes to chromosomal condensation. Cell Cycle. 2011;10:2317–2322. doi: 10.4161/cc.10.14.15913. [DOI] [PubMed] [Google Scholar]
- 41.Nielsen S.J., Schneider R., Bauer U-M., Bannister A.J., Morrison A., O’Carroll D., Firestein R., Cleary M., Jenuwein T., Herrera R.E., et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 42.Chu L., Zhu T., Liu X., Yu R., Bacanamwo M., Dou Z., Chu Y., Zou H., Gibbons G.H., Wang D., et al. SUV39H1 orchestrates temporal dynamics of centromeric methylation essential for faithful chromosome segregation in mitosis. J. Mol. Cell Biol. 2012;4:331–40. doi: 10.1093/jmcb/mjs023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krouwels I.M., Wiesmeijer K., Abraham T.E., Molenaar C., Verwoerd N.P., Tanke H.J., Dirks R.W. A glue for heterochromatin maintenance: stable SUV39H1 binding to heterochromatin is reinforced by the SET domain. J. Cell Biol. 2005;170:537–549. doi: 10.1083/jcb.200502154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alabert C., Groth A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012;13:153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- 45.Casas-Delucchi C.S., Cardoso M.C. Epigenetic control of DNA replication dynamics in mammals. Nucleus. 2011;2:370–382. doi: 10.4161/nucl.2.5.17861. [DOI] [PubMed] [Google Scholar]
- 46.Probst A.V., Dunleavy E., Almouzni G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- 47.Collins N., Poot R.A., Kukimoto I., Garcia-Jimenez C., Dellaire G., Varga-Weisz P.D. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat. Genet. 2002;32:627–632. doi: 10.1038/ng1046. [DOI] [PubMed] [Google Scholar]
- 48.Prasanth S.G., Shen Z., Prasanth K.V., Stillman B. Human origin recognition complex is essential for HP1 binding to chromatin and heterochromatin organization. Proc. Natl. Acad. Sci. U.S.A. 2010;107:15093–15098. doi: 10.1073/pnas.1009945107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peixoto P., Castronovo V., Matheus N., Polese C., Peulen O., Gonzalez A., Boxus M., Verdin E., Thiry M., Dequiedt F., et al. HDAC5 is required for maintenance of pericentric heterochromatin, and controls cell-cycle progression and survival of human cancer cells. Cell Death Differ. 2012;19:1239–1252. doi: 10.1038/cdd.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rowbotham S.P., Barki L., Nerves-Costa A., Santos F., Dean W., Hawkes N., Choudhary P., Will W.R., Webster J., Oxley D., et al. Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell. 2011;42:285–296. doi: 10.1016/j.molcel.2011.02.036. [DOI] [PubMed] [Google Scholar]
- 51.Schwaiger M., Kohler H., Oakeley E.J., Stadler M.B., Schubeler D. Heterochromatin protein (HP1) modulates replication timing of the Drosophila genome. Genome Res. 2010;20:771–780. doi: 10.1101/gr.101790.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quivy J.-P., Roche D., Kirschner D., Tagami H., Nakatani Y., Almouzni G. A CAF-1 dependent pool of HP during heterochromatin duplication. EMBO J. 2004;23:3516–3526. doi: 10.1038/sj.emboj.7600362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quivy J.-P., Gerard A., Cook A.J.L., Roche D., Almouzni G. The HP-p150/CAF-1 interaction is required for pericentric heterochromatin replication and S-phase progression in mouse cells. Nat. Struct. Mol. Biol. 2008;15:972–979. doi: 10.1038/nsmb.1470. [DOI] [PubMed] [Google Scholar]
- 54.Kim H.S., Rhee D.K., Jang Y.K. Methylations of histone H3 lysine 9 and lysine 36 are functionally linked to DNA replication checkpoint control in fission yeast. Biochem. Biophys. Res. Commun. 2008;368:419–425. doi: 10.1016/j.bbrc.2008.01.104. [DOI] [PubMed] [Google Scholar]
- 55.Fischle W., Tseng B.S., Dormann H.L., Ueberheide B.M., Garcia B.A., Shabanowtz J., Hunat D.F., Funabiki H., Allis C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 56.Liu W., Tanasa B., Tyurina O.V., Zhou T.Y., Gassmann R., Liu W.T., Ohgi K.A., Benner C., Garcia-Bassets I., Aggarwal A.K., et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.