

Gymnodimine (1, Figure 1) is a member of the spirocyclic imine family of marine toxins initially isolated from oysters collected off the coast of New Zealand. The gross structure was initially reported by Yasumoto in 1995[i] and subsequently, Munro and Blunt reported the relative and absolute stereochemistry elucidated through X-ray crystallographic analysis of a reduced, N-acylated derivative.[ii] This toxin is produced by the dinoflagellate Karenia selliforms (formerly Gymnodinium selliforme) and is active in the mouse bioassay for neurotoxic shellfish poisoning.[iii] Recently, gymnodimine was found to sensitize neurons to the effects of okadaic acid[iv] and there is evidence that it binds to a subset of muscle nicotinic acetylcholine receptors.[v] Two additional analogs, differing only by an allylic oxidation at the C17–C18 olefin, were isolated and named gymnodimine B (2) and C (3), respectively.[vi] Other members of this growing family of spirocyclic imine toxins include the pinnatoxins,[vii] spirolides,[viii] pteriatoxins,[ix] prorocentrolide,[x] and spiro-prorocentrimine.[xi]
Figure 1.
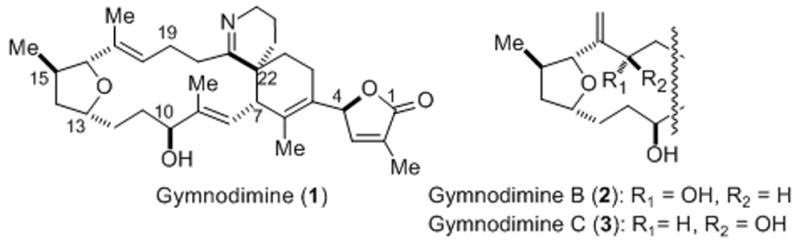
Structures of known members of the gymnodimine family of spirocyclic imine marine toxins.
This family of spirocyclie-containing marine toxins has inspired intense synthetic efforts[xii] that have culminated in total or formal syntheses of the pinnatoxins and pteriatoxins.[xiii] However, the total synthesis of gymnodimine still remains elusive.[xiv] The seemingly simpler architecture of gymnodimine compared to other members of this family conceals subtle, challenging structural elements, in particular the known labile butenolide adding to the challenge of a total synthesis.[xv] Herein, we describe the first total synthesis of (−)-gymnodimine that provides suitable intermediates for eventual production of an enzyme-linked immunosorbent assay (ELISA) for gymnodimine detection and also further mode of action studies.[xvi]
Our synthetic plan called for a convergent coupling of the spirolactam 5 with a hypothetical, dual reactivity, tetrahydrofuran 4 (Figure 2). A Nozaki–Hiyama–Kishi (NHK) macrocyclization,[xvii] was initially envisioned for the proposed merging of C9 and C10 (gymnodimine numbering) but ultimately led a Barbier-type macrocyclization. The proposed formation of the C20–C21 bond through nucleophilic opening of a δ-lactam by an sp3 carbanion is rare, especially in this complex setting and even less frequently in a macrocyclization.[xviii] The fragile butenolide would be annulated at a late stage by a vinylogous Mukaiyama aldol reaction of a hypothetical furanone anion 6 to a ketone at C5 following unmasking of the silylenol ether of spirolactam 5.
Figure 2.
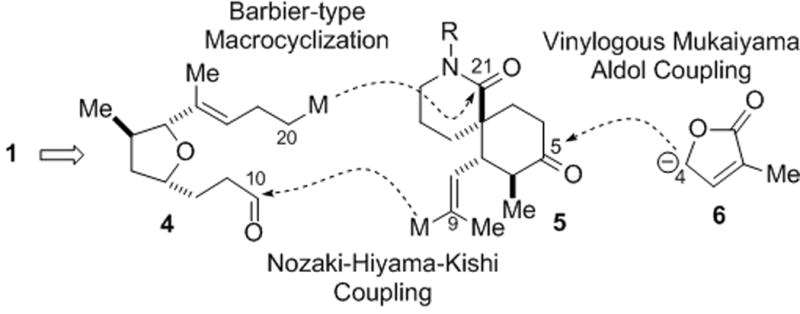
Retrosynthetic strategy toward gymnodimine (1) showing principal disconnections. M = metal.
Our initial strategy for fragment coupling called for a NHK macrocyclization following joining of the iodotetrahydrofuran 10, available in three steps from the previously described ether 7,[xivb] and the optically active spirolactam 11 (95% ee),[xix] previously obtained via a catalytic, asymmetric Diels–Alder reaction (Scheme 1).[xivi] After some experimentation, we found optimal conditions for a Barbier-type fragment coupling involving halogen-lithium exchange in the presence of the N-tosyl lactam electrophile[xx] providing adduct 12 in 92% yield, while generation of the alkyl lithium and subsequent addition of the N-tosyl lactam gave greatly inferior results (17%). This was a crucial precedent for the eventual solution for macrocyclization (vide infra) since numerous attempts towards a NHK macrocyclization from iodoolefins derived from 12 were unsuccessful. At this juncture, we elected to switch the order of coupling and investigate a rather unconventional strategy involving a Barbier-type macrocyclization.[xxi]
Scheme 1.
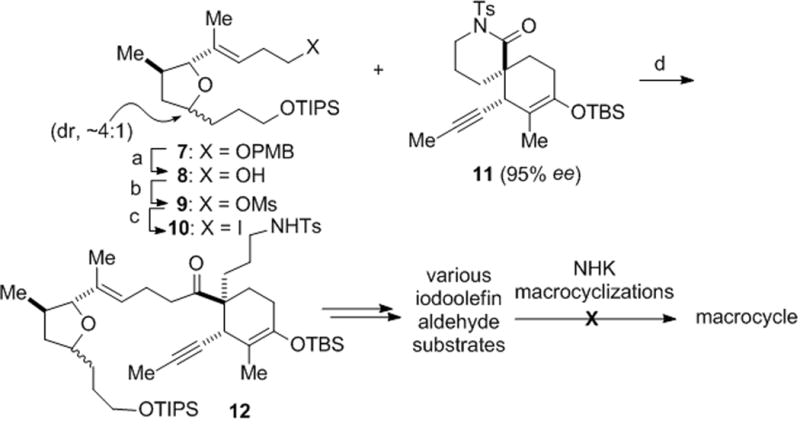
Reagents and conditions. a) Nao, NH3(l), THF, −78 °C, 92%; b) MsCl, Et3N, CH2Cl2, 92%; c) n-Bu4NI, THF, 66 °C, 91%; d) t-BuLi, Et2O, −78 °C; then 11, 17%; or 10 and 11, t-BuLi, Et2O, −78 °C, 92%. PMB = para-methoxybenzyl, THF = tetrahydrofuran, Ms = methanesulfonyl, TIPS = triisopropylsilyl.
The synthesis of the required tetrahydrofuran aldehyde 14b commenced with deprotection of PMB ether 13a[xivb] and conversion to chloride 13c by treatment with PPh3/CCl4 in warm DMF (Scheme 2). Following selective hydroboration of the terminal olefin, the intermediate alcohol 14a was oxidized with Dess–Martin periodinane to provide aldehyde 14b.[xxii]
Scheme 2.
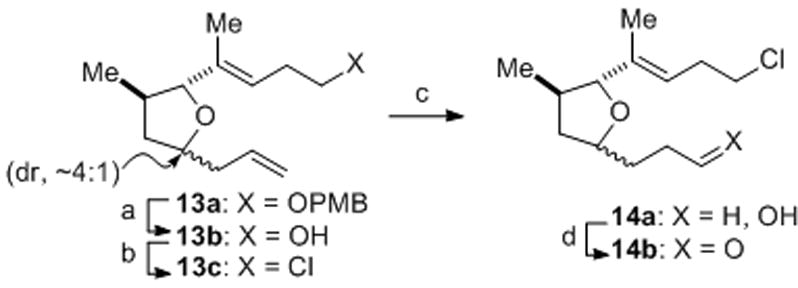
Reagents and conditions: a) Nao, NH3(l), THF, −78 °C, 92%; b) PPh3, CCl4, DMF, 65 °C, 85%; c) 9-BBN, THF; NaOH, H2O2, 98%; d) Dess–Martin periodinane, NaHCO3, CH2Cl2, 71%. DMF = N,N-dimethylforamide, 9-BBN = 9-borabicyclo[3.3.1]nonane.
The synthesis of the required vinyl iodide partner 16 for the projected Barbier macrocyclization began once again with optically active spirolactam 11 (Scheme 3). Functionalization of the internal acetylene in 11 proved to be rather challenging. Among all the protocols examined, only Pd-catalyzed hydrostannylation[xxiii] gave the corresponding vinyl stannane 15 and use of a non-polar solvent as reported by Semmelhack[xxiv] gave optimal conversion to stannane 15. Stannane-iodide exchange at low temperature then afforded the sensitive vinyl iodide 16 in 76% yield.
Scheme 3.
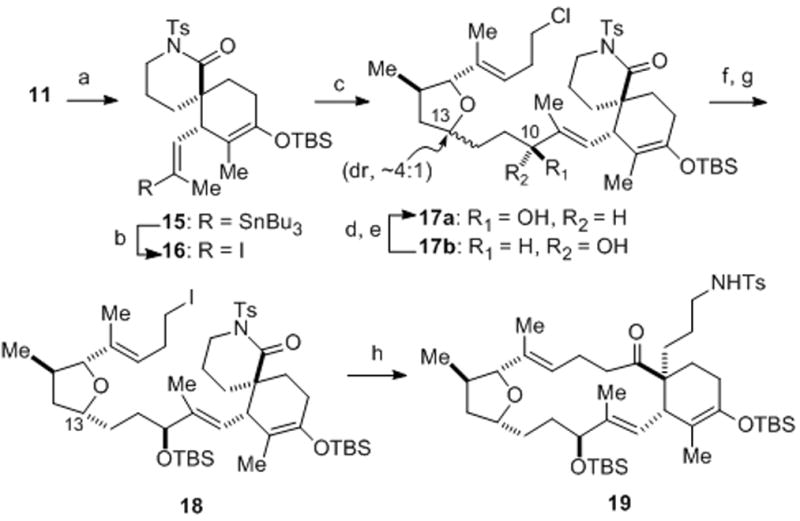
Reagents and conditions: a) PdCl2(PPh3)2, n-Bu3SnH, THF/hexanes (1:7), 85%; b) I2, CH2Cl2, −78 °C, 76%; c) 14b, CrCl2/0.5 mol% NiCl2, DMF/THF (1:1), 97%, 17a:17b = 1.3:1; d) Dess–Martin periodinane, NaHCO3, CH2Cl2, 88%; e) (R)-Me-CBS, catecholborane, CH2Cl2, 0 °C, 80%, dr = 6:1; f) Et3N, TBSOTf, CH2Cl2, −78 °C, 86%; g) NaI, acetone, 65 °C, 99%; h) t-BuLi, Et2O, 23 °C, 56–61%. (R)-Me-CBS = (R)-methyl-oxazaborolidine, Tf = trifluoromethanesulfonyl.
Aldehyde 14b and vinyl iodide 16 were coupled under standard NHK conditions, providing allylic alcohols 17a/b as a diastereomeric mixture (1.3:1, β/α-epimers at C10) and the C10 epimers were readily separable (Scheme 3). The undesired α-epimer 17b could be converted to 17a via an oxidation-reduction sequence using the Itsuno–Corey reduction protocol (dr, 6:1) enabling greater material throughput.[xxv] Subsequent protection of the hydroxyl group and Finkelstein reaction furnished alkyl iodide 18, the required intermediate for the crucial macrocyclization which could be separated from the undesired C13-epimer at this stage. The low temperature conditions (−78 °C) developed for the intermolecular Barbier-type coupling (cf. Scheme 1) were disappointing in this instance providing a mixture of deiodinated t-butyl ketone derived from quenching of the alkyllithium and t-BuLi addition to the δ-lactam. Surprisingly, performing the reaction in an identical manner but adding t-BuLi to the iodo N-tosyl lactam 18 at ambient temperature (23 °C) rather than −78 °C gave macrocycle 19 reproducibly on scales up to ~100 mg in 56–61% yields. While both conformational effects and relative rates of halogen-metal exchange,[xxvi] macrocyclization, t-BuLi addition to the N-tosyl lactam, and elimination of t-butyl iodide must all play a role in this process, further understanding of this intriguing process must await additional studies.
At this stage, it was necessary to switch the robust N-tosyl group to a more labile trifluoroacetamide utilizing our recently developed protocol for this purpose (Scheme 4).[xxvii] The silyl groups of macrocycle 20 were then cleaved under acidic conditions, furnishing the crystalline hydroxy ketone 21, which enabled confirmation of the relative stereochemistry of the macrocycle by single crystal X-ray analysis (inset, Scheme 4).
Scheme 4.
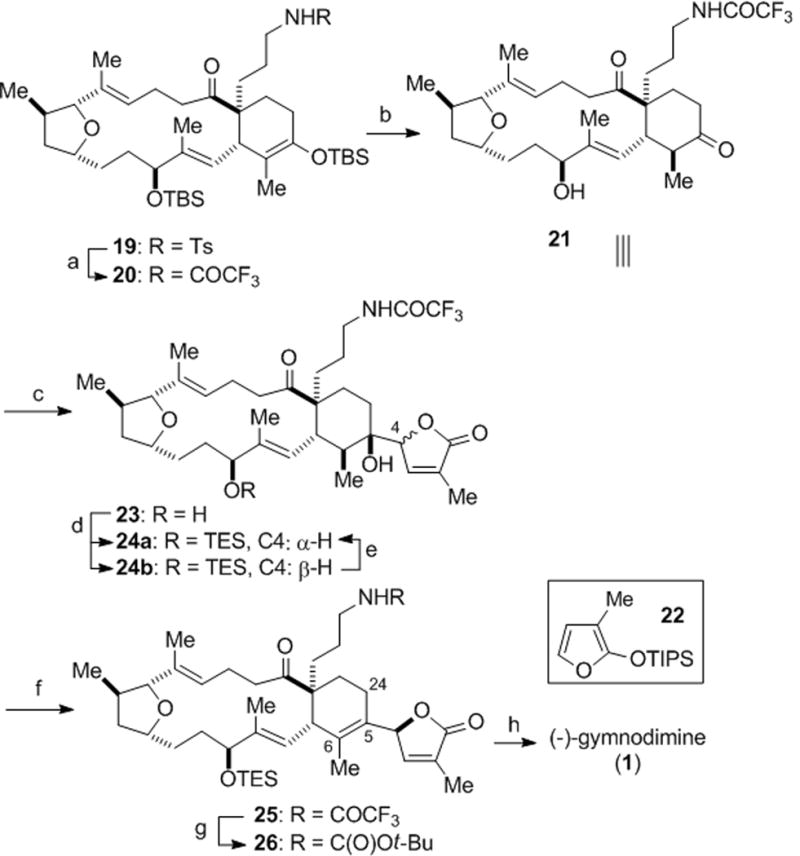
Reagents and conditions. a) Et3N, (CF3CO)2O, CH2Cl2, 0 °C; then SmI2, 23 °C, 73%; b) p-TSA, CH2Cl2/THF/MeOH, 84%; c) TiCl4, 22, CH2Cl2, 61% (dr, 1:1); d) TESCl, imidazole, DMAP, CH2Cl2, 23 °C, 76% (dr, 1:1, 24a/b); e) DBU, CH2Cl2, 60% (dr,2:1, 24a/b); f) Et3N, SOCl2, CH2Cl2, −78 °C, 82% (Δ5,6/Δ5,24, 3 :1); g) (Boc)2O, Et3N, DMAP, CH2Cl2, then H2NNH2, 99%; h) TFA, CH2Cl2, 68%. (Inset: ORTEP representation of X-ray structure of ketone 21). p-TSA = para-toluenesulfonic acid, TES = triethylsilyl, DMAP = 4-dimethylaminopyridine, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, Boc = tert-butoxycarbonyl, TFA = trifluoroacetic acid
For butenolide annulation, we employed our recently described strategy via a vinylogous Mukaiyama aldol reaction.[xxviii] Brief exposure (1 min) of a mixture of the macrocyclic ketone 21 and silyloxyfuran 22[xxix] to TiCl4 at 23 °C provided butenolide 23 in good yield as a ~1:1 mixture of two diastereomers (epimeric at C4; single stereochemistry at C5, Scheme 4).[xxx] The lack of diastereoselectivity at C4 during this transformation is to a great extent offset by the conciseness of this direct vinylogous Mukaiyama aldol addition strategy for butenolide annulation. The epimeric tertiary alcohols 24a/b were readily separated after alcohol protection. It was found that the undesired diastereomer 24b could be epimerized to a 2:1 mixture of the diastereomeric butenolides 24a/b upon treatment with DBU at ambient temperature. Dehydration of the tertiary alcohol 24a (Et3N, SOCl2) afforded the desired tetrasubstituted olefin 25 as the predominant regioisomer (Δ5,6/Δ5,24, 3:1). Application of mild basic conditions for cleavage of the trifluoroacetamide 25 led to degradation of the butenolide, in agreement with the findings of Miles that the butenolide of gymnodimine is unstable under both neutral and mild alkaline conditions.[xv] Attempted acid hydrolysis also proved unsuitable for this highly functionalized substrate. Eventually, a solution was found involving N-Boc protection and mild trifluoroacetamide cleavage using a modified Burk protocol.[xxxi] Careful treatment of the derived Boc-amine 26 with trifluoroacetic acid led to both t-butylcarbamate and silylether cleavage. Finally, cyclization to the cyclic imine under vacuum led to (−)-gymnodimine (1) as evidenced by correlation of spectral data of the synthetic material to that of the natural product.[xxxii] Using an identical synthetic sequence, C4-epi-gymnodimine (C4-epi-1) was also synthesized from the diastereomeric butenolide alcohol 24b (not shown) for comparison and provided further evidence that alcohol 24a possessed the natural configuration at C4.[xxxii]
In conclusion, the first total synthesis of (−)-gymnodimine was achieved in a highly convergent fashion, featuring an unusual Barbier-type macrocyclization strategy at ambient temperature. Also, a late stage appendage of the chiral butenolide via a vinylogous Mukaiyama aldol addition to a highly useful macrocyclic ketone 21 provides convenient avenues for synthesis of gymnodimine derivatives for further mode of action studies and hapten synthesis. The latter studies are directed towards development of a robust ELISA assay for detection of gymnodimine and congeners in the marine environment and will be reported in due course.
Footnotes
In Memory of John L. Hogg
The work was supported by NIH (GM52964) and the Welch Foundation (A-1280). We thank Dr. Ziad Moussa for assistance with scale-up. We thank Dr. Joseph Reibenspies and Dr. Nattamai Bhuvanesh (TAMU) for X-ray structure analyses and Profs. John W. Blunt and Murray H.G. Munro for providing characterization data for natural gymnodimine.
Supporting Information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- i.Seki T, Satake M, Mackenzie L, Kaspar HF, Yasumoto T. Tetrahedron Lett. 1995;36:7093. [Google Scholar]
- ii.Stewart M, Blunt JW, Munro MHG, Robinson WT, Hannah DJ. Tetrahedron Lett. 1997;38:4889. [Google Scholar]
- iii.Miles CO, Wilkins AL, Stirling DJ, MacKenzie AL. J Agric Food Chem. 2000;48:1373. doi: 10.1021/jf991031k. [DOI] [PubMed] [Google Scholar]
- iv.Dragunow M, Trzoss M, Brimble MA, Cameron R, Beuzenberg V, Holland P, Mountfort D. Environ Toxicol Pharmacol. 2005;20:305. doi: 10.1016/j.etap.2005.02.008. [DOI] [PubMed] [Google Scholar]
- v.Kharrat R, Servent D, Girard E, Ouanounou G, Amar M, Marrouchi R, Benoit E, Molgo J. J Neurochem. 2008;107:952. doi: 10.1111/j.1471-4159.2008.05677.x. [DOI] [PubMed] [Google Scholar]
- vi.Miles CO, Wilkins AL, Stirling DJ, MacKenzie AL. J Agric Food Chem. 2003;51:4838. doi: 10.1021/jf030101r. [DOI] [PubMed] [Google Scholar]
- vii.Uemura D, Chou T, Haino T, Nagatsu A, Fukuzawa S, Zheng S, Chem H. J Am Chem Soc. 1995;117:1155. [Google Scholar]
- viii.Falk M, Burton IW, Hu T, Walter JA, Wright JLC. Tetrahedron. 2001;57:8659. and references therein. [Google Scholar]
- ix.Takada N, Umemura N, Suenaga K, Uemura D. Tetrahedron Lett. 2001;42:3495. [Google Scholar]
- x.Torigoe K, Murata M, Yasumoto T, Iwashita T. J Am Chem Soc. 1988;110:7876. [Google Scholar]
- xi.Lu CK, Lee G-H, Huang R, Chou H-N. Tetrahedron Lett. 2001;42:1713. [Google Scholar]
- xii.O’Connor PD, Brimble MA. Nat Prod Rep. 2007;24:869. doi: 10.1039/b700307m. [DOI] [PubMed] [Google Scholar]
- xiii.For recent syntheses and lead references to pinnatoxin synthesis, see:; a) Stivala CE, Zakarian A. J Am Chem Soc. 2008;130:3774. doi: 10.1021/ja800435j. [DOI] [PubMed] [Google Scholar]; b) Nakamura S, Kikuchi F, Hashimoto S. Angew Chem Int Ed. 2008;47:7091. doi: 10.1002/anie.200802729. [DOI] [PubMed] [Google Scholar]; For syntheses of pteriatoxins, see:; c) Matsuura F, Peters R, Anada M, Harried SS, Hao J, Kishi Y. J Am Chem Soc. 2006;128:7463. doi: 10.1021/ja0618954. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hao J, Matsuura F, Kishi Y, Kita M, Uemura D, Asai N, Iwashita T. J Am Chem Soc. 2006;128:7742. doi: 10.1021/ja061893j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xiv.a) Ishihara J, Miyakawa J, Tsujimoto T, Murai A. Synlett. 1997:1417. [Google Scholar]; b) Yang J, Cohn ST, Romo D. Org Lett. 2000;2:763. doi: 10.1021/ol005510c. [DOI] [PubMed] [Google Scholar]; c) Ahn Y, Cardenas GI, Yang J, Romo D. Org Lett. 2001;3:751. doi: 10.1021/ol0155081. [DOI] [PubMed] [Google Scholar]; d) Tsujimoto T, Ishihara J, Horie M, Murai A. Synlett. 2002:399. [Google Scholar]; e) White JD, Wang G, Quaranta L. Org Lett. 2003;5:4109. doi: 10.1021/ol030101c. [DOI] [PubMed] [Google Scholar]; f) White JD, Wang G, Quaranta L. Org Lett. 2003;5:4983. doi: 10.1021/ol035939e. [DOI] [PubMed] [Google Scholar]; g) Johannes JW, Wenglowsky S, Kishi Y. Org Lett. 2005;7:3997. doi: 10.1021/ol051553n. [DOI] [PubMed] [Google Scholar]; h) Brimble MA, Crimmins D, Trzoss M. ARKIVOC. 2005;1:39. [Google Scholar]; i) Kong K, Moussa Z, Romo D. Org Lett. 2005;7:5127. doi: 10.1021/ol051840r. [DOI] [PubMed] [Google Scholar]
- xv.Miles CO, Hawkes AD, MacKenzie AL, Munday R, Towers NP, Prinsep MR. Marine Biotoxin Science Workshop, New Zealand. 1999:94. [Google Scholar]
- xvi.For some recent examples, see:; a) Oguri H, Hirama M, Tsumuraya T, Fujii I, Maruyama M, Uehara H, Nagumo Y. J Am Chem Soc. 2003;125:7608. doi: 10.1021/ja034990a. [DOI] [PubMed] [Google Scholar]; b) Forsyth CJ, Xu J, Nguyen ST, Samdal IA, Briggs LR, Rundberget T, Sandvik M, Miles CO. J Am Chem Soc. 2006;128:15114. doi: 10.1021/ja066971h. and references therein. [DOI] [PubMed] [Google Scholar]
- xvii.a) Takai K, Tagashira M, Kuroda T, Oshima K, Utimoto K, Nozaki H. J Am Chem Soc. 1986;108:6048. doi: 10.1021/ja00279a068. [DOI] [PubMed] [Google Scholar]; b) Jin H, Uenishi J, Christ WJ, Kishi Y. J Am Chem Soc. 1986;108:5644. [Google Scholar]; c) For an review, see:; Fürstner A. Chem Rev. 1999;99:991. doi: 10.1021/cr9703360. [DOI] [PubMed] [Google Scholar]
- xviii.Giovannini A, Savoia D, Umani-Ronchi A. J Org Chem. 1989;54:228. [Google Scholar]
- xix.The absolute configuration of this spirolactam was previously determined by X-ray analysis (anomalous dispersion) of a derivative, see ref. xivi.
- xx.For recent precedents to halogen-metal exchange in the presence of electrophiles, see:; a) Crimmins MT, King BW. J Am Chem Soc. 1998;120:9084. [Google Scholar]; b) Charest MG, Lerner CD, Brubaker JD, Siege DR, Myers AG. Science. 2005;308:395. doi: 10.1126/science.1109755. [DOI] [PubMed] [Google Scholar]
- xxi.For related reactions on simpler substrates, see:; a) Cooke MP, Jr, Houpis IN. Tetrahedron Lett. 1985;26:4987. [Google Scholar]; b) Souchet M, Clark RD. Synlett. 1990:151. [Google Scholar]
- xxii.Dess DB, Martin JC. J Org Chem. 1983;48:4155. [Google Scholar]
- xxiii.a) Zhang HX, Guibé F, Balavoine G. J Org Chem. 1990;55:1857. [Google Scholar]; b) For a review, see:; Smith ND, Mancuso J, Lautens M. Chem Rev. 2000;100:3257. doi: 10.1021/cr9902695. [DOI] [PubMed] [Google Scholar]
- xxiv.a) Semmelhack MF, Hooley RJ. Tetrahedron Lett. 2003;44:5737. [Google Scholar]; b) Yuan Y, Men H, Lee C. J Am Chem Soc. 2004;126:14720. doi: 10.1021/ja0447154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxv.Corey EJ, Helal CJ. Angew Chem Int Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- xxvi.Halogen-metal exchange is known to be an extremely fast process proceeding in the presence of MeOH in some cases, see:; a) Bailey WF, Punzalan ER. J Org Chem. 1990;55:5404. [Google Scholar]; b) Negishi E, Swanson DR, Roussett CJ. J Org Chem. 1990;55:5406. [Google Scholar]; c) Bailey WF, Patricia JJ, Nurmi TT, Wang W. Tetrahedron Lett. 1986;27:1861. [Google Scholar]; d) Clayden J. Organolithiums: Selectivity for Synthesis. Pergamon Press; 2002. [Google Scholar]
- xxvii.Moussa Z, Romo D. Synlett. 2006:3294. [Google Scholar]
- xxviii.a) Kong K, Romo D. Org Lett. 2006;8:2909. doi: 10.1021/ol060534q. [DOI] [PubMed] [Google Scholar]; b) For a review of silyloxyfuran, vinylogous Mukaiyama aldol reactions, see:; Casiraghi G, Zanardi F, Appendino G, Rassu G. Chem Rev. 2000;100:1929. doi: 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]
- xxix.Martin SF, Barr KJ. J Am Chem Soc. 1996;118:3299. [Google Scholar]
- xxx.The stereochemical outcome is consistent with simpler models confirmed by X-ray analysis. See ref. xxviiia for further information.
- xxxi.Burk MJ, Allen JG. J Org Chem. 1997;62:7054. While installation of a Boc group rather than the trifluoroacetamide on the primary sulfonamide 19 was successful, attempted cleavage of the sulfonamide failed and points to the necessity of the electron withdrawing trifluoroacetamide group for successful reductive cleavage (see ref. xxvii). [Google Scholar]
- xxxii.See Supporting Information for details.