


Abstract
Poly(ADP-ribose) polymerase-1 (PARP-1) is an abundant nuclear enzyme in mammalian cells. The enzyme synthesizes polymers of ADP-ribose from the coenzyme NAD+ and plays multifaceted roles in cellular responses to genotoxic stress, including DNA repair. It had been shown that mouse fibroblasts treated with a DNA methylating agent in combination with a PARP inhibitor exhibit higher cytotoxicity than cells treated with methylating agent alone. This lethality of the PARP inhibitor is dependent on apurinic/apyrimidinic (AP) sites in the DNA and the presence of PARP-1. Here, we show that purified PARP-1 is capable of forming a DNA-protein cross-link (DPC) by covalently attaching to the AP site. This DPC formation is specific to the presence of the natural AP site in DNA and is accompanied by a single-strand DNA incision. Cellular studies confirm the formation of PARP-1 DPCs during alkylating agent-induced base excision repair (BER) and formation of DPCs is enhanced by a PARP inhibitor. Using an N-terminal and C-terminal truncated PARP-1 we show that a polypeptide fragment comprising the zinc 3 and BRCT sub-domains is sufficient for DPC formation. The covalent attachment of PARP-1 to AP site-containing DNA appears to be a suicidal event when BER is overwhelmed or disrupted.
INTRODUCTION
PARP-1 is an abundantly expressed chromatin associated nuclear enzyme that has been implicated in a range of cellular processes, including detection of DNA damage, DNA repair, chromatin remodeling and regulation of transcription (1–4). Mammalian PARP-1 is a member of a superfamily of enzyme isoforms that have different primary structures, but share homology in the domain that synthesizes poly(ADP-ribose) (PAR) from the coenzyme nicotinamide adenine dinucleotide (NAD+) (5–7). PARP-1 is believed to be a sensor and ‘first responder’ for DNA lesions, especially those containing strand breaks (4,7,8). PARP-1 becomes activated for PAR synthesis upon binding to DNA lesions and adducts itself (PARylation) as well as other proteins involved in DNA metabolism (2). PARylation serves a signaling role for protein–protein interactions in DNA metabolism and for self-regulation of DNA binding by PARP-1 (9,10).
PARP-1 is composed of three main functional domains within a 113 kDa polypeptide chain; the amino-terminal DNA-binding domain (DBD), a central auto-modification domain (AD) and the carboxy-terminal catalytic domain (CAT) (11). The DBD is comprised of two homologous zinc finger sub-domains and a recently characterized third zinc finger sub-domain (12–14). The AD contains a protein–protein interaction sub-domain corresponding to the breast cancer gene 1 protein (BRCA1), carboxyl-terminus (BRCT) and acceptor amino acids for covalent attachment of PAR (2,15). The function of a conserved segment of 80–90 residues between the AD and CAT, known as the WGR (tryptophan, glycine and arginine-rich) region, is unknown (15). However, structural and biochemical studies have suggested that the WGR region, along with the zinc finger sub-domains, promotes binding to DNA lesions (15,16). The loop between the BRCT and WGR regions is known to be required for double-stranded DNA binding (17). Recently, Mansoorabadi et al. proposed a structural model in which the double-stranded DNA binding loop region is positioned to bind DNA adjacent to a lesion site (18). The CAT sub-domain of PARP-1 is responsible for synthesis of PAR from NAD+, transfer of PAR units onto acceptor amino acids and elongation of PAR chains (11).
Although the biological functions of PARP-1 remain under investigation, the enzyme has been implicated in DNA base excision repair (BER) (8,19). In mammalian cells, BER is considered the primary defense against simple DNA lesions, such as base loss, single-strand breaks (SSBs) and smaller base adducts that arise from a variety of exogenous and endogenous sources (20–22). In the case of base damage, BER is initiated by a damage-specific DNA glycosylase, which removes the damaged base, resulting in an abasic (AP) site in the DNA (23). PARP-1 recognizes this AP site and becomes activated for PAR synthesis following incision of the AP site by AP endonuclease 1 (APE1), which yields the 5′-deoxyribose phosphate (5′-dRP) group in a single nucleotide gap (24,25). It was proposed that PARylation enhances recruitment of repair factors to sites of DNA damage and that the negative charge conferred by self-PARylation facilitates PARP-1 dissociation from DNA, enabling the repair process to proceed (26–28).
In mouse embryonic fibroblasts undergoing alkylating agent-induced BER, inhibition of PARP activity by small molecule inhibitors that are analogs of the nicotinamide compound NAD+, which is the substrate for PARP-1, results in extreme cytotoxicity (9,29). All these inhibitors including, 3-amino benzamide (3-AB), 4-amino-1,8-napthalimde (4-AN), olaparib (AZD2281), veliparib (ABT-888) and MK-4827 inhibit PARP-1 activity by competing with its substrate NAD+ (30). This effect of PARP inhibition correlates with the presence of the 5′-dRP group in the BER intermediate (31–33). While the underlying mechanism of this cell killing phenotype is not clear, important clues have emerged. For example, PARP-1 protein expression and inhibitor treatment during S-phase of the cell cycle are required for the extreme cell killing phenotype, and treatment with PARP inhibitor results in more DNA-bound and chromatin-bound PARP-1, respectively, than in untreated cells (34,35). Further, direct measurement of genomic DNA strand breaks by pulse field gel electrophoresis revealed more strand breaks in PARP inhibitor-treated cells than in untreated cells (36). Based on these findings, it is proposed that inhibited DNA-bound PARP-1 hinders DNA repair and blocks DNA replication, resulting in replication fork collapse and formation of double-strand breaks (DSBs) (28,36–39).
In light of these observations, it is important to understand the interactions of PARP-1 with AP site-containing BER intermediates. We have shown previously that purified PARP-1 has AP lyase enzymatic activity against an AP site-containing BER substrate and that the reaction occurs through the Schiff base chemistry and strand incision by β-elimination (24,40). In these experiments, the covalent cross-link between purified PARP-1 and an AP site-containing DNA was measured after sodium borohydride (NaBH4) reduction of the enzyme–DNA complex (24,40). Here, we further examined covalent cross-linking of purified PARP-1 to various alternate DNA substrates. In the course of these experiments, we obtained the surprising result that PARP-1 forms a covalent DNA-protein cross-link (DPC) with some DNA substrates even without NaBH4 reduction. This intrinsic DPC formation was found to be explicit for the intact natural AP site or incised natural AP site in DNA. Formation of the PARP-1 genomic DPC also was observed in cell extracts and in mouse fibroblast cells in culture. The implications of the PARP-1 DPC are discussed.
MATERIALS AND METHODS
Materials
Synthetic oligodeoxyribonucleotides were from The Midland Certified Reagent Co. (Midland, TX). Cordycepin [α-32P]ddATP (5000 Ci/mmol) was from GE Healthcare (Piscataway, NJ). [γ-32P]ATP (7000 Ci/mmol) was from MP Biomedicals (Irvine, CA). Optikinase and terminal deoxynucleotidyl transferase were from USB Corp. (Cleveland, OH) and Fermentas Inc. (Hanover, MD), respectively. Purified mouse anti-human PARP-1 was from BD Biosciences PharMingen (San Jose, CA). The secondary antibody, anti-mouse IgG (H+L) conjugated to affinity purified horseradish peroxidase, was from Bio-Rad Laboratories (Hercules, CA). Protease inhibitors complete (EDTA-free) were from Roche Molecular Diagnostics Corp. (Indianapolis, IN). QIAEX II silica suspension was from Qiagen (Valencia, CA).
Recombinant proteins
Recombinant human DNA polymerase β (pol β) was over-expressed and purified as described previously (41). Human APE1, uracil DNA glycosylase (UDG) with 84 amino acids deleted from the amino-terminus, and amino-terminus His-tagged human PARP-1 (polypeptide I, amino acid residues 1–1014) were purified as described previously (42–45). Wild-type human PARP-1 fragments containing the amino acids 234–1014 (polypeptide II, Zn3-CAT), 234–518 (polypeptide III, Zn3-BRCT) and 353–662 (polypeptide IV, BRCT-WGR) were amplified from cDNA. These PCR products were then ligated into the directional TOPO cloning vector pENTR containing a TEV tag on the amino-terminus (pENTR™/TEV/D-TOPO cloning kit, Life Technologies) according to the manufacturer's protocol and transformed into bacteria (One Shot TOP10 competent Escherichia coli, Life Technologies, Grand Island, NY). Single bacterial clones were selected and verified by DNA sequencing. The verified PARP-1 fragment entry clones were then transferred by recombination to the pDEST17 expression vector containing an amino-terminal His-tag. The purified CAT (Polypeptide V, amino acid residues 654–1014) fragment of PARP-1 was a gift from Dr. Chen Qui (Laboratory of Structural Biology, NIEHS).
Protein purification
Full-length wild-type human PARP-1 and the fragments (I to IV) were purified using the protocol of Langelier et al. (44). Briefly, the proteins were expressed in One Shot TOP10 E. coli cells according to the manufacture's protocol by inducing with 1 mM IPTG, 0.1% arabinose and 0.5 mM ZnCl2 for 18 h at 15°C. The recombinant protein was first purified using a HisTrap HP Column (GE HealthCare Life Sciences, Piscataway, NJ) and eluted with 250 mM imidazole. Fractions containing recombinant protein were pooled and buffer exchanged into 50 mM Hepes, pH 7.4 and 50 mM NaCl. This exchanged protein was applied onto Mono S HR 10/10 (GE HealthCare Life Sciences) and eluted with a gradient of 50–500 mM NaCl. Fractions containing the recombinant protein were pooled, concentrated and loaded onto a HiPrep 26/60 Sephacryl S-200 High Resolution gel filtration column. Fractions containing recombinant protein were pooled, concentrated and stored at −80°C.
5′- and 3′-end labeling of DNA for cross-linking assays
Dephosphorylated 34-mer oligodeoxyribonucleotide (5′-CTGCAGCTGATGCGCUGTACGGATCCCCGGGTAC-3′) containing a uracil residue at position 16- or a 19-mer (pUGTACGGATCCCCGGGTAC-3′) were either 3′- or 5′-end labeled with terminal deoxynucleotidyl transferase and Cordycepin [α-32P] ddATP or with Optikinase and [γ-32P]ATP. The 34-mer (5′GTACCCGGGGATCCGTACGGCGCATCAGCTGCAG-3′) template was annealed with 32P-labeled oligonucleotides by heating the solution at 90°C for 3 min and allowing the solution to slowly cool to 25°C. In the case 5′ uracil-containing 19-mer oligonucleotide, the 34-mer template was annealed with 15-mer (5′-CTGCAGCTGATGCGC-3′) and 19-mer 32P-labeled oligonucleotides. Unincorporated Cordycepin [α-32P] ddATP or [γ-32P]ATP was removed using a MicroSpin™ G-25 column (GE HealthCare) according to the manufacturer's suggested protocol.
Preparation of AP DNA substrate
The 32P-labeled duplex oligonucleotide was treated with human UDG to generate AP sites in the intact or nicked DNA. Typically, 400 nM DNA substrate was pretreated in a reaction mixture containing 25 mM NaPO4 buffer, pH 7.0, 25 mM NaCl and 40 nM UDG. The reaction mixture was incubated for 30 min at 30°C. After this incubation, the reaction mixture was divided into two equal portions. One portion of the reaction was supplemented with 10 mM MgCl2 and 50 nM APE1 and incubated for 20 min at 37°C to generate incised AP site-containing DNA. Due to the labile nature of the incised AP site-containing DNA, the DNA substrate was prepared just before performing the NaBH4 trapping experiment.
Intrinsic and NaBH4 cross-linking of purified human PARP-1 and DNA
NaBH4 cross-linking of PARP-1 and DNA was performed with 5′- or 3′-end 32P-labeled DNA pretreated with UDG, essentially as described previously (46) with a slight modification of the reaction conditions. Briefly, the reaction mixture (10 μl) containing 25 mM NaPO4 buffer, pH 7.0, 25 mM NaCl, 10 mM EDTA, 200 nM 32P-labeled duplex DNA (34 base pair) and 200–500 nM purified PARP-1 (24) was assembled on ice. Reaction mixtures were then supplemented with water or 2 mM NaBH4. In some reaction mixtures purified PARP-1 was replaced with 10 μg cell extract, as indicated. The reaction mixture was incubated for 60 min on ice and followed by 15 min at room temperature. After incubation, the reaction was terminated by addition of 10 μl of SDS-PAGE gel-loading dye. The Nu-PAGE Bis-Tris gel (4–12%) and MOPS running buffer systems (Life Technologies) were used to separate DPC complexes. A Typhoon PhosphorImager (GE HealthCare) was used for scanning the gels. The PARP-1 (113.1 kDa) construct used contained the N-terminal His-tag.
Iodoacetamide treatment of PARP-1
PARP-1 (10 μM) was incubated with 50 mM iodoacetamide in a reaction mixture (10 μl) containing 25 mM Na PO4 buffer, pH 7.0 and 25 mM NaCl for 30 min at room temperature in the dark. The reaction was terminated by adding 60 mM DTT, and the excess amounts of iodoacetamide and DTT were removed by Zebra micro spin desalting column 7K MWCO (Thermo Scientific) according to the manufacturer's suggested protocol. The final volume of each desalted sample was measured to calculate the concentration of PARP-1, and the recovery was ascertained by SDS-PAGE analysis. Appropriate dilutions were used for cross-linking experiments with PARP-1 and 32P-labeled DNA.
Proteolysis of the cross-linked PARP-1-DNA complex with trypsin
After intrinsic cross-linking of polypeptides I and II with 32P-labeled AP site-containing DNA as above, the pH of the of reaction mixture was adjusted to 8.0 by adding 1 M Tris-HCl, pH 8.0, to a final concentration of 100 mM. An aliquot was withdrawn before adding trypsin to the reaction mixture containing the PARP-1 and DNA complex. To the remaining reaction mixture, trypsin was added at a 1:5 weight ratio of trypsin to polypeptide, and the mixture was incubated for 20 min at 25°C. The reaction was terminated by adding 100 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, SDS-PAGE sample buffer, and boiling for 5 min. The digested products were separated by Nu-PAGE Bis-Tris gel (4–12%), and a PhosphorImager was used to detect radiolabeled cross-linked peptides. Then, the same gel was stained for proteins detection with SimplyBlue SafeStain solution (Life Technologies).
Cell culture
PARP-1+/+ and PARP-1−/− spontaneously immortalized mouse fibroblasts were obtained from Dr. Josianne Ménissier-de Murcia (CNRS, Illkirch-Graffenstaden, France). They were cultured at 37°C in a 10% CO2 incubator in Dulbecco's modified Eagle's medium containing L-glutamine and 10% fetal bovine serum (HyClone, Logan, UT).
Isolation of DPCs by the DNAzol-silica method
PARP-1+/+ mouse fibroblasts were treated for 1 h with methyl methanesulfonate (MMS; 5 mM) in the absence or presence of 4-AN (10 μM) or with 4-AN alone for a total of 2 h. Cells were washed in phosphate buffered saline (PBS), then isolated by scraping. For isolation of DPCs the method of Barker et al. (47) was followed with slight modifications. Briefly, nuclei were lysed by the addition of 500 μl DNAzol per 6 × 107 cells and vortexing briefly. After lysis, 2 ml pre-warmed (65°C) 10 mM Tris-HCl, pH 7.0, was added and the DNA was sheared by passing the suspension through a 21-gauge needle and then through a 25-gauge needle three times each. 5 M NaCl (9.6 ml) was added to a final concentration of 4 M, and the mixture was incubated at 37°C in a shaking water bath for 20 min. 8 M urea (12 ml) was added to a final concentration of 4 M, and the incubation was continued for 20 min at 37°C in a shaking water bath. After this incubation, an equal volume (24 ml) of 100% ethanol was added and the sample mixed by inversion. The QIAEX II silica slurry (0.5 ml per 6 × 107 nuclei) was added to each sample, and the samples were gently rocked for 40 min at room temperature to allow DNA to bind. Silica particles were then collected by centrifugation, and the supernatant fraction was carefully removed and discarded. The silica particles were washed four times with 50% ethanol and collected by centrifugation. The DPC complexes were eluted from silica by adding 2 ml of 8 mM NaOH and incubation at 65°C for 5 min. The elution process was repeated and the supernatant fractions were combined. An aliquot (10 μl) from each sample was saved for DNA analysis (Supplementary Figure S5). For DNA digestion, the supernatant fractions were supplemented with 1 ml of 5x digestion buffer (50 mM MgCl2, 50 mM ZnCl2, 0.5 M sodium acetate, pH 5.0), 5 units of DNase I and 5 units of S1 nuclease. The samples were then incubated at 37°C for 1 h, and the digestion of DNA was stopped by transferring samples to 65°C for 10 min. After this heat inactivation, 100% ice-cold trichloroacetic acid (TCA) was added to each sample to a final concentration of 15%, and the samples were incubated for 60 min on ice for precipitation of DPCs. All samples were centrifuged at 12,000 rpm for 15 min at 4°C. The pellet containing DPCs was washed twice with 15% ice-cold TCA and twice with 100% ice-cold acetone. The pellet was air-dried and dissolved in 10 μl 1 M Tris-HCl, pH 8.0 and 90 μl SDS-PAGE gel buffer for immunoblotting analysis of PARP-1.
Immunoblotting
Approximately equal amounts (adjusted by DNA content, Supplementary Figure S5) of each DPC sample (∼ 30 μl) isolated by the DNAzol-silica method as described above, and purified PARP-1 (50 ng) were separated by Nu-PAGE 4–12% Bis-Tris mini-gel and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.1% (v/v) Tween 20 (TBS-T) and then probed with anti-PARP-1 antibody as indicated in the figure legends (Figure 7c). Goat anti-mouse IgG conjugated to horseradish peroxidase (1:10,000 dilution) was used as secondary antibody, and immobilized horseradish peroxidase activity was detected by enhanced chemiluminescence.
Figure 7.
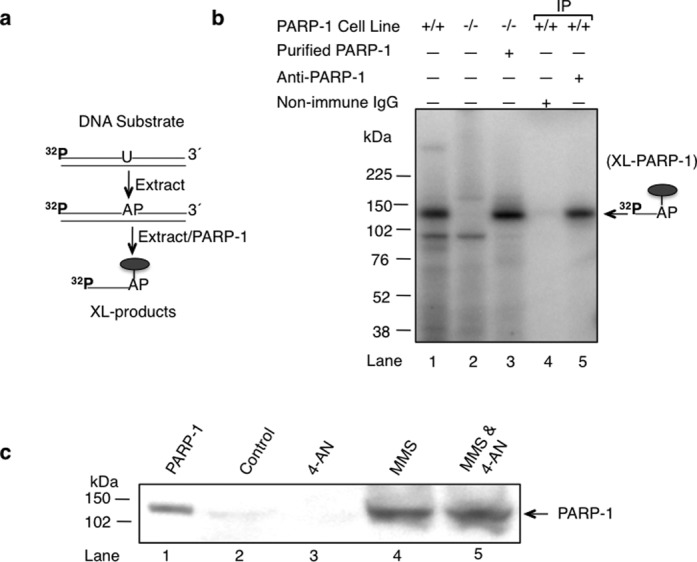
Intrinsic cross-linking of PARP-1 to AP site-containing DNA in vivo. (a) The reaction scheme of covalent cross-linking of PARP-1 in extracts and the schematic representation of the 32P-labeled uracil-containing DNA substrate are illustrated. (b) Intrinsic cross-linking of PARP-1 to AP site DNA in cell extracts. Mouse fibroblast cell extracts from wild-type (PARP-1+/+) cells (lanes 1, 4 and 5) and PARP-1 null (PARP-1−/−) cells (lanes 2 and 3) were incubated with 32P-5′-end labeled uracil-containing DNA substrate (depicted in the panel a) without NaBH4. The reaction mixture in lane 3 was supplemented with purified PARP-1 (100 nM). After incubation, the reaction mixtures in lanes 4 and 5 were subjected to immunoprecipitation analysis with non-immune IgG or anti-PARP-1 IgG, as indicated. The cross-linked products were analyzed as in Figure 1. The symbol ( ) represents PARP-1. The migration position of the incised DNA strand adducted to PARP-1 is indicated. (c) Intrinsic covalent cross-linking of PARP-1 to genomic DNA in mouse fibroblasts treated as indicated. DPCs were isolated as described under Materials and Methods. Samples were from control untreated cells (lane 2), 4-AN-treated cells (lane 3), MMS-treated cells (lane 4) or cells treated with the MMS and 4-AN combination (lane 5). Samples were analyzed by SDS-PAGE and immunoblotting with anti-PARP-1 antibody. Lane 1 corresponds to a sample of purified PARP-1 used as a positive control. A representative image of three experiments is shown.
) represents PARP-1. The migration position of the incised DNA strand adducted to PARP-1 is indicated. (c) Intrinsic covalent cross-linking of PARP-1 to genomic DNA in mouse fibroblasts treated as indicated. DPCs were isolated as described under Materials and Methods. Samples were from control untreated cells (lane 2), 4-AN-treated cells (lane 3), MMS-treated cells (lane 4) or cells treated with the MMS and 4-AN combination (lane 5). Samples were analyzed by SDS-PAGE and immunoblotting with anti-PARP-1 antibody. Lane 1 corresponds to a sample of purified PARP-1 used as a positive control. A representative image of three experiments is shown.
DNA labeling and fiber-spread analysis
PARP-1+/+ or PARP-1−/− fibroblasts were treated with 3 mM MMS for 1 h in the presence of the PARP-1 inhibitor 4-AN (10 μM), as described (34). The cells were harvested with trypsin, washed twice with PBS and collected, and DNA fibers were prepared as previously described (48). The resulting DNA spreads were air-dried, fixed in 3:1 methanol/acetic acid for 2 min then stored at ∼ 20°C for at least 24 h. The slides containing the DNA fibers were then washed once with PBS (pH 7.4) plus 0.1% Tween-20 (PBS-T) and twice with PBS (pH 7.4). Next, slides were blocked with 5% BSA in PBS for 1 h at room temperature in a humidified chamber. All antibodies were diluted in 5% BSA in PBS-T. Slides were first incubated for 1 h at room temperature with mouse anti-human PARP-1 (1:333) (BD Biosciences PharMingen). Slides were then washed three times with PBS, blocked with 5% BSA in PBS for 30 min at room temperature and incubated for 30 min with the secondary antibody AlexaFluor 594 rabbit anti-mouse (1:333) (Life Technologies). Next, the slides were washed once with PBS-T, and twice with PBS and blocked with 5% BSA. Tertiary antibody, AlexaFluor 594 goat anti-rabbit (1:333) (Life Technologies), was added and the slides washed three times with PBS. After the last wash, the slides were incubated with the DNA stain YOYO-1 iodide (Life Technologies) (1 μM in PBS) for 5 min, in order to visualize all of the DNA fibers on the slide, and then washed three times with PBS. The slides were then mounted with Pro-long Gold Anti-fade (Life Technologies). The method used for making DNA fiber spreads is reliant upon using 0.5% SDS and 50 mM EDTA to gently strip off any non-covalently bound protein. This ‘stripping’ step is similar to that employed for DNA fiber-based replication studies, except in the latter case an HCl incubation step was used to denature the DNA fibers to allow antibody access to nucleotide analogs incorporated into the DNA fibers. SDS/EDTA is effective in stripping off proteins from the DNA. In previous studies (P. Chastain, unpublished observation) of stripped DNA fibers, we could not detect any of the histone core proteins remained associated with the DNA.
Visualization of PARP-1 in DNA fibers and processing of data
DNA fibers were visualized with an Olympus FV400 confocal microscope under magnification × 630. At least 600 fibers and five images were scored for each repeat; at least 100,000,000 nts per experimental repeat were analyzed. Enumeration of the PARP-1 (PARP-1 per megabase (Mb)) within the DNA fibers was performed using the Computer Aided Scoring and Analysis (CASA) software described previously (49). CASA software recognizes the DNA fibers, scans them for PARP-1 signal (red dots) and creates an Excel file with data for each fiber identified.
RESULTS
Intrinsic cross-linking of purified PARP-1 to AP DNA
PARP-1 is known to interact with AP site-containing BER intermediates, and the interaction includes Schiff base formation between a primary amine in PARP-1 and the C1′ atom of deoxyribose in the AP site. NaBH4 reduction of the Schiff base-containing complex results in formation of a covalent bond between PARP-1 and DNA (24,25,40). To further characterize the PARP-1 and AP site interaction, we initially performed NaBH4 cross-linking experiments with purified PARP-1 and AP site-containing DNA (Figure 1 and Supplementary Figure S1). PARP-1 was incubated with an AP site-containing 5′-end labeled DNA substrate in phosphate buffer at pH 7, and the complex was analyzed without or with NaBH4 reduction (Figure 1a, lanes 2 and 3, respectively). Representative images of the full gel are shown (Supplementary Figure S1). Under these conditions, two species of PARP-1 and DNA cross-linked products were observed with NaBH4 reduction (Figure 1a, lane 3). Surprisingly, a cross-linked product also was observed without NaBH4 reduction (Figure 1a, lane 2). The migration of this cross-linked product was similar to that of the faster migrating product observed with NaBH4 reduction (Figure 1a, lane 3). Our interpretation of these results is as follows: during the PARP-1 AP lyase reaction (24), a primary amine nucleophile in the enzyme attacks the C1′ atom of deoxyribose in the AP site yielding the Schiff base intermediate. Reduction of this Schiff base with NaBH4 results in covalent cross-linking of PARP-1 to DNA (Figure 1a and c, lane 3, the slower migrating species corresponding to PARP-1 cross-linked to the full-length DNA strand). When the AP lyase reaction proceeds through the β-elimination of DNA from the C3′ position of deoxyribose, a DNA-PARP-1 cross-link (DPC) is formed at the dRP group, corresponding to the faster migrating species (Figure 1a and c, lanes 2 and 3, i.e. PARP-1 cross-linked to the incised DNA strand). This cross-linked species is formed in reaction mixtures both with and without NaBH4 reduction (Figure 1a).
Figure 1.
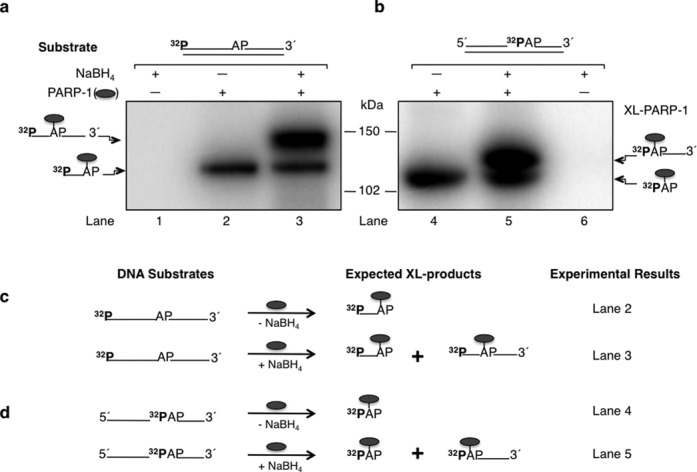
Cross-linking of purified PARP-1 to AP site-containing DNA. Phosphorimages of cross-linked PARP-1 to 32P-labeled AP site-containing DNA are shown in panels (a) and (b). Schematic representations of DNA substrates are shown above the phosphorimages. (a) Substrates were 5′-end 32P-labeled 34 bp DNA with a natural AP site or (b) 5′-end 32P-labeled dRP in a 34 bp DNA with a nick. Purified PARP-1 and 32P-labeled AP site DNA were incubated on ice without (-) or with (+) NaBH4 (see Materials and Methods). Lanes 1 and 6 in panels (a) and (b) represent DNA substrate incubations without PARP-1 as controls. The cross-linking products were analyzed by SDS-PAGE and followed by phosphorimaging. The migration positions of PARP-1 cross-linked to the full-length DNA strand (∼ 127 kDa), C3′ incised DNA strand (∼ 121 kDa), nicked DNA strand (∼ 122 kDa) and labeled dRP (∼ 116 kDa) are indicated. Panels (c) and (d) represent the substrates used in panels (a) and (b), respectively, and the expected cross-linked products under reaction conditions without (-) or with (+) NaBH4 are indicated. The symbol () represents PARP-1, AP represents the AP site in the DNA, XL-PARP-1 means the cross-linked PARP-1 and 32P indicates the position of radiolabel on the DNA.
To further analyze the covalent cross-linking phenomenon of PARP-1 to AP site-containing DNA without NaBH4 reduction, we utilized a nicked BER intermediate containing a 32P-dRP group (AP site) at the 5′-margin in a nick (Figure 1b). This AP site DNA substrate containing a 5′-dRP group was subjected to cross-linking with PARP-1, as in Figure 1(a). For the reaction mixture with NaBH4 reduction, the results revealed formation of two labeled species of PARP-1 cross-linked products and only one cross-linked product without NaBH4 reduction (Figure 1b, lanes 5 and 4, respectively). These results can be explained in a fashion similar to the results shown in Figure 1(a): NaBH4 reduction of the Schiff base intermediate results in a cross-linked enzyme–DNA complex (Figure 1b and d, lane 5, i.e. the slower migrating species corresponding to PARP-1 and the DNA strand). However, the lyase reaction proceeding through β-elimination of the DNA from the C3′ position of deoxyribose, produces an enzyme-dRP group linkage (Figure 1b and d, lanes 4 and 5, i.e. the faster migrating species). The gel migration positions of these cross-linked products were consistent with a linkage between one molecule each of PARP-1 and dRP (Figure 1b, faster migrating species) or the DNA strand (Figure 1b, slower migrating species). Formation of the faster migrating species was observed in reaction mixtures both with and without NaBH4 reduction.
Association of PARP-1 cross-linking with β-elimination and strand incision
To confirm that the intrinsic cross-linking of PARP-1 to dRP/AP site-containing DNA is associated with β-elimination, we conducted cross-linking experiments with AP site-containing DNA substrates labeled at the 3′-end, one with an intact strand and the other with a 5′-dRP group at a nick (Figure 2). For comparison, the reaction mixtures in lanes 1 and 2 of Figure 2(a) and (b) represent repeats of the experiments described in Figure 1(a) and (b) where the expected results were observed. However, for the incubations with the 3′-end labeled DNA along with NaBH4 reduction, only the slower migrating species was observed, corresponding to the intact DNA strand and PARP-1 (Figure 2a, lane 4). No labeled cross-linked product was observed for the reaction mixture without NaBH4 (Figure 2a, lane 3). These results are consistent with the lyase reaction proceeding through β-elimination of the DNA from the C3′ position of deoxyribose in the AP site; a labeled DNA-PARP-1 complex is not observed since the radiolabel is on the 3′-end of the DNA strand (see Figure 2c). To confirm this interpretation, we used a nicked BER intermediate labeled at the 3′-end. In the absence of NaBH4, no labeled cross-linked product was observed with this 3′-end labeled substrate (Figure 2b, lane 3); the slower migrating labeled cross-linked product was observed with NaBH4 reduction (Figure 2b, lane 4). From the results in Figures 1 and 2, we conclude that formation of a covalent bond between PARP-1 and the AP site deoxyribose is closely associated with β-elimination mediated strand incision at C3′.
Figure 2.
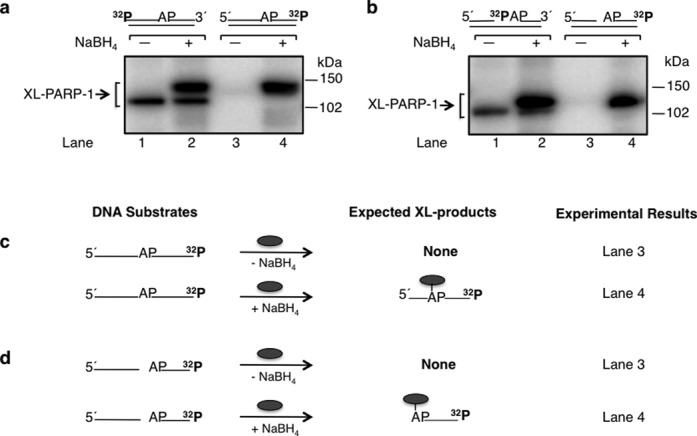
Cross-linking of purified PARP-1 to AP site-containing DNA. Phosphorimages of cross-linked PARP-1 to 32P-labeled AP site-containing DNA are shown in panels (a) and (b). Schematic representations of DNA substrates are shown above the phosphorimages. (a) Substrates were 5′-end or 3′-end 32P-labeled 34 bp DNA with an AP site or (b) 5′-end 32P-labeled 34 bp DNA with an AP site in a nick (i.e. 5′-dRP) or 3′-end 32P-labeled 34 bp DNA with an AP site in a nick (i.e. 5′-dRP). The substrates in lanes 1 and 2 of panels (a) and (b) were the same as in panels (a) and (b) of Figure 1 and incubations were included for comparison. Purified PARP-1 and 32P-labeled DNA were incubated on ice without (-) or with (+) NaBH4 (see Materials and Methods). The cross-linking products were analyzed by SDS-PAGE and followed by phosphorimaging. The migration positions of the PARP-1 cross-linked with the full-length DNA strand, C3′ incised DNA strand, nicked DNA strand and labeled dRP are indicated. Panels (c) and (d) represent the substrates used in panels (a) and (b), respectively, and the expected labeled products under reaction conditions without (-) or with (+) NaBH4 are indicated; only one DNA strand is shown. The symbol () represents PARP-1, AP represents the AP site in the DNA and 32P indicates the position of radiolabel on the DNA.
Zinc 3-BRCT sub-domain and intrinsic cross-linking of PARP-1 to AP DNA
We next evaluated the covalent bond formation between PARP-1 and AP site-containing DNA as a function of PARP-1 domain structure (Figure 3). Different segments of the PARP-1 primary structure were expressed and purified, as described in Supplementary Information and Figure 3. Robust cross-linking was observed (Figure 3) with the full-length construct polypeptide I and also with the construct truncated in the zinc 1 and zinc 2 sub-domains, i.e. polypeptide II. Similar cross-linking also was observed with the polypeptide III construct comprising the zinc 3 and BRCT sub-domains. In contrast, the constructs corresponding to the AD (polypeptide IV) and CAT sub-domains failed to cross-link. These results suggest that the zinc 3 sub-domain is sufficient for the cross-linking observed with polypeptides, I, II and III.
Figure 3.
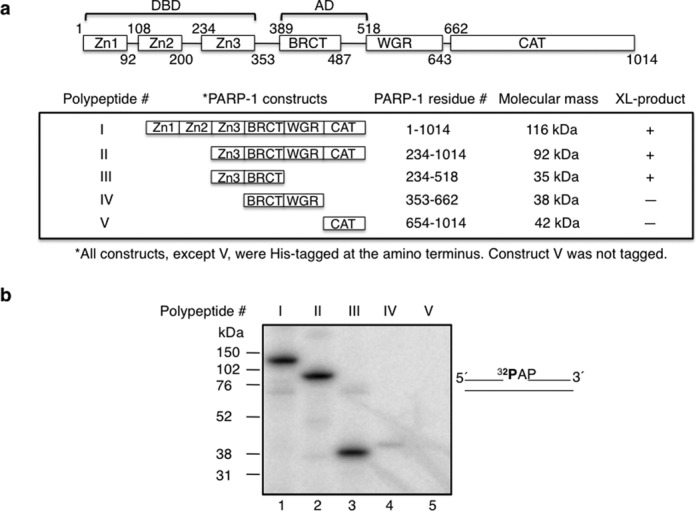
Cross-linking of various PARP-1 domain constructs to AP site-containing DNA. (a) Schematic representation of human PARP-1 sub-domains. Zn1, Zn2, Zn3, BRCT, WGR, and CAT represent, zinc finger 1, zinc finger 2, zinc finger 3, BRCA C-terminus, tryp-gly-arg-rich region, and carboxy-terminal catalytic sub-domains of PARP-1, respectively. PARP-1 residues of each construct are indicated. BRCT is located within the region of AD of PARP-1. DBD consists of Zn1, Zn2 and Zn3 sub-domains of PARP-1. Various PARP-1 polypeptides (I–V) representing full-length PARP-1, Zn3-CAT, Zn3-BRCT, BRCT-WGR, and CAT, respectively, were expressed in E. coli and purified to near homogeneity as described under Materials and Methods. The residue numbers of each construct and the relative molecular mass (kDa) of each construct are indicated. All the constructs were His-tagged at the amino terminus, except CAT sub-domain that has no tag. The intrinsic cross-linking reactivity to AP-DNA is indicated by positive (+) or negative (-) sign. (b) Intrinsic cross-linking of purified PARP-1 polypeptides (I–V) to AP site-containing DNA. 5′-end 32P-labeled 34 bp DNA with an AP site in a nick (i.e. 5′-dRP), shown next to the phosphorimage, was used for cross-linking. PARP-1 polypeptide (I–V) and 32P-labeled DNA were incubated on ice without NaBH4 as in Figure 1(a). The cross-linking products were analyzed by SDS-PAGE and followed by phosphorimaging. The relative positions of marker proteins are indicated.
We next performed experiments to evaluate the intrinsic covalent cross-linking of the labeled DNA probe to polypeptides I and II. The cross-linked DNA–polypeptide complexes were subjected to trypsin proteolysis to examine domains involved in the intrinsic covalent cross-linking. Trypsin proteolysis of the cross-linked DNA–polypeptide complexes revealed several radiolabeled protein fragments (Figure 4). A prominent peptide migrating between the molecular mass markers of 52 and 38 kDa was observed (Figure 4b) and appeared strongly radiolabeled (Figure 4a, lanes 3 and 6). The migration of this radiolabeled tryptic fragment was similar to that of recombinant polypeptide III (Figure 4a and b, lanes 3, 6 and 7). These results are consistent with covalent cross-linking of DNA in a region of polypeptides I and II that corresponds to polypeptide III (residues 234 to 518) studied in Figure 3. A similar tryspsin proteolysis pattern was observed with polypeptide I and II that were not subjected to cross-linking (data not shown). Amino-terminal sequence analysis for the prominent fragment migrating between the 52 and 38 kDa markers indicated that it corresponded to the zinc 3 and BRCT sub-domains (Supplementary Figure S2).
Figure 4.
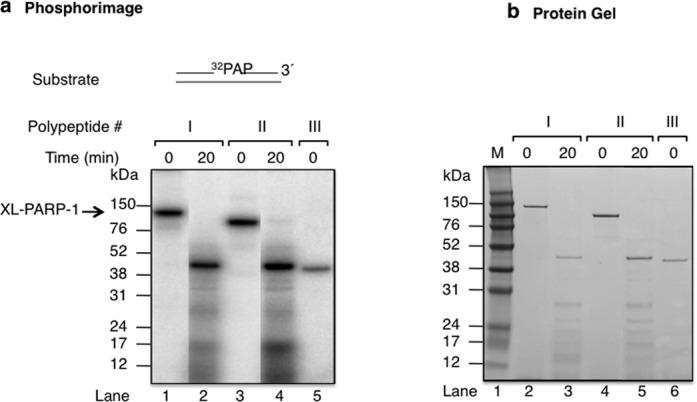
Proteolysis of cross-linkined polypetides I and II with trypsin. (a) Phosphorimage illustrating the proteolysis of the PARP-1-DNA cross-linked complex with trypsin. Purified polypeptides I and II and 32P-labeled AP site DNA were incubated on ice without NaBH4 under similar reaction conditions as in Figure 1 (lanes 1–4). After the intrinsic cross-linking, the pH of the reaction mixture was adjusted to 8.0 by adding 1 M Tris-HCl, pH 8.0, to a final concentration of 100 mM. The covalently cross-linked complex was then treated with trypsin at a 1:5 ratio of trypsin to complex (w/w), and the mixture was incubated at 25ºC for 20 min. The proteolysis products were analyzed as in Figure 1. Cross-linked polypeptide III was used as reference (lane 5). (b) Photograph of the Coomassie blue-stained gel in panel (a). Schematic representation of a 5′-end 32P-labeled 34 bp DNA with an incised natural AP site is shown above the phosphorimage in (a).
Specificity of intrinsic cross-linking of PARP-1 to AP DNA
When the same cross-linking protocol was performed with two other known dRP/AP lyase BER enzymes, pol β and 8-oxoguanine DNA glycosylase 1 (46,50,51), we failed to observe cross-linked products without NaBH4 reduction, but cross-linking was observed with NaBH4, as expected (Supplementary Figure S3a). To verify the requirement of an AP site in DNA, various alternate DNA substrates with and without the AP site were evaluated (Supplementary Figure S3b). The results revealed intrinsic cross-linking of PARP-1 with the substrate containing the AP site (Supplementary Figure S3c, lanes 1 and 2), as expected, whereas the other DNA molecules that either did not have an AP site or had a reduced AP site analog (THF) failed to show any cross-linking to PARP-1 (Supplementary Figure S3c, lanes 3–10).
Modification of exposed cysteine residues reduces intrinsic DNA cross-linking of PARP-1
For the intrinsic covalent cross-linking to occur between PARP-1 and AP site-containing DNA, the Schiff base formed between a primary amine in PARP-1 and the C1′ atom of deoxyribose in the AP site must be reduced to generate a covalent bond between PARP-1 and DNA. Therefore, the presence of a hydride ion-generating group in the enzyme in proximity to the Schiff base may serve as a reducing agent. PARP-1 has two cysteine residues, Cys256 in the zinc 3 domain and Cys845 in the CAT, that are solvent exposed (52). To examine the possibility that these cysteine residues serve in an oxidation-reduction capacity in the PARP-1 and AP site cross-linking, we first treated PARP-1 with iodoacetamide and then used the treated enzyme in cross-linking experiments, as above. Iodoacetamide covalently reacts with exposed thiol group of cysteine so that the protein cannot form disulfide bonds. The results revealed that iodoacetamide-treated PARP-1 had reduced cross-linking as compared to that of untreated PARP-1, while the concentration of protein in both samples was similar (Figure 5a). In a second approach, we altered the exposed cysteine residues to alanine to produce single C256A and double C256A/C845A mutant forms of PARP-1. The intrinsic covalent cross-linking property of the altered forms was compared with wild-type PARP-1. The results revealed a significant reduction in the cross-linking activity of the altered forms compared with that of wild-type PARP-1 (see Figure 5b). These results suggested that cysteine mediated disulfide bond formation might serve in the Schiff base reduction involved in the intrinsic cross-linking of PARP-1.
Figure 5.
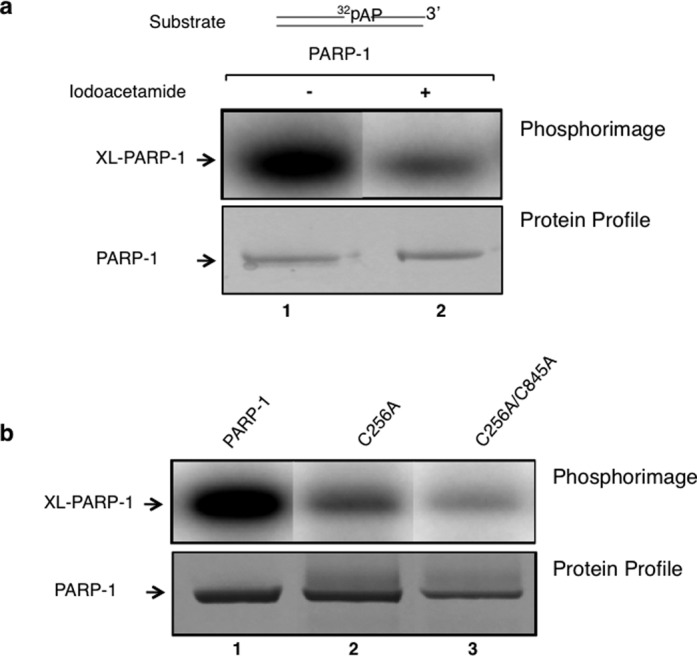
Site-directed alterations and iodoacetamide treatment of PARP-1 reduce intrinsic covalent cross-linking of PARP-1. (a) A phosphorimage of cross-linked PARP-1 to 32P-labeled AP site-containing DNA is shown. PARP-1 (10 μM) was incubated with 50 mM iodoacetamide in a reaction mixture (10 μl) containing 25 mM NaPO4 buffer, pH 7.0 and 25 mM NaCl for 30 min at room temperature in the dark. Then, the addition of 60 mM DTT was used to terminate the reaction. The excess amounts of iodoacetamide and DTT were removed with a Zebra micro spin desalting column. The intrinsic cross-linking reaction was performed with untreated PARP-1 (lane 1) or iodoacetamide-treated PARP-1 (lane 2). After capturing the phosphorimage of the 32P-labeled DNA-PARP-1 complex (upper panel), the same gel was stained with Coomassie-blue for proteins (lower panel). (b) Photographs of the phosphorimage and the Coomassie-stained gel are shown. The cross-linking reaction was performed with wild-type (lane 1), single mutant C256A (lane 2) or with a double mutant C256A/C845A (lane 3) of PARP-1 as in panel (a). The results revealed a significant reduction in cross-linking of PARP-1 with the iodoactamide-treated sample or with the alteration of solvent exposed cysteine residues 256 and 845 to alanines.
PARylation has no effect on cross-linking
To investigate whether PARylation of PARP-1 alters formation of the DPC, we conducted cross-linking experiments in the presence of NAD. Note that in the experiments described so far, NAD was not present in the cross-linking reaction mixtures. With an intact AP site-containing substrate, addition of NAD to the reaction mixture after cross-linking caused a change in the gel migration pattern of the labeled DNA and PARP-1 complex, consistent with PARylation of the cross-linked PARP-1/DNA complex (Figure 6a, lanes 1 and 2). This was confirmed by including poly(ADP-ribose) glycohydrolase (PARG) in the incubation along with NAD; the use of PARG enabled comparison of PARP-1 labeling with and without NAD (Figure 6a, lanes 1 and 3), and this revealed that cross-linking was similar in each case. PARG alone did not alter the cross-linked PARP-1 (Figure 6a, lane 4). Next, NAD was included in the cross-linking incubation, and then one portion of the reaction mixture was supplemented with PARG (Figure 6b). The reaction mixture prior to addition of PARG showed PARylation of the cross-linked PARP-1/DNA complex (Figure 6b, lane 1); this was confirmed by addition of PARG (Figure 6b, lane 2). Similar cross-linking was observed in incubations with and without NAD (Figure 6a and b). In addition, this experiment indicated that the strand break that accompanies cross-linking was sufficient to support activation of PARP-1 for PAR synthesis. Finally, experiments were conducted with the alternate substrate representing the pre-incised AP site-containing single-nucleotide gap with the 5′-dRP group; similar results were obtained with this substrate (Figure 6c and d). In summary, PARP-1 self-PARylation had no significant effect on cross-linking and conversely, cross-linking had no significant effect on PARylation. Addition of PARP-1 inhibitor (4-AN) to the cross-linking solution has no effect on the cross-linking of PARP-1 (Supplementary Figure S4).
Figure 6.
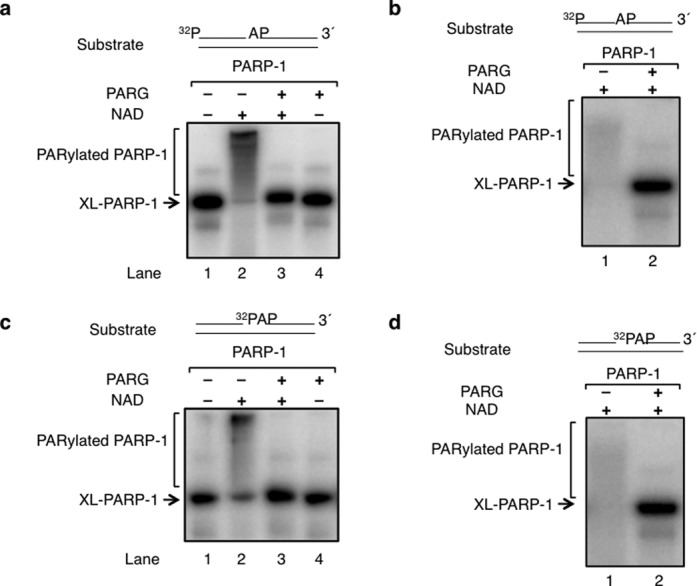
PARylation is independent of cross-linking of PARP-1. (a) A reaction mixture (40 μl) containing purified PARP-1 and 32P-labeled AP site-containing DNA was incubated on ice without NaBH4 under similar reaction conditions as in Figure 1. After this incubation, the reaction mixture was divided into four equal portions and each reaction mixture was supplemented as follows: lane 1, buffer; lane 2, 100 μM NAD; lane 3, 100 μM NAD and 100 ng PARG; and lane 4, 100 ng PARG. All the reaction mixtures also contained 7.5 mM MgCl2. Incubation was 15 min at 30°C. (b) A reaction mixture (20 μl) containing purified PARP-1, 32P-labeled AP site-containing DNA, 100 100 μM NAD and 7.5 mM MgCl2 was incubated as in panel (a). After this incubation, the reaction mixture was divided into two equal portions and each reaction mixture was supplemented as follows: lane 1, buffer, and lane 2, 100 ng PARG. Incubation was 15 min at 30°C. (c) and (d) The experiments were repeated as described in panels (a) and (b), except the DNA substrate had a nick and was 32P-labeled in the nick as depicted above the phosphorimage. The migration positions of cross-linked PARP-1 (XL-PARP-1) and PARylated PARP-1 are indicated.
Intrinsic cross-linking of PARP-1 to AP-DNA in cell extracts
Having observed intrinsic cross-linking of purified PARP-1 to AP site-containing DNA in vitro, we next tested for similar cross-linking using whole cell extract prepared from wild-type mouse fibroblasts (PARP-1+/+) and PARP-1 null mouse fibroblasts (PARP-1−/−). The extracts were incubated with 5′-end labeled uracil-containing DNA (Figure 7a), where endogenous uracil-DNA glycosylase produced the AP site, and incubations were without NaBH4 reduction. A prominent radiolabeled cross-linked product, consistent with PARP-1 labeling (band with molecular mass of ∼ 118 kDa), was observed in wild-type cell extract (Figure 7b, lane 1), but not in PARP-1 null cell extract (Figure 7b, lane 2). The migration of this complex was confirmed to be consistent with PARP-1 when the extract from PARP-1 null cells was supplemented with purified PARP-1 (Figure 7b, lane 3). In addition, immunoprecipitation of the reaction mixture in lane 1 with anti-PARP-1 IgG revealed a radiolabeled ∼ 118 kDa complex (Figure 7b, lane 5) that was not observed with non-immune IgG (Figure 7b, lane 4). The mass of the labeled complex was consistent with one molecule each of the ∼113 kDa PARP-1 monomer plus the DNA strand (∼ 5 kDa) after incision at C3′.
DNA-PARP-1 cross-link (DPC) formation in genomic DNA in mouse fibroblasts
It is known that mouse fibroblast cells exhibit strong cytotoxicity when treated with a monofunctional DNA methylating agent in combination with a PARP inhibitor (32,34,35). These methylating agents form lesions that are repaired by BER, and the repair intermediates are recognized by PARP-1 (24). To evaluate PARP-1 cross-linking to DNA in vivo, cells were treated with the alkylating agent MMS and the PARP inhibitor 4-AN, either alone or as the MMS and 4-AN combination (34,36). Then, the levels of PARP-1 DPCs were measured with the DNAzol-silica method used previously for measurement of Top1 DPCs (47,53,54). Samples of DNA cross-linked proteins were isolated, separated by SDS-PAGE, transferred to a nitrocellulose membrane and probed with antibody against PARP-1. Results of this analysis (Figure 7c) revealed negligible amounts of PARP-1 DPCs in the control samples, corresponding to either untreated cells (Figure 7c, lane 2) or cells treated with 4-AN alone (Figure 7c, lane 3). Among other points, this confirmed that the sample preparation method stripped proteins from DNA in the absence of covalent attachment. Therefore, the PARP-1 DPC observed in Figure 7(c) represents PARP-1 molecules covalently attached to DNA as opposed to simply DNA bound PARP-1 or chromatin-bound PARP-1, as was observed previously (34,35). Our results showed similar PARP-1 DPC formation in both the sample from cells treated with MMS and the sample from cells treated with the MMS and 4-AN combination (Figure 7c, lanes 4 and 5, respectively). These results indicated that PARP-1 is cross-linked to DNA in vivo when cells are treated with a highly cytotoxic dose (5 mM) of the alkylating agent MMS. In light of the present results, covalent attachment of PARP-1 to DNA during BER could also be a mechanism of PARP-1 immobilization. Furthermore, these results suggest that molecules of PARP-1 can be covalently attached to BER intermediates in cells during BER process, especially when the repair process becomes overwhelmed by a DNA damaging agent, is inhibited or is genetically deficient.
Visualization of PARP-1 in DNA fibers during BER
To visualize PARP-1 covalently attached to genomic DNA in cells undergoing BER, a form of DNA fiber-spread analysis was performed (48,49). The method was adjusted to detect protein covalently attached to DNA using antibody staining. As illustrated in Figure 8(a), abundant PARP-1 was observed along DNA fibers generated from cells treated with the MMS and 4-AN combination. Considerably, more PARP-1 was observed in these DNA fibers than those isolated from untreated control cells (Figure 8b). Under similar conditions, no PARP-1 signal was observed with PARP-1 null fibroblasts, as expected (Supplementary Figure S6a). However, wild-type fibroblasts treated with MMS also showed PARP-1 on DNA fibers; with cells treated with 4-AN alone very little PARP-1 was detected on DNA fibers (Supplementary Figure S6b). These results corroborate the findings described above (Figure 7c) indicating that PARP-1-mediated DPCs are formed during the response to MMS-induced DNA damage.
Figure 8.
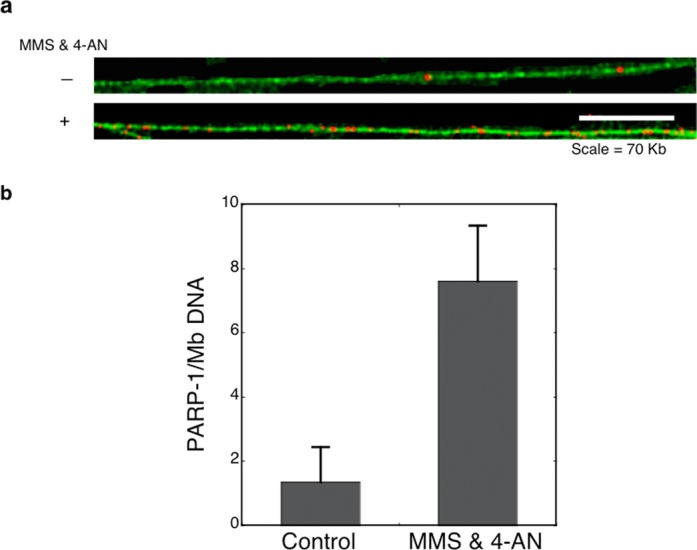
DNA fiber-spread analysis of PARP-1 covalently attached to genomic DNA. Shown is detection and enumeration of PARP-1 immunostaining in DNA fiber spreads isolated from either control untreated PARP-1+/+ cells or cells treated with the MMS and 4-AN combination. (a) Representative images of DNA fiber stained with YOYO-1 green fluorescent dye. PARP-1 sites were detected with anti-PARP-1 antibody followed by secondary and tertiary antibodies that were labeled with red fluorescence. Scale bar = 70 Kb. (b) Number of PARP-1 sites per Mb (106 nts) as determined fiber-spread analysis for cells under normal culture conditions and after exposure to MMS and 4-AN. Fiber analysis values were determined from analysis of two repeat experiments and from two different slides for each repeat. At least 200 Mb were analyzed per experimental condition. Error bars represent standard error of the mean.
DISCUSSION
We have shown that PARP-1 forms a covalent bond with DNA corresponding to BER intermediates containing the dRP/AP site; the covalent bond formation is accompanied by strand incision. The intrinsic covalent attachment of PARP-1 to DNA could have important biological implications, since these DPCs would pose a threat to genome stability and cell viability (55). For example, we found that in cells undergoing BER after alkylating agent treatment, the repair process enables PARP-1 to become cross-linked to DNA most likely at repair intermediates containing the 2′-deoxyribose.group (Figures 7 and 8). Should a replication fork collide with the PARP-1 DPC, replication fork progression may stall leading to fork collapse and DSB formation (34,56). Thus, the formation of these potentially lethal PARP-1 DPCs could have implications for the cell killing phenotype associated with MMS and PARP inhibitor treatment.
It had been proposed that PARP-1 remains bound on repair intermediates in PARP inhibitor treated cells, since under normal conditions PARylated PARP-1 is released from the repair intermediate or product by virtue of poly(ADP-ribose)/DNA charge repulsion (28,57). In light of the results described here, however, covalent attachment of PARP-1 to DNA likely accounts for at least a portion of the observed PARP-1 immobilization. On the other hand, a similar level of PARP-1 DPCs also were observed in MMS treated cells undergoing BER in the absence of PARP inhibitor (Figure 7c). The PARP-1 DPC level was much greater in cells treated with a cytotoxic dose of alkylating agent than in normal untreated cells (Figure 7c). Therefore, it is likely that under cellular conditions where the BER system becomes overwhelmed by DNA damage or interruption of the repair process by a PARP inhibitor, PARP-1-mediated DPCs are formed, and these DPCs represent an abortive repair event in cells.
In light of the above results, we asked whether cross-linked PARP-1 could be PARylated, and the results revealed that the cross-linked enzyme is fully capable of self-PARylation (Figure 6a and c). Conversely, PARylation did not block cross-linking (Figure 6b and d). The biological implications of these results are interesting to consider: it appears that PARP-1 under normal conditions is rapidly PARylated upon APE1 incision of AP DNA in the cell. However, cross-linking of PARP-1 is dependent on the presence of the AP site in DNA. For instance, if the AP site and 5′-dRP group are removed by APE1 and pol β, respectively, PARP-1 will be PARylated but will not be cross-linked. Under conditions where AP sites/5′-dRP groups persist in the DNA, PARP-1 may form cross-linked abortive repair complex; these conditions could be due to an overwhelming amount of DNA damage, inhibition of PARP-1 activity by PARP-1 inhibitors, imbalance in the repair enzymes, or mutations in repair enzymes, among others.
The cross-linking of PARP-1 to AP site-containing DNA must involve the redox (oxidation-reduction) capacity that is intrinsic to the protein. The cross-linking at the C1′ carbon of the deoxyribose group was accompanied by β-elimination at the C3′ carbon and strand incision. From our results it appears that these steps are connected. Therefore, dual lesions develop in the DNA during this process, resulting in protein adduction and strand incision. The Schiff base formed between a primary amine in PARP-1 and the C1′ atom of deoxyribose in the AP site must be reduced to make a covalent bond between PARP-1 and DNA. Hence, the presence of a hydride ion-generating groups in the enzyme in proximity to the Schiff base may serve in a reducing function and protein in turn will be oxidized. The results of iodoacetamide treatment of PARP-1 were consistent with such a possibility (Figure 5). This notion was further supported by a strong reduction in covalent cross-linking of altered forms of PARP-1 where the solvent exposed cysteine residues Cys256 and Cys845 were changed to Ala256 and Ala845.
Interestingly, the zinc finger-containing proteins that were considered as static structures are now well recognized as redox sensors, in which zinc release is coupled to conformational changes that control various enzymatic activities, DNA-binding interactions and molecular chaperone activity (58–60). In these proteins, reversible disulfide bond formation involves reactivity of cysteine thiol groups that are in close proximity resulting in structural rearrangements to affect the function of the protein. A combination of such features is found in cysteine-coordinating zinc centers in many proteins including PARP-1 (59). It is quite reasonable to assume that cysteine in the zinc finger 3 sub-domain of PARP-1 may have such a role, along with Cys256; more work is required to fully understand how such a redox mechanism occurs in coordination with Schiff base chemistry in PARP-1.
The adducted PARP-1 at the 3′ end of the incised DNA strand represents an abortive repair complex, as has been observed in the case of the abortive Top1 re-ligation reaction (61). In an attempt to investigate how the abortive PARP-1 complex could be repaired in cells, we treated the DNA-PARP-1 cross-linked complex with AP endonuclease 1 and tyrosyl-DNA phosphodiesterase 1, as both enzymes have AP site incision activity. These experiments failed to show any incision activity at the AP site (data not shown), and it appears that these enzymes had difficulty in processing such a long peptide. Therefore, it may be interesting to investigate proteasome-mediated degradation of cross-linked PARP-1 in cells treated with MMS and PARP-1 inhibitors (62).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank W.A. Beard, E.D. Brown, R.E. London, W.C. Copeland and A.S. Serianni for critical reading and helpful discussion of the manuscript.
FUNDING
Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences [Z01ES050158, Z01 ES050159] (in part). Funding for open access charge: Laboratory of Structural Biology, NIEHS.
Conflict of interest. None declared.
REFERENCES
- 1.Kraus W.L. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008;20:294–302. doi: 10.1016/j.ceb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Amours D., Desnoyers S., D'Silva I., Poirier G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 3.Lord C.J., Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber V., Dantzer F., Ame J.C., de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 5.Amé J.C., Spenlehauer C., de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 6.Ménissier de Murcia J., Ricoul M., Tartier L., Niedergang C., Huber A., Dantzer F., Schreiber V., Amé J.C., Dierich A., LeMeur M., et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22:2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hassa P.O., Hottiger M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 8.Dantzer F., de La Rubia G., Ménissier de Murcia J., Hostomsky Z., de Murcia G., Schreiber V. Base excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- 9.Carrozza M.J., Stefanick D.F., Horton J.K., Kedar P.S., Wilson S.H. PARP inhibition during alkylation-induced genotoxic stress signals a cell cycle checkpoint response mediated by ATM. DNA Repair. 2009;8:1264–1272. doi: 10.1016/j.dnarep.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horton J.K., Stefanick D.F., Kedar P.S., Wilson S.H. ATR signaling mediates an S-phase checkpoint after inhibition of poly(ADP-ribose) polymerase activity. DNA Repair. 2007;6:742–750. doi: 10.1016/j.dnarep.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruf A., Ménissier de Murcia J., de Murcia G., Schulz G.E. Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proc. Natl. Acad. Sci. U.S.A. 1996;93:7481–7485. doi: 10.1073/pnas.93.15.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eustermann S., Videler H., Yang J.C., Cole P.T., Gruszka D., Veprintsev D., Neuhaus D. The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. J. Mol. Biol. 2011;407:149–170. doi: 10.1016/j.jmb.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tao Z., Gao P., Hoffman D.W., Liu H.W. Domain C of human poly(ADP-ribose) polymerase-1 is important for enzyme activity and contains a novel zinc-ribbon motif. Biochemistry. 2008;47:5804–5813. doi: 10.1021/bi800018a. [DOI] [PubMed] [Google Scholar]
- 14.Pion E., Bombarda E., Stiegler P., Ullmann G.M., Mely Y., de Murcia G., Gerard D. Poly(ADP-ribose) polymerase-1 dimerizes at a 5’ recessed DNA end in vitro: a fluorescence study. Biochemistry. 2003;42:12409–12417. doi: 10.1021/bi0344432. [DOI] [PubMed] [Google Scholar]
- 15.Altmeyer M., Messner S., Hassa P.O., Fey M., Hottiger M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009;37:3723–3738. doi: 10.1093/nar/gkp229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langelier M.F., Planck J.L., Roy S., Pascal J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012;336:728–732. doi: 10.1126/science.1216338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huambachano O., Herrera F., Rancourt A., Satoh M.S. Double-stranded DNA binding domain of poly(ADP-ribose) polymerase-1 and molecular insight into the regulation of its activity. J. Biol. Chem. 2011;286:7149–7160. doi: 10.1074/jbc.M110.175190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mansoorabadi S.O., Wu M., Tao Z., Gao P., Pingali S.V., Guo L., Liu H.W. Conformational activation of poly(ADP-ribose) polymerase-1 upon DNA binding revealed by small-angle X-ray scattering. Biochemistry. 2014;53:1779–1788. doi: 10.1021/bi401439n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Vos M., Schreiber V., Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem. Pharmacol. 2012;84:137–146. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 20.Klungland A., Rosewell I., Hollenbach S., Larsen E., Daly G., Epe B., Seeberg E., Lindahl T., Barnes D.E. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindahl T. DNA repair enzymes. Annu. Rev. Biochem. 1982;51:61–87. doi: 10.1146/annurev.bi.51.070182.000425. [DOI] [PubMed] [Google Scholar]
- 22.Lindahl T., Wood R.D. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 23.Demple B., Herman T., Chen D.S. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl. Acad. Sci. U.S.A. 1991;88:11450–11454. doi: 10.1073/pnas.88.24.11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khodyreva S.N., Prasad R., Ilina E.S., Sukhanova M.V., Kutuzov M.M., Liu Y., Hou E.W., Wilson S.H., Lavrik O.I. Apurinic/apyrimidinic (AP) site recognition by the 5’-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1) Proc. Natl. Acad. Sci. U.S.A. 2010;107:22090–22095. doi: 10.1073/pnas.1009182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavrik O.I., Prasad R., Sobol R.W., Horton J.K., Ackerman E.J., Wilson S.H. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J. Biol. Chem. 2001;276:25541–25548. doi: 10.1074/jbc.M102125200. [DOI] [PubMed] [Google Scholar]
- 26.El-Khamisy S.F., Masutani M., Suzuki H., Caldecott K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mortusewicz O., Amé J.C., Schreiber V., Leonhardt H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007;35:7665–7675. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satoh M.S., Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 29.Horton J.K., Stefanick D.F., Naron J.M., Kedar P.S., Wilson S.H. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest after DNA methylating agent exposure. J. Biol.Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- 30.Ferraris D.V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010;53:4561–4584. doi: 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- 31.Horton J.K., Watson M., Stefanick D.F., Shaughnessy D.T., Taylor J.A., Wilson S.H. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008;18:48–63. doi: 10.1038/cr.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horton J.K., Wilson S.H. Predicting enhanced cell killing through PARP inhibition. Mol. Cancer Res. 2013;11:13–18. doi: 10.1158/1541-7786.MCR-12-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heacock M., Poltoratsky V., Prasad R., Wilson S.H. Evidence for abasic site sugar phosphate-mediated cytotoxicity in alkylating agent treated Saccharomyces cerevisiae. PLoS One. 2012;7:e47945. doi: 10.1371/journal.pone.0047945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kedar P.S., Stefanick D.F., Horton J.K., Wilson S.H. Increased PARP-1 association with DNA in alkylation damaged, PARP-inhibited mouse fibroblasts. Mol. Cancer Res. 2012;10:360–368. doi: 10.1158/1541-7786.MCR-11-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murai J., Huang S.Y., Das B.B., Renaud A., Zhang Y., Doroshow J.H., Ji J., Takeda S., Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heacock M.L., Stefanick D.F., Horton J.K., Wilson S.H. Alkylation DNA damage in combination with PARP inhibition results in formation of S-phase-dependent double-strand breaks. DNA Repair. 2010;9:929–936. doi: 10.1016/j.dnarep.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gassman N.R., Stefanick D.F., Kedar P.S., Horton J.K., Wilson S.H. Hyperactivation of PARP triggers nonhomologous end-joining in repair-deficient mouse fibroblasts. PLoS One. 2012;7:e49301. doi: 10.1371/journal.pone.0049301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woodhouse B.C., Dianova II, Parsons J.L., Dianov G.L. Poly(ADP-ribose) polymerase-1 modulates DNA repair capacity and prevents formation of DNA double strand breaks. DNA Repair. 2008;7:932–940. doi: 10.1016/j.dnarep.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 39.Pachkowski B.F., Tano K., Afonin V., Elder R.H., Takeda S., Watanabe M., Swenberg J.A., Nakamura J. Cells deficient in PARP-1 show an accelerated accumulation of DNA single strand breaks, but not AP sites, over the PARP-1-proficient cells exposed to MMS. Mutat. Res. 2009;671:93–99. doi: 10.1016/j.mrfmmm.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prasad R., Williams J.G., Hou E.W., Wilson S.H. Pol β associated complex and base excision repair factors in mouse fibroblasts. Nucleic Acids Res. 2012;40:11571–11582. doi: 10.1093/nar/gks898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beard W.A., Wilson S.H. Purification and domain-mapping of mammalian DNA polymerase β. Methods Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- 42.Slupphaug G., Eftedal I., Kavli B., Bharati S., Helle N.M., Haug T., Levine D.W., Krokan H.E. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry. 1995;34:128–138. doi: 10.1021/bi00001a016. [DOI] [PubMed] [Google Scholar]
- 43.Strauss P.R., Beard W.A., Patterson T.A., Wilson S.H. Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem. 1997;272:1302–1307. doi: 10.1074/jbc.272.2.1302. [DOI] [PubMed] [Google Scholar]
- 44.Langelier M.F., Planck J.L., Servent K.M., Pascal J.M. Purification of human PARP-1 and PARP-1 domains from Escherichia coli for structural and biochemical analysis. Methods Mol. Biol. 2011;780:209–226. doi: 10.1007/978-1-61779-270-0_13. [DOI] [PubMed] [Google Scholar]
- 45.Gagnon S.N., Desnoyers S. Single amino acid substitution enhances bacterial expression of PARP-4D214A. Mol. Cell. Biochem. 2003;243:15–22. doi: 10.1023/a:1021645327079. [DOI] [PubMed] [Google Scholar]
- 46.Piersen C.E., Prasad R., Wilson S.H., Lloyd R.S. Evidence for an imino intermediate in the DNA polymerase β deoxyribose phosphate excision reaction. J. Biol. Chem. 1996;271:17811–17815. doi: 10.1074/jbc.271.30.17811. [DOI] [PubMed] [Google Scholar]
- 47.Barker S., Murray D., Zheng J., Li L., Weinfeld M. A method for the isolation of covalent DNA-protein crosslinks suitable for proteomics analysis. Anal. Biochem. 2005;344:204–215. doi: 10.1016/j.ab.2005.06.039. [DOI] [PubMed] [Google Scholar]
- 48.Chastain P.D., 2nd, Nakamura J., Rao S., Chu H., Ibrahim J.G., Swenberg J.A., Kaufman D.G. Abasic sites preferentially form at regions undergoing DNA replication. FASEB J. 2010;24:3674–3680. doi: 10.1096/fj.09-145276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stewart J.A., Wang F., Chaiken M.F., Kasbek C., Chastain P.D., 2nd, Wright W.E., Price C.M. Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J. 2012;31:3537–3549. doi: 10.1038/emboj.2012.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prasad R., Beard W.A., Strauss P.R., Wilson S.H. Human DNA polymerase β deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J. Biol. Chem. 1998;273:15263–15270. doi: 10.1074/jbc.273.24.15263. [DOI] [PubMed] [Google Scholar]
- 51.Zharkov D.O., Rosenquist T.A., Gerchman S.E., Grollman A.P. Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem. 2000;275:28607–28617. doi: 10.1074/jbc.M002441200. [DOI] [PubMed] [Google Scholar]
- 52.Clark N.J., Kramer M., Muthurajan U.M., Luger K. Alternative modes of binding of poly(ADP-ribose) polymerase 1 to free DNA and nucleosomes. J. Biol. Chem. 2012;287:32430–32439. doi: 10.1074/jbc.M112.397067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barker S., Weinfeld M., Zheng J., Li L., Murray D. Identification of mammalian proteins cross-linked to DNA by ionizing radiation. J. Biol. Chem. 2005;280:33826–33838. doi: 10.1074/jbc.M502477200. [DOI] [PubMed] [Google Scholar]
- 54.Barker S., Weinfeld M., Murray D. DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat. Res. 2005;589:111–135. doi: 10.1016/j.mrrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 55.Connelly J.C., Leach D.R. Repair of DNA covalently linked to protein. Mol. Cell. 2004;13:307–316. doi: 10.1016/s1097-2765(04)00056-5. [DOI] [PubMed] [Google Scholar]
- 56.Pines A., Mullenders L.H., van Attikum H., Luijsterburg M.S. Touching base with PARPs: moonlighting in the repair of UV lesions and double-strand breaks. Trends Biochem. Sci. 2013;38:321–330. doi: 10.1016/j.tibs.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 57.Lindahl T., Satoh M.S., Poirier G.G., Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- 58.Maret W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid. Redox Signal. 2006;8:1419–1441. doi: 10.1089/ars.2006.8.1419. [DOI] [PubMed] [Google Scholar]
- 59.Cremers C.M., Jakob U. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 2013;288:26489–26496. doi: 10.1074/jbc.R113.462929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilcox D.E., Schenk A.D., Feldman B.M., Xu Y. Oxidation of zinc-binding cysteine residues in transcription factor proteins. Antioxid. Redox Signal. 2001;3:549–564. doi: 10.1089/15230860152542925. [DOI] [PubMed] [Google Scholar]
- 61.Pourquier P., Pilon A.A., Kohlhagen G., Mazumder A., Sharma A., Pommier Y. Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps. Importance of DNA end phosphorylation and camptothecin effects. J. Biol. Chem. 1997;272:26441–26447. doi: 10.1074/jbc.272.42.26441. [DOI] [PubMed] [Google Scholar]
- 62.Debethune L., Kohlhagen G., Grandas A., Pommier Y. Processing of nucleopeptides mimicking the topoisomerase I-DNA covalent complex by tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 2002;30:1198–1204. doi: 10.1093/nar/30.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.