

Abstract
Background and Purpose
Bryostatin, a potent protein kinase C (PKC) activator, has demonstrated therapeutic efficacy in preclinical models of associative memory, Alzheimer's disease, global ischemia, and traumatic brain injury. In this study, we tested the hypothesis that administration of bryostatin provides a therapeutic benefit in reducing brain injury and improving stroke outcome using a clinically relevant model of cerebral ischemia with tissue plasminogen activator (tPA) reperfusion in aged rats.
Methods
Acute cerebral ischemia was produced by reversible occlusion of the right middle cerebral artery (MCAO) in 18-20 month old female Sprague-Dawley rats using an autologous blood clot with tPA-mediated reperfusion. Bryostatin was administered at 6 h post-MCAO then at 3, 6, 9, 12, 15, and 18 d after MCAO. Functional assessment was conducted at 2, 7, 14, and 21 d after MCAO. Lesion volume and hemispheric swelling/atrophy were performed at 2, 7, and 21 d post-MCAO. Histological assessment of PKC isozymes was performed at 24 h post-MCAO.
Results
Bryostatin-treated rats showed improved survival post-MCAO, especially during the first 4 d. Repeated administration of bryostatin post-MCAO resulted in reduced infarct volume, hemispheric swelling/atrophy, and improved neurological function at 21 d post-MCAO. Changes in PKC alpha expression and PKC epsilon expression in neurons were noted in bryostatin-treated rats at 24 h post-MCAO.
Conclusions
Repeated bryostatin administration post-MCAO protected the brain from severe neurological injury post-MCAO. Bryostatin treatment improved survival rate, reduced lesion volume, salvaged tissue in infarcted hemisphere by reducing necrosis and peri-infarct astrogliosis, and improved functional outcome following MCAO.
Keywords: ischemic stroke, neuroprotection, neurovascular unit, αPKC, εPKC, aging
INTRODUCTION
Thrombolytic treatment with recombinant tissue plasminogen activator (tPA) remains the only FDA approved drug for treatment of acute ischemic stroke. However, less than 3% of people suffering an ischemic stroke receive tPA due to increased risk of secondary cerebral hemorrhage and edema formation1. Based on the aging population and increased stroke burden, an unmet need exists to develop alternative approaches to treat acute ischemic stroke, either independently of tPA or in an effort to increase the number of patients eligible for tPA.
Preclinical studies of proposed therapeutics for ischemic stroke have largely failed to consider the greatest risk factor for stroke, age2. Previous studies suggest that aged rats represent a more clinically relevant model of ischemic stroke when compared to younger animals3,4. These studies illustrate an increased severity of ischemic injury and altered neural injury progression3-6. Thus, use of aged animals may increase clinical relevance and the likelihood of bench-to-bedside therapeutic translation.
Protein kinase C (PKC) plays a critical role in storage of associative memory and related synaptogenesis in normal animals7; reducing the accumulation of A Beta, synaptic loss, cognitive deficits, and neurodegeneration in preclinical models of Alzheimer's disease8,9; and protection of neurons against ischemic pathology10,11 and traumatic brain injury in rodents12. PKC isozymes alpha (α) and epsilon (ε) regulate synaptogenic and anti-apoptotic signaling pathways13, as well as critical functional pathways in the multicellular response to ischemiareperfusion injury14. Individual PKC isozymes play differing, and sometimes opposing, roles in injury response that often depend on cell types and degree of pathophysiology15,16. Observations that PKC activation mediates both protective and harmful messages results from different PKC isoforms being activated by different signals that play concentration- and time-dependent roles in the development of neuronal damage and regeneration7,17.
Bryostatin, an ultrapotent PKC activator, may provide substantial benefit in the treatment of acute ischemic stroke. Studied extensively as an anti-tumorogenic agent, recent studies demonstrate that administration of bryostatin following global ischemic insult resulted in curative neurogenesis, synaptogenesis, and cognitive enhancement11. The purpose of this study was to investigate the pharmacological potential of repeated administration of bryostatin to improve outcome following acute ischemic stroke in aged rats.
MATERIALS & METHODS
Animals and Treatment Protocol
Sixty-six female Sprague-Dawley rats (18-20 months old) were purchased from Hilltop Laboratories (Scottdale, PA) and housed under 12-h:12-h light-dark conditions with food and water ad libitum. All work involving rats was approved by the West Virginia University Animal Care and Use Committee. For study 1, rats were randomly divided into two treatment groups (N=56). Group 1 underwent a 2 h MCAO with tPA-mediated (5 mg/kg; i.v.) reperfusion as previously described2,18, and at 6 h was administered bryostatin (2.5 mg/kg; i.v.) with subsequent doses every 3 d for a total of 7 doses over 21 d. Group 2 served as the control group and underwent the same procedures except were administered 0.9% saline at 6 h post-MCAO instead of bryostatin. Rats were assayed at 2, 7, and 21 d following MCAO. A second set of rats (N=10; 6 saline and 4 bryostatin treated rats) underwent MCAO with bryostatin/saline treatment at 6 h and were then euthanized at 24 h to visualize changes in PKC isozyme (α and ε) expression in neurons, endothelial cells, and astrocytes. Ischemia and reperfusion were defined as a minimum of 80% decrease in cerebral blood flow at onset of ischemia and return to 80% of baseline blood flow measurement, respectively. Rats not achieving these standards were excluded from the study2,18.
Neurological functional assessment
The modified Neurological Severity Scores (mNSS) was used to assess neurological function at 2, 7, 14, and 21 d after MCAO (n=7 saline and 10 bryostatin-treated rats). The mNSS is a composite score of motor, sensory, balance and reflex measures ranging from scores of 1 to 17, with higher scores indicating greater neurological injury19.
TUNEL Staining, Immunohistochemistry, and Infarct Volume Measurement
TUNEL staining was performed at 7 (n=5 rats per treatment) and 21 (n=7 saline and 10 bryostatin-treated rats) d after MCAO according to manufacturer's instructions (Roche Applied Science). After mounting and nuclear staining with 4'-6-diamidino-2-phenylindole (DAPI), slices were visualized for apoptotic tissue.
Using alternating slices from the same brain, immunohistochemistry using a modified ABC procedure was performed with anti-GFAP (1:1000) for evaluation of astrocyte morphology (n=7 saline and 10 bryostatin treated rats)6. At 2 (n=4 rats per treatment), 7 (n=5 rats per treatment), and 21 (n=7 saline and 10 bryostatin-treated rats) d following MCAO, lesion volumes were determined using cresyl violet staining. Degree of cerebral hemispheric swelling/atrophy at 2, 7, and 21 d following MCAO was calculated using the formula: cerebral hemispheric swelling/atrophy = (LV-RV)/LVx100%, where LV was volume of left hemisphere (mm3) and RV was volume of right hemisphere (mm3).
At 24 h following MCAO, tri-labeled confocal microscopy was performed to determine immunolocalization of εPKC (1:100) and αPKC (1:100) with neurons (1:150), endothelial cells (1:100), and astrocytes (1:100) at 24 h following MCAO (n=4 rats per treatment). Nuclear staining was performed on sections using DAPI and secondary antibodies were incubated at 1:200 dilution.
Expression levels of DNA damage (TUNEL), εPKC, and αPKC were evaluated based on immunofluorescence intensity by an observer blinded to treatment group. Confocal images were quantified using ImageJ as previously described9.
Water maze test
Spatial acquisition was determined using 6 training days with a probe test on day 7. Training began the 15th day post-MCAO and the probe test was conducted at 21 d post-MCAO (n=3 saline and 4 bryostatin-treated rats). Learning trials were conducted over 6 consecutive days with 4 trials per day. Time interval between trials was 60 s and maximum swim time to find platform was 120 s. Start location used N, S, E, and W designations for the start of each trial. Start locations for each trial were semi-randomly selected so that all directions were used for each rat on each training day. Each training session and probe test was conducted by investigators blinded to treatment. During the training period and probe test, latency speed and distance traveled to platform and 300% of platform was measured.
Statistical analysis
All data were compiled and analyzed by an investigator blinded to treatment and presented as mean±SEM. Physiologic parameters, functional data, and immunofluorescence intensity were compared by Student's t-test or analysis of variance grouped by treatment (saline vs bryostatin) and/or days post-MCAO (2, 7, and 21 d). Mortality data were compared using Fisher's exact test. P<0.05 was considered statistically significant.
RESULTS
Administration of bryostatin following MCAO in aged rats improved survival rate & neurological function
A total of 66 rats were used in this study. Fifty-six rats were evaluated for effects of bryostatin on ischemic brain injury over 21 d after MCAO. Two rats from this cohort, both saline-treated, were excluded from study for not meeting ischemia/reperfusion criteria. Survival rates were documented daily from 1 to 21 d (Figure 1A). Administration of bryostatin post-MCAO improved survival rates through 21 d with positive effects most evident during the first 4 d and a significant increase in survival achieved from 2 to 17 d post-MCAO. Although survival decreased in bryostatin-treated rats after day 4, improved survival was demonstrated at 7 d (68% bryostatin, n=13, versus 41% saline, n=7, treated rats), 14 d (59% bryostatin, n=11, versus 41% saline, n=7, treated rats), and 21 d (53% bryostatin, n=10, versus 41% saline, n=7, treated rats) post-MCAO. The dosing schedule used in this study was selected based on prioir preclinical studies showing cognitive enhancement in rats (hongpaisan and Alkon, 2007), therapeutic efficacy in young rats following global ischemia (Sun and Alkon, 2008), and neuroprotection in transgenic mice displaying Alzheimer's disease-like symptoms. Based on the results of this study, future studies will need to use dose-response measurements to determine the optimal dosing schedule for acute ischemic stroke in aged rats. Functional assessment revealed a significant improvement in mNSS scores at 21 d post-MCAO in bryostatin-treated rats (2.6±0.3; n=10) compared to saline-treated rats (3.7±0.2; n=7) (Figure 1B). No difference (p>0.05) in mNSS scores was observed at 2 (n=15 saline and 18 bryostatin treated rats), 7 (n=7 saline and 13 bryostatin treated rats), and 14 (n=7 saline and 11 bryostatin treated rats) d post-MCAO. Spatial acquisition was measured using the Morris water maze test. Results showed no difference (p>0.05) between treatment groups in latency speed or distance traveled to platform during the 6 training days. However, a significant improvement in latency speed (200±10 cm/s; n=3) and distance traveled (13.2±0.8 m; n=3) to platform was measured in bryostatin-treated rats as compared to saline-treated rats (300±10 cm/s and 15.9±0.4 m, respectively; n=4) on probe day (Figure 1C & 1D). When the platform was increased to 300% of the test platform, no difference (p>0.05) in latency speed and distance traveled was observed between treatment groups (Figure 1C & 1D). Both saline- and bryostatin-treated rats demonstrated a significant decrease in distance traveled to find the 300% platform as compared to the test platform (Figure 1D). No difference (p>0.05) in latency speed between treatment groups was measured (Figure 1C).
Figure 1.
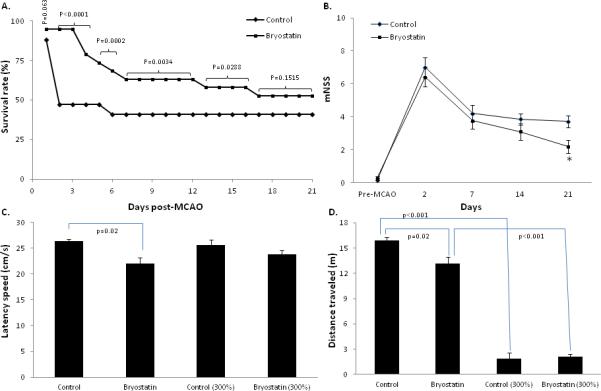
A.) Administration of bryostatin increased the survival rate of aged rats from 2 to 17 d post-MCAO and tPA reperfusion. B.) Neurological function was improved in bryostatin-treated aged rats at 21 d post-MCAO. No difference in neurological score between saline and bryostatin treated rats was measured at 2, 7, and 14 d post-MCAO. (C.) Latency speed and (D.) distance traveled were measured at 21 d post-MCAO. Results showed a reduction in latency speed and distance traveled in bryostatin-treated rats as compared to saline-treated rats. Both treatment groups demonstrated a reduction in distance traveled when using the 300% platform. Data presented as mean ± SEM.
Bryostatin resulted in diminished lesion volume post-MCAO
Quantification of lesion volume for cortex, striatum and total cerebral hemisphere was measured at 2, 7, and 21 d post-MCAO using cresyl violet stained coronal sections. No difference (p>0.05) in lesion volume was observed at 2 d post-MCAO (Figure 2A). At 7 d, bryostatin administration resulted in a significant reduction in lesion volume in the striatum and total hemisphere as compared to saline-treated rats (Figure 2A). At 21 d, a significant decrease in lesion volume was observed in cortex, striatum, and total hemisphere as compared to saline-treated rats (Figure 2A). Measurements of cortical hemispheric swelling or atrophy revealed a significant decrease in hemispheric swelling at 2 and 7 d after MCAO (Figure 2B). At 21 d following MCAO, the swelling phase of ischemic brain injury had subsided and atrophy of the saline-treated rats was visually evident by cavitation and necrosis of the infarcted hemisphere (Figure 3B & 3D). A significant decrease in atrophy was measured in bryostatin-treated rats at 21 d following MCAO (Figure 2C).
Figure 2.
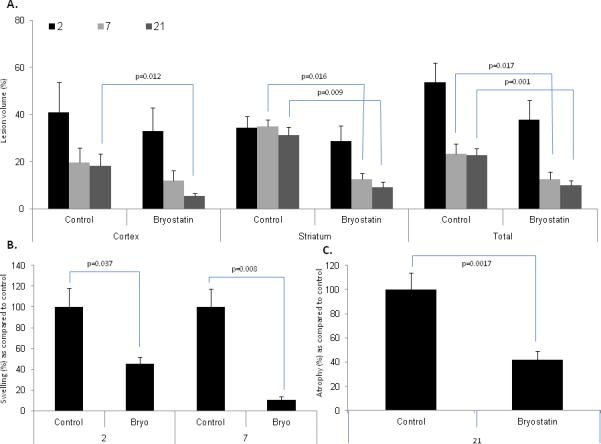
A.) Repeated administration of bryostatin decreased lesion volume in striatum and total hemisphere at 7 post-MCAO. At 21 d following MCAO, lesion volume was reduced in the cortex, striatum, and total hemisphere. No difference in lesion volume was measured at 2 d post-MCAO. B.) Bryostatin reduced hemispheric swelling at 2 and 7 d following MCAO. C.) Bryostatin reduced cerebral atrophy in the ischemic hemisphere as compared to saline-treated rats at 21 d post-MCAO. Data presented as mean ± SEM.
Figure 3.
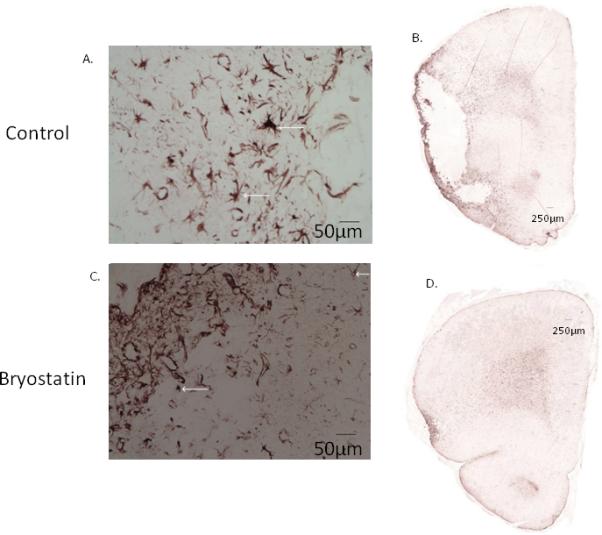
A.) Representative photomicrographs of GFAP-staining positive cells at 7 and 21 d following MCAO. Astrogliosis was observed in saline-treated brain at 7 and 21 d following MCAO, bryostatin mitigated the degree of astrogliosis. B.) At 21 d after MCAO, cavitation and necrosis was evident in saline-treated rats. Administration of bryostatin decreased lesion volume and reduced atrophy as compared to saline-treated rats. Cell nuclei were stained using DAPI.
Administration of bryostatin decreases astrogliosis and reduces cellular apoptosis in aged rats following at 7 and 21 d following MCAO
A significant reduction in TUNEL-positive cells, representing those cells undergoing apoptosis due to DNA fragmentation, were detected in the peri-infarct region surrounding the infarct in bryostatin-treated rats at 7 (75±6% of control) and 21 (43±3% of control) d after MCAO (Figure 4). This finding was reinforced by a profound reduction in reactive astrocytes, identified by GFAP expression, observed in bryostatin-treated rats, in which not only was astrogliosis reduced but the degree of tissue destruction was markedly attenuated (Figure 3). At both 7 and 21 d, astrocytic hypertrophy with enlarged processes was evident in tissue surrounding the necrotic zone around the infarct in saline-treated aged rats, consistent with the astrocytic response. Bryostatin attenuated this response and reduced tissue damage in the peri-infarct area (Figure 3).
Figure 4.
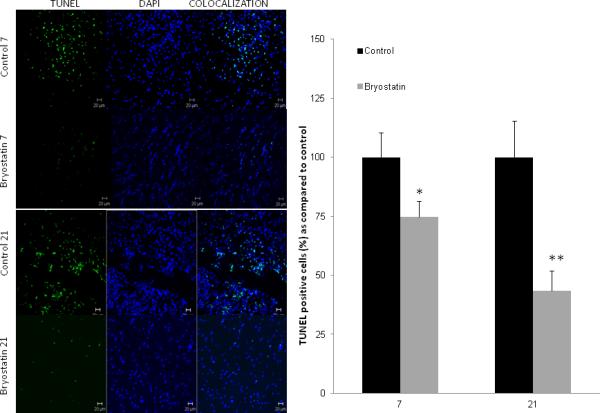
Comparison of TUNEL staining in aged rats showed a marked reduction in TUNEL positive cells in bryostatin-treated rats at 7 and 21 d post-MCAO as compared to saline-treated rats. Cell nuclei were stained using DAPI.
αPKC expression was reduced in neurons in the penumbral region of bryostatin treated rats at 24 h following MCAO
Colocalization of αPKC with neurons (NeuN), endothelial cells (CD31), and astrocytes (GFAP) was investigated in penumbra surrounding the infarct in aged rats at 24 h post-MCAO (n=4 rats per treatment group). αPKC immunoreactivity was observed in neurons and endothelial cells (Figure 5A & 6A). Expression of αPKC was diffusely localized throughout the cytoplasm of the cell. No colocalization of αPKC was shown with GFAP-positive astrocytes (data not shown). Rats treated with bryostatin showed a significant decrease in immunoreactivity of αPKC in neurons at 24 h following MCAO (Figure 5A & 5C). No change (p=0.23) in αPKC expression was observed in endothelial cells at 24 h following MCAO (Figure 6A & 6C).
Figure 5.
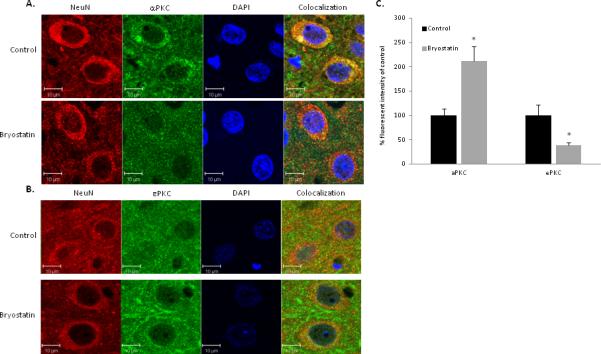
Representative images of tri-color confocal immunofluorescent microscopy demonstrating colocalization of NeuN with αPKC (A) and εPKC (B) in the penumbra of the infarcted hemisphere of saline and bryostatin treated aged rats at 24 h after MCAO. (C) In neurons, a 68% decrease in αPKC expression and a 202% increase in εPKC expression were measured at 24 h following MCAO in bryostatin treated rats as compared to saline-treated rats. Data expressed as mean ± SEM, * p<0.05. Cell nuclei were stained using DAPI.
Figure 6.
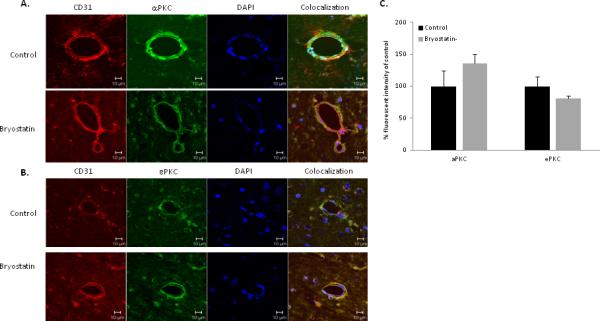
Representative images of tri-color confocal immunofluorescent microscopy demonstrating colocalization of CD31 (endothelial cells) with αPKC (A) and εPKC (B) in the penumbra of the infarcted hemisphere of saline and bryostatin treated aged rats at 24 h after MCAO. (C) In endothelial cells, no difference (p>0.05) in αPKC and εPKC expression were observed at 24 h following MCAO between bryostatin- and saline-treated rats. Data expressed as mean ± SEM, *p<0.05. Cell nuclei were stained using DAPI.
Bryostatin increased εPKC within neurons in the penumbra at 24 h following MCAO and tPA reperfusion
Colocalization of εPKC with NeuN and CD31 in the penumbral area at 24 h after MCAO was investigated (n=4 rats per treatment group). εPKC immunoreactivity was observed in neurons and endothelial cells (Figure 5B & 6B). Expression of εPKC was punctate along cytoplasmic edge of the plasma membrane of the cell. No colocalization of εPKC was shown with GFAP-positive astrocytes. Rats treated with bryostatin showed a significant increase in immunoreactivity of εPKC in neurons at 24 h following MCAO (Figure 5B & 5C).
DISCUSSION
The primary finding of this study was that administration of bryostatin after MCAO with tPA reperfusion decreased mortality over 21 d, with marked improvement during the first 4 d. Additionally, we are the first to report that bryostatin administration improved recovery from ischemic brain injury and functional outcome up to 21 d after MCAO in aged animals. Use of bryostatin attenuated both necrotic and apoptotic cell death. The reduced hemispheric swelling/atrophy observed at 2 and 7 d following MCAO and the significant attenuation of ipsilateral hemispheric atrophy seen in aged rats treated with bryostatin at 21 d suggests the potential for decreased inflammation and reduced cellular death following MCAO and tPA reperfusion. A marked improvement in neurological function was observed in bryostatin-treated rats at 21 d following MCAO. In this study, we also evaluated changes in post-stroke cognition using the Morris water maze test. Results showed that bryostatin-treated rats had improved spatial cognition as compared to saline-treated rats at 21 d following MCAO. Extinguishment of the cognitive deficit in saline-treated rats in the 300% platform trial suggests that discrimination of environment is improved in aged rats treated with bryostatin.
Effects measured in this study were associated with changes in expression and localization of PKC isozymes in neurons and endothelial cells within the peri-infarct region. Regulation of PKC activity is the primary reported mechanism of action for bryostatin. Inhibition of PKC isozymes blocked the synaptogenic and cognitive-enhancing effects of bryostatin on normal animals7. Therefore, it is plausible that pathological mechanisms initiated following ischemic brain injury involve PKC-mediated pathways20,21. Results from this study clearly show increased εPKC expression and a modest decrease in αPKC expression in neurons of bryostatin treated rats at 24 h post-MCAO. Roles of PKC activity following ischemic stroke are complex and poorly defined. What is known is that PKC activity mediates protective and damaging signaling cascades during the injury and reparative phases of ischemic stroke15,16; suggesting that timing, localization, and the PKC isozyme activated play critical, and sometimes contrasting, roles in injury response and progression of neuronal damage and regeneration following stroke7,17. Previous studies report that increased εPKC activity initiates proliferative and cell survival signaling pathways following ischemic stroke and reperfusion14,22,23, while increased αPKC activity exacerbates neuronal damage following post-stroke brain injury through initiation of anti-proliferative and pro-apoptotic pathways24; thus, while still inconclusive, our results suggest that changes in PKC activity in endothelial cells and neurons may play a vital role in the acute neuroprotective actions of bryostatin. It should be noted that while bryostatin initially activates both PKC α and ε, it only causes prolonged binding of εPKC to the critically required anchoring protein, RACK 1 that is required for prolonged activation (unpublished data).
Of particular interest, especially during the acute phase of stroke injury is the role different PKC isozymes play in alterations of endothelial cell function during pathophysiological conditions. Increased PKC activity has been shown to affect structural and functional integrity of the BBB following ischemic brain injury25 with αPKC and βPKC increasing BBB opening through alterations in tight junction protein localization and εPKC enhancing BBB integrity by upregulating the expression of claudin 526-28. Our study showed that αPKC and εPKC were localized to both neurons and cerebral endothelial cells. No colocalization of εPKC or αPKC was observed with GFAP-positive astrocytes; however, that does not mean that changes in PKC activity do not have a significant influence on astrocyte reactivity as demonstrated by our study, which showed a reduction in astrogliosis and tissue damage in bryostatin treated rats. In fact, in relation to chronic brain response following ischemic injury, attenuation of glial response may end up being the most significant finding of this study. A recent study shows that PKC activation leads to a significant decrease in calcium wave propagation between astrocytes by attenuating astroglial gap junction communication29. Gaining a better understanding of the critical mediators and timing of this response will be a primary focus of follow-up studies. A limitation of this study and immunofluorescent imaging, in general, was the effect of bryostatin on PKC activation. Future studies will probe for changes in PKC activity by measuring cytosolic to membrane translocation and phosphorylation of specific PKC isozymes at different times post-ischemia.
In conclusion, administration of bryostatin following MCAO and tPA reperfusion has neuroprotective effects on the magnitude of ischemic brain injury. A notable finding in this study was the marked improvement in survival observed in bryostatin-treated rats, especially during the first week following ischemic insult. While mortality increased in the bryostatin treated group during the 21 d, it must be emphasized that PKC activator treatment, depending on dose and duration, causes three sequential consequences: activation, down regulation (and thus inhibition), and de novo protein synthesis. Thus, careful selection of treatment dosing and duration has a profound impact on PKC-mediated phosphorylation of down-stream substrates and associated therapeutic benefits30. The beneficial effects of bryostatin administration following stroke is most likely due to attenuation of inflammatory cell activity; thus, bryostatin may act as a potent adjuvant with other proven methods such as hypothermia31 for reduction of the inflammatory reaction during the acute phase of ischemia/reperfusion injury. In summary, bryostatin-treated rats displayed reduced ischemic brain injury following MCAO that was characterized by increased survival, reduced infarct volume, decreased hemispheric swelling/atrophy, and improved neurological function.
Acknowledgments
SOURCES OF FUNDING
This study was supported by a grant from NIH NINDS [RO1 NS061954] to J.D.H.
Footnotes
CONFLICT OF INTEREST/DISCLOSURE
Authors have no conflict of interest or disclosures regarding results in this manuscript
REFERENCES
- 1.Katzan IL, Hammer MD, Hixson ED, Furlan AJ, Abou-Chebl A, Nadzam DM. Utilization of intravenous tissue plasminogen activator for acute ischemic stroke. Arch Neurol. 2004;61:346–350. doi: 10.1001/archneur.61.3.346. [DOI] [PubMed] [Google Scholar]
- 2.Rosen CL, Dinapoli VA, Nagamine T, Crocco T. Influence of age on stroke outcome following transient acute ischemia. J Neurosurg. 2005;103:687–694. doi: 10.3171/jns.2005.103.4.0687. [DOI] [PubMed] [Google Scholar]
- 3.DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29:753–764. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Badan I, Buchhold B, Hamm A, Gratz M, Walker LC, Platt D, et al. Accelerated glial reactivity to stroke in aged rats correlates with reduced functional recovery. J Cereb Blood Flow Metab. 2003;23:845–854. doi: 10.1097/01.WCB.0000071883.63724.A7. [DOI] [PubMed] [Google Scholar]
- 6.Dinapoli VA, Benkovic SA, Li X, Kelly KA, Miller DB, Rosen CL, et al. Age exaggerates proinflammatory cytokine signaling and truncates signal transducers and activators of transcription 3 signaling following ischemic stroke in the rat. Neuroscience. 2010;170:633–644. doi: 10.1016/j.neuroscience.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hongpaisan J, Alkon DL. A structural basis for enhancement of long-term associative memory in single dendritic spines regulated by pkc. Proc Natl Acad Sci U S A. 2007;104:19571–19576. doi: 10.1073/pnas.0709311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Etcheberrigaray R, Tan M, Dewachter I, Kuiperi C, Van der Auwera I, Wera S, et al. Therapeutic effects of pkc activators in alzheimer's disease transgenic mice. Proc Natl Acad Sci U S A. 2004;101:11141–11146. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hongpaisan J, Sun MK, Alkon DL. Pkc epsilon activation prevents synaptic loss, abeta elevation, and cognitive deficits in alzheimer's disease transgenic mice. J Neurosci. 2011;31:630–643. doi: 10.1523/JNEUROSCI.5209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun MK, Hongpaisan J, Alkon DL. Postischemic pkc activation rescues retrograde and anterograde long-term memory. Proc Natl Acad Sci U S A. 2009;106:14676–14680. doi: 10.1073/pnas.0907842106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun MK, Hongpaisan J, Nelson TJ, Alkon DL. Poststroke neuronal rescue and synaptogenesis mediated in vivo by protein kinase c in adult brains. Proc Natl Acad Sci U S A. 2008;105:13620–13625. doi: 10.1073/pnas.0805952105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zohar O, Lavy R, Zi X, Nelson TJ, Hongpaisan J, Pick CG, et al. Pkc activator therapeutic for mild traumatic brain injury in mice. Neurobiol Dis. 2011;41:329–337. doi: 10.1016/j.nbd.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Quattrone A, Pascale A, Nogues X, Zhao W, Gusev P, Pacini A, et al. Posttranscriptional regulation of gene expression in learning by the neuronal elav-like mrna-stabilizing proteins. Proc Natl Acad Sci U S A. 2001;98:11668–11673. doi: 10.1073/pnas.191388398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bright R, Mochly-Rosen D. The role of protein kinase c in cerebral ischemic and reperfusion injury. Stroke. 2005;36:2781–2790. doi: 10.1161/01.STR.0000189996.71237.f7. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto S, Shamloo M, Matsumoto E, Isshiki A, Wieloch T. Protein kinase c-gamma and calcium/calmodulin-dependent protein kinase ii-alpha are persistently translocated to cell membranes of the rat brain during and after middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2004;24:54–61. doi: 10.1097/01.WCB.0000095920.70924.F5. [DOI] [PubMed] [Google Scholar]
- 16.Skaper SD, Facci L, Strijbos PJ. Neuronal protein kinase signaling cascades and excitotoxic cell death. Ann N Y Acad Sci. 2001;939:11–22. doi: 10.1111/j.1749-6632.2001.tb03606.x. [DOI] [PubMed] [Google Scholar]
- 17.Reshef A, Sperling O, Zoref-Shani E. Activation and inhibition of protein kinase c protect rat neuronal cultures against ischemia-reperfusion insult. Neurosci Lett. 1997;238:37–40. doi: 10.1016/s0304-3940(97)00841-0. [DOI] [PubMed] [Google Scholar]
- 18.Dinapoli VA, Rosen CL, Nagamine T, Crocco T. Selective mca occlusion: A precise embolic stroke model. J Neurosci Methods. 2006;154:233–238. doi: 10.1016/j.jneumeth.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32:1005–1011. doi: 10.1161/01.str.32.4.1005. [DOI] [PubMed] [Google Scholar]
- 20.Popa-Wagner A, Buga AM, Kokaia Z. Perturbed cellular response to brain injury during aging. Ageing Res Rev. 2011;10:71–79. doi: 10.1016/j.arr.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Selvatici R, Marino S, Piubello C, Rodi D, Beani L, Gandini E, et al. Protein kinase c activity, translocation, and selective isoform subcellular redistribution in the rat cerebral cortex after in vitro ischemia. J Neurosci Res. 2003;71:64–71. doi: 10.1002/jnr.10464. [DOI] [PubMed] [Google Scholar]
- 22.Bright R, Sun GH, Yenari MA, Steinberg GK, Mochly-Rosen D. Epsilonpkc confers acute tolerance to cerebral ischemic reperfusion injury. Neurosci Lett. 2008;441:120–124. doi: 10.1016/j.neulet.2008.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao CT, Li K, Li JT, Zheng W, Liang XJ, Geng AQ, et al. Pkcdelta regulates cortical radial migration by stabilizing the cdk5 activator p35. Proc Natl Acad Sci U S A. 2009;106:21353–21358. doi: 10.1073/pnas.0812872106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirai Y, Adachi N, Saito N. Protein kinase cepsilon: Function in neurons. FEBS J. 2008;275:3988–3994. doi: 10.1111/j.1742-4658.2008.06556.x. [DOI] [PubMed] [Google Scholar]
- 25.Willis CL, Meske DS, Davis TP. Protein kinase c activation modulates reversible increase in cortical blood-brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab. 2010;30:1847–1859. doi: 10.1038/jcbfm.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleegal MA, Hom S, Borg LK, Davis TP. Activation of pkc modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am J Physiol Heart Circ Physiol. 2005;289:H2012–2019. doi: 10.1152/ajpheart.00495.2005. [DOI] [PubMed] [Google Scholar]
- 27.Krizbai I, Szabo G, Deli M, Maderspach K, Lehel C, Olah Z, et al. Expression of protein kinase c family members in the cerebral endothelial cells. J Neurochem. 1995;65:459–462. doi: 10.1046/j.1471-4159.1995.65010459.x. [DOI] [PubMed] [Google Scholar]
- 28.Sonobe Y, Takeuchi H, Kataoka K, Li H, Jin S, Mimuro M, et al. Interleukin-25 expressed by brain capillary endothelial cells maintains blood-brain barrier function in a protein kinase cepsilon-dependent manner. J Biol Chem. 2009;284:31834–3184229. doi: 10.1074/jbc.M109.025940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enkvist MO, McCarthy KD. Activation of protein kinase C blocks astroglial gap junction communication and inhibits the spread of calcium waves. J Neurochem. 1992;59:519–26. doi: 10.1111/j.1471-4159.1992.tb09401.x. [DOI] [PubMed] [Google Scholar]
- 30.Alkon DL, Sun MK, Nelson TJ. Pkc signaling deficits: A mechanistic hypothesis for the origins of alzheimer's disease. Trends Pharmacol Sci. 2007;28:51–60. doi: 10.1016/j.tips.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Joseph C, Buga AM, Vintilescu R, Balseanu AT, Moldovan M, Junker H, et al. Prolonged gaseous hypothermia prevents the upregulation of phagocytosis-specific protein annexin 1 and causes low-amplitude EEG activity in the aged rat brain after cerebral ischemia. J Cereb Blood Flow Metab. 2012;32:1632–42. doi: 10.1038/jcbfm.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]