

Abstract
KCNJ5 mutations were recently described in primary hyperaldosteronism (PH or Conn’s syndrome). The frequency of these mutations in PH and the way KCNJ5 defects cause disease remain unknown. A total of 53 patients with PH have been seen at the National Institutes of Health over the last 12 years. Their peripheral and tumor DNAs (the latter from 16 that were operated) were screened for KCNJ5 mutations; functional studies on the identified defects were performed after transient transfection. Only two mutations were identified, and both in the tumor DNA only. There were no germline sequencing defects in any of the patients except for known synonymous variants of the KCNJ5 gene. One mutation was the previously described c.G451C alteration; the other was a novel one in the same codon: c.G451A; both lead to the same amino acid substitution (G151R) in the KCNJ5 protein. Functional studies confirmed previous findings that both mutations caused loss of channel selectivity and a positive shift in the reversal potential. In conclusion, the KCNJ5 protein was strongly expressed in the zona glomerulosa of normal adrenal glands but showed variable expression in the aldosterone-producing adenomas with and without mutation. The rate of KCNJ5 mutations among patients with PH and/or their tumors is substantially lower than what was previously reported. The G151R amino acid substitution appears to be the most frequent one so far detected in PH, despite additional nucleotide changes. The mutation causes loss of this potassium channel’s selectivity and may assist in the design of new therapies for PH.
Introduction
Primary hyperaldosteronism (PH) is now considered as one of the more common causes of secondary hypertension (HTN). While older data suggested that PH is rare, with an estimated prevalence of <1% of all patients with HTN, subsequent data indicated that it may actually be present in as many as 5–15% of patients with HTN. It may be present in an even greater percentage of patients with treatment-resistant HTN (Mulatero et al. 2004, Douma et al. 2008). The two principal causes of PH are bilateral adrenal hyperplasia, which accounts for approximately two-thirds of the cases, and aldosterone-producing adenomas (APAs), which constitute about one-third of the cases (Schirpenbach & Reincke 2007). Less frequent causes include unilateral hyperplasia, aldosterone-producing carcinoma, hereditary glucocorticoid-remediable aldosteronism (also known as familial hyperaldosteronism type I), and familial hyperaldosteronism type II.
Recently, Choi et al. (2011) have reported two different somatic mutations in the KCNJ5 gene encoding the potassium channel in eight out of 22 APAs and a third heterozygous germline mutation in a family (a father and two daughters) with aldosterone-producing adrenal hyperplasia. Using electrophysiological studies, they also demonstrated that these mutations result in loss of channel selectivity, with increased sodium conductance, leading to membrane depolarization, opening of voltage-activated Ca2+ channels, activation of calcium signaling pathway, and finally, increased aldosterone production (Choi et al. 2011). KCNJ5 encodes the G-protein-coupled inwardly rectifying K+ channel (Kir3.4) that exists as homotetramer or heterotetramer with Kir3 (Ishihara et al. 2009). Recently, KCNJ5 missense mutations have been identified in a patient with atrial fibrillation (Calloe et al. 2007) and in a family with autosomal dominant long QT syndrome (Yang et al. 2010).
In this study, we report the sequencing results from 53 patients with PH that have been admitted to the National Institutes of Health (NIH) Clinical Research Center over the last 12 years. Unlike what Choi et al. has found, the frequency of KCNJ5 mutations in this population was rather low. The data have implication for genetic counseling and the care of patients with PH.
Subjects and methods
Patients and tumor samples
The study was approved by the Institutional Review Board of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. All participants gave written informed consent. Blood DNA from 53 patients (41 females and 12 males) diagnosed with PH over the last 12 years was tested; DNA samples from 16 APAs that were removed from these patients were also studied. PH was diagnosed based on the criteria published from the Endocrine Society (Funder et al. 2008). When APA was considered as the most likely diagnosis, unilateral adrenalectomy was performed. When possible, tissue was collected at surgery and processed for routine histopathology and immunohistochemistry (IHC) following formalin fixation and paraffin embedment; additional fragments were frozen at −70 °C until nucleic acid extraction. DNA was extracted from blood, frozen, and archived tissue samples using standard methods (Qiagen).
Sequencing studies
Mutation analysis for exons, exon–intron boundaries, and flanking intronic regions was performed for the KCNJ5 gene; the two exons of KCNJ5 were amplified and sequenced by PCR-based bidirectional Sanger sequencing. Primers were designed using the online PCR primer design tools Primer 3. Exon 2 was divided into three parts (1, 2, and 3) as it spans 947 bp (Supplementary Table 1, see section on supplementary data given at the end of this article). All amplified samples were examined by agarose gel electrophoresis to confirm successful amplification of each exon. Direct sequencing of the purified fragments was then done using the Genetic Sequencer ABI3100 Applied Biosystems apparatus (Applied Biosystems Inc, Foster City, CA, USA). Sequences were analyzed using Vector NTI 10 Software (Invitrogen).
Immunohistochemistry
IHC was performed at Histoserve, Inc. (Germantown, MD, USA) using standard procedures. Slides used for KCNJ5 staining came from patient ADT 74.02 harboring a KCNJ5 mutation and from an APA of a patient negative for any known KCNJ5 mutation. The following primary antibody was used: KCNJ5, ab81114, rabbit polyclonal IgG (Abcam, Inc.).
Electrophysiology
HEK 293T cells were co-transfected with either wild-type KCNJ5 (0.5 μg) or its mutations and KCNJ3 (0.5 μg) in 35 mm dishes using Fugene 6 system (Roche Applied Science). Empty vector was used as a control. Recordings were made 16 h after transfection. Coverslips containing transfected HEK 293T cells were submerged in the recording chamber and exposed to a continuous flow of extracellular recording solution (ACSF) consisting of the following (mM): 140 NaCl, 5 KCl, 1.8 CaCl2, 1.8 MgCl2, and 10 HEPES. BaCl2 (1 mM) was used to assess channel selectivity. Choline (140 mM) was substituted for 140 mM NaCl in the presence of BaCl2. An infrared differential interference contrast videomicroscopy system (Zeiss Instruments, Oberkochen, Germany; Diagnostic Instruments, Sterling Heights, MI, USA) was used to visualize cells. Patch pipettes were pulled from thick-walled borosilicate glass (Warner Instruments) to achieve a tip resistance of 2–5 MΩ. Pipettes were filled with an internal solution containing 140 mM KCl, 5 mM HEPES, 1 mM EGTA, 1 mM CaCl2, and 4 mM MgCl2. Whole-cell voltage clamp recordings were made using a Multiclamp 700B amplifier (Molecular Devices Sunnyvale, CA, USA) and Clampex 10.1 Software (Molecular Devices). Signals were digitized at 10 kHz with a Digidata 1440A (Molecular Devices) and filtered at 2 kHz. Currents were elicited by delivering a second long pulse from −100 to 60 mV in 20 mV increments from a 0 mV holding. This protocol was repeated three times for each cell. Leak currents were recorded from empty vector controls and subtracted from each recording. A linear fit between averaged least negative and positive currents from each cell was used to estimate the reversal potential. All recordings were performed at room temperature. Recordings were analyzed using Clampfit 10.1 (Molecular Devices) and Microsoft Excel. Statistical significance was evaluated using Student’s t-test (unpaired, two tails). The original KCNJ5 constructs were kindly provided by Dr Lifton, Departments of Genetics and Internal Medicine, Howard Hughes Medical Institute, Yale University School of Medicine, USA.
Results
DNA and allelic studies
We identified two somatic mutations of the KCNJ5 gene among 16 APAs that we sequenced, both at position 451 bp. In one case, the G>A substitution at nucleotide G 451 (Fig. 1a) is the same reported by Choi et al. (2011). However, in the second case, we identified a G>C substitution at the same position (Fig. 1b), which, as far as we know, is a novel mutation. Both of them caused the substitution of glycine (G) to an arginine (R) at residue 151(G151R). No germline mutations were detected in the 53 patients that we sequenced. We identified several synonymous variations in both exons 2 and 3 of the gene (Table 1).
Figure 1.

Chromatograms showing the genomic DNA sequence of KCNJ5 exon 2 of (a) peripheral blood (b) DNA and tumor DNA of patients ADT 74.02 (a) and ADT 78.03 (b), who carried the c.G451A and c.G151RC mutations in their APAs. These changes were identified only in tumor DNA and not in peripheral DNA.
Table 1.
Germline and somatic variations identified in 53 patients with aldosterone-producing adenomas (APAs) and in 16 matched APAs
| Variations | Number of cases | Type |
|---|---|---|
| c.G451A, p.151G>R | 1 | Somatic |
| c.G451C, p.151G>R | 1 | Somatic |
| c.171T>C (rs6590357) | 53 | Germline |
| c.810T>G (rs7118824) | 53 | Germline |
| c.834T>C (rs7118833) | 53 | Germline |
| c.844C>G (rs7102584) | 53 | Germline |
| c.938-10 G>A (rs4937391) | 10 | Germline |
Electrophysiological studies
We examined the properties of normal and mutant KCNJ5 channels by co-expressing them with KCNJ3 in HEK 293T cells. In whole-cell voltage clamp recordings, we measured currents at voltages ranging between −100 and +60 mV. We observed an inwardly rectifying current with a reversal potential of −21.0±3.7 mV in the wild-type channels (Fig. 2). This current was eliminated in the presence of barium (data not shown). As previously reported by Choi et al. (2011) mutations T158A, G151R_2 (G>A), and L168R caused a loss of channel selectivity (i.e. decrease in barium inhibition) and a positive shift in the reversal potential. In addition, the newly discovered mutation G151R_1 (G>C) showed similar loss of channel selectivity and less negative reversal potentials; while the substitution of choline for sodium in the bath solution did not affect the wild-type current, it significantly reduced the current in all mutant channels (data not shown). These findings indicate that the mutations near the channel pore lead to increased sodium conductance and loss of K+ ion selectivity.
Figure 2.
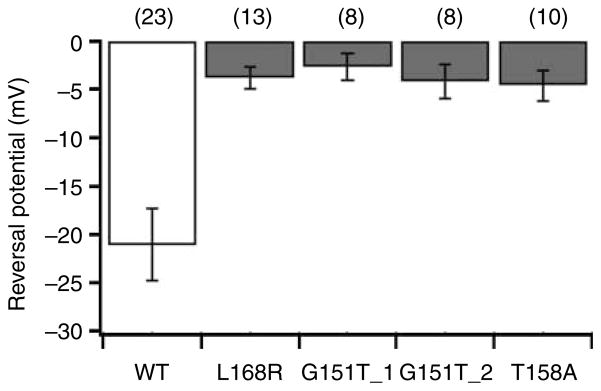
KCNJ5 mutations result in depolarization of the current reversal potential. Reversal potentials are plotted for WT KCNJ5/KCNJ3 channels and each KCNJ5 mutation/KCNJ3. Normal sequence (wild-type) channels (open bar) show a negative reversal potential. Mutant channels (gray bars) show less negative reversal potentials (P<0.05 in each case), suggesting a loss of ion selectivity. Numbers in parentheses are the number of cells recorded. G151R1:G/A, G151R2:G/C.
Immunohistochemistry
Staining of APAs from patients with and without a KCNJ5 mutation demonstrated a selective staining of the glomerulosa cells in the outermost cell layers of the normal adrenal cortex but not in the center of the tumor (Fig. 3a); staining was positive and strong both in the zona glomerulosa and in the APA tumor sample from a patient without a KCNJ5 mutation (Fig. 3b).
Figure 3.
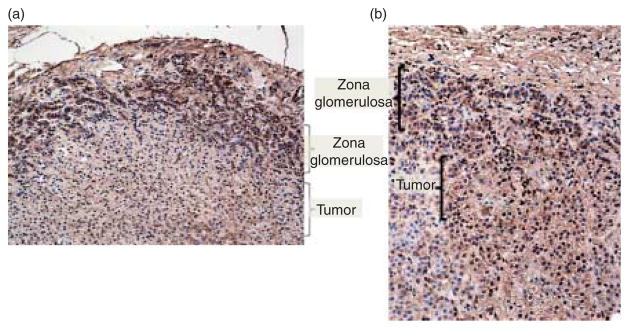
KCNJ5 staining of adrenal samples from patients with and without a KCNJ5 mutation. Significant staining with the KCNJ5 antibody was observed at the outermost cells (zona glomerulosa) of the normal adrenal cortex but not in the tumor (a); staining was positive both in the zona glomerulosa and in the tumor cells from a patient with an APA but without a KCNJ5 mutation (b).
Clinical data on patients with PH and somatic KCNJ5 mutations
The clinical characteristics of the patients identified with the two mutations are as follows: i) case 1 (ADT 74.02): the patient’s tumor carried the novel mutation c.G451A, p.151G>R; she was a 47-year-old female who was found to have hypokalemia (2.7 mmol/l) on routine blood tests. Her blood pressure was found to be elevated at 140/92 mmHg. After the hypokalemia was corrected with potassium supplements, she underwent screening for PH. She had a high aldosterone to plasma renin activity (PRA) ratio (ARR) of 70. Confirmatory testing with a normal saline suppression test was performed; at 4 h, the levels of aldosterone remained elevated at 1.80 nmol/l. A computed tomography scan of the adrenals showed a 1.5 cm left adrenal mass. Adrenal venous sampling (AVS) was performed that showed lateralization of aldosterone production on the left side. The patient underwent a left laparoscopic adrenalectomy. Histology of the excised gland revealed a 1.5 cm ‘golden yellow’ adenoma. Postoperatively her blood pressure decreased to 126/76 mmHg and her potassium normalized without potassium supplementation. One month after adrenalectomy, the aldosterone levels and PRA returned to normal. ii) Case 2 (ADT 78.03): in this patient’s tumor, the previously reported KCNJ5 mutation was identified (c.G451C, p.151G>R). She was a 36-year-old Vietnamese female who was diagnosed with PH during pregnancy and suffered a spontaneous abortion at 6 months of gestation. She was found to have proteinuria; a kidney biopsy was performed that revealed focal segmental glomerulosclerosis. She required four antihypertensive agents at maximum doses to control her blood pressure. Computed tomography of the adrenals revealed a 2.3 cm left adrenal mass. Biochemical work up showed a high ARR of 796, suppressed PRA, and hypokalemia. Confirmatory testing was performed with a normal saline suppression test that showed nonsuppressed aldosterone levels. AVS was performed which showed lateralization of aldosterone production on the left side. The patient underwent a left laparoscopic adrenalectomy. Histology of the excised gland showed a homogeneous yellow adenoma measuring 2.5×1.5×1.5 cm. Post-operatively the hypokalemia resolved and her blood pressure improved, requiring a single agent (instead of four) for adequate blood pressure control. At her 2-month follow-up visit, her PRA and aldosterone levels had returned to normal.
Discussion
We report the sequencing data of KCNJ5 gene in 53 patients diagnosed with PH; 16 of the 53 patients had an APA and underwent surgery. We sequenced all 53 germline DNA samples and the 16 matched APAs. We did not identify any germline mutations except for synonymous variations (Table 1). However, we identified two somatic mutations, both at the G451 nucleotide changing 151 G>R. Choi et al. reported the G>A substitution at position c.451(4); the G>C substitution at the same position is a novel mutation. Despite the fact that both mutations result in the same amino acid change (G>R), we wanted to confirm that it had the same functional effect on ion function after cDNA transfection. We performed electrophysiological studies that proved that this mutation has the same effect on ion selectivity and membrane depolarization as the other reported mutations. The G151 amino acid lies in a highly conserved position through species and is the first glycine of the GYG motif of the K+ channel selectivity filter. It has been shown that mutations in and near K+channel selectivity filter can alter channel selectivity and produce nonselective cation channels (Dibb et al. 2003). The fact that both our mutations concentrated on amino acid G151 indicates that this residue is a hot spot of mutation in APAs.
In their report, Choi et al. identified eight somatic mutations in 22 tumors (36.36%). We detected two somatic mutations in 16 tumors (12.5%). The difference in the rate of mutations found in KCNJ5 between the two studies is quite interesting. One explanation could be selection bias, as cases referred to the NIH are often complicated and maybe due to germline (rather than somatic) mutations; however, as most of our patients are sporadic, we do not expect a high rate of germline defects consistent with other cohorts (Zennaro & Jeunemaitre 2011).
According to Choi et al. (2011), the patients harboring mutations in KCNJ5 were young (under the age of 35 years), had a tumor size >2 cm, and usually presented with hypokalemia. These features were also present in our patients: both patients were young at presentation (36 and 45 years respectively), the tumors were between 1.5 and 2.5 cm, and both patients presented with severe hypokalemia.
Previous studies have shown a relatively low rate of genetic aberrations in APAs compared with adrenocortical cancers (Sidhu et al. 2002). Similarly, in the 22 adenomas analyzed by Choi et al. (2011), the rate of allelic losses was insignificant. On our genomic sequencing results, we identified both alleles (normal and mutant) in approximately equal intensity.
Recently, KCNJ5 missense mutations have been identified in a patient with atrial fibrillation and in a family with autosomal dominant long QT syndrome (Calloe et al. 2007, Yang et al. 2010). There are no reports regarding heart rhythm and conductivity in the patients harboring a somatic or germline KCNJ5 mutation. Evaluation of the effect of KCNJ5 mutations on heart rhythm and conductivity in the patients harboring a germline mutation probably deserves further investigation.
In conclusion, we have identified two somatic KCNJ5 mutations in 16 human APAs studied, one of them is novel. Both mutations result in a depolarization of current reversal potential and loss of channel selectivity. The prevalence of KCNJ5 mutations in PH was much lower in the NIH cohort than the one originally reported by Choi et al. Screening for KCNJ5 mutations in a larger cohort will give a better estimation of the prevalence of somatic mutations of this particular gene in APAs. KCNJ5 defects may lead the way for new, molecularly designed therapies for PH.
Supplementary Material
Acknowledgments
Funding
This work was entirely supported by the Intramural Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD).
Footnotes
This is linked to the online version of the paper at http://dx.doi.org/10.1530/ERC-12-0022.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Calloe K, Ravn LS, Schmitt N, Sui JL, Duno M, Haunso S, Grunnet M, Svendsen JH, Olesen SP. Characterizations of a loss-of-function mutation in the Kir3.4 channel subunit. Biochemical and Biophysical Research Communications. 2007;364:889–895. doi: 10.1016/j.bbrc.2007.10.106. [DOI] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768–772. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibb KM, Rose T, Makary SY, Claydon TW, Enkvetchakul D, Leach R, Nichols CG, Boyett MR. Molecular basis of ion selectivity, block, and rectification of the inward rectifier Kir3.1/Kir3.4 K(+) channel. Journal of Biological Chemistry. 2003;278:49537–49548. doi: 10.1074/jbc.M307723200. [DOI] [PubMed] [Google Scholar]
- Douma S, Petidis K, Doumas M, Papaefthimiou P, Triantafyllou A, Kartali N, Papadopoulos N, Vogiatzis K, Zamboulis C. Prevalence of primary hyperaldosteronism in resistant hypertension: a retrospective observational study. Lancet. 2008;371:1921–1926. doi: 10.1016/S0140-6736(08)60834-X. [DOI] [PubMed] [Google Scholar]
- Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, Young WF, Jr, Montori VM Endocrine Society. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism. 2008;93:3266–3281. doi: 10.1210/jc.2008-0104. [DOI] [PubMed] [Google Scholar]
- Ishihara K, Yamamoto T, Kubo Y. Heteromeric assembly of inward rectifier channel Kir2.1 with Kir3.4. Biochemical and Biophysical Research Communications. 2009;380:832–837. doi: 10.1016/j.bbrc.2009.01.179. [DOI] [PubMed] [Google Scholar]
- Mulatero P, Stowasser M, Loh KC, Fardella CE, Gordon RD, Mosso L, Gomez-Sanchez CE, Veglio F, Young WF., Jr Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. Journal of Clinical Endocrinology and Metabolism. 2004;89:1045–1050. doi: 10.1210/jc.2003-031337. [DOI] [PubMed] [Google Scholar]
- Schirpenbach C, Reincke M. Primary aldosteronism: current knowledge and controversies in Conn’s syndrome. Nature Clinical Practice. Endocrinology & Metabolism. 2007;3:220–227. doi: 10.1038/ncpendmet0430. [DOI] [PubMed] [Google Scholar]
- Sidhu S, Marsh DJ, Theodosopoulos G, Phillips J, Bambach CP, Campell P, Magarey CJ, Russel CF, Schulte KM, Roher HD, et al. Comparative genomic hybridization analysis of adrenocortical tumors. Journal of Clinical Endocrinology and Metabolism. 2002;87:3467–3474. doi: 10.1210/jc.87.7.3467. [DOI] [PubMed] [Google Scholar]
- Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, Olesen SP, Rasmussen HB, Ellinor PT, Gao L, et al. Identification of a Kir3.4 mutation in congenital long QT syndrome. American Journal of Human Genetics. 2010;86:872–880. doi: 10.1016/j.ajhg.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zennaro MC, Jeunemaitre X. Mutations in KCNJ5 gene cause hyperaldosteronism. Circulation Research. 2011;108:1417–1418. doi: 10.1161/RES.0b013e318224a359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.