

Abstract
The laboratory mouse is the most widely used mammalian model organism in biomedical research. The 2.6 × 109 bases of the mouse genome possess a high degree of conservation with the human genome1, so a thorough annotation of the mouse genome will be of significant value to understanding the function of the human genome. So far, most of the functional sequences in the mouse genome have yet to be found, and the cis-regulatory sequences in particular are still poorly annotated. Comparative genomics has been a powerful tool for the discovery of these sequences2, but on its own it cannot resolve their temporal and spatial functions. Recently, ChIP-Seq has been developed to identify cis-regulatory elements in the genomes of several organisms including humans, Drosophila melanogaster and Caenorhabditis elegans3–5. Here we apply the same experimental approach to a diverse set of 19 tissues and cell types in the mouse to produce a map of nearly 300,000 murine cis-regulatory sequences. The annotated sequences add up to 11% of the mouse genome, and include more than 70% of conserved non-coding sequences. We define tissue-specific enhancers and identify potential transcription factors regulating gene expression in each tissue or cell type. Finally, we show that much of the mouse genome is organized in to domains of coordinately regulated enhancers and promoters. Our results provide a resource for the annotation of functional elements in the mammalian genome and for the study of mechanisms regulating tissue-specific gene expression.
We identified the genomic localizations of RNA polymerase II (polII), the insulator-binding protein CCCTC-binding factor (CTCF) and three chromatin modification marks, histone H3 lysine 4 trimethylation (H3K4me3), histone H3 lysine 4 monomethylation (H3K4me1) and H3 lysine 27 acetylation (H3K27ac), in 13 adult tissues, four embryonic tissues and two primary cell lines (Fig. 1a, b) by performing chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-Seq)6 (Supplementary Tables 1 and 2). Enrichment of H3K4me3 or polII binding signals is indicative of an active promoter, whereas the presence of H3K4me1 or H3K27ac outside promoter regions can be used as marks for enhancers7–11. CTCF binding is considered a mark for potential insulator elements12. In a subset of tissue and cell types, we also performed ChIP-Seq on the co-activator protein p300 and used its promoter-distal binding sites to train an enhancer prediction tool on the basis of chromatin signatures13. We determined the transcriptome in each tissue and cell type through RNA-Seq experiments, using a protocol that can detect both the abundance and strand of origin of RNA transcripts14 (Supplementary Fig. 1). By analysing the genomic occupancy of the above chromatin marks and transcription factors (Supplementary Methods), we identified 295,676 non-redundant cis-regulatory sequences, including 53,834 putative promoters, 234,764 potential enhancers and 111,062 CTCF-binding sites (Fig. 1b). With an estimated span of 1,000 base pairs for each element, the combined length of these putative cis regulatory sequences is 295.6 million base pairs, or 11% of the mouse genome.
Figure 1. Identification of cis-regulatory elements in the mouse genome.

a, UCSC genome browser views of ChIP-Seq and RNA-Seq data for mESC, heart and liver (chromosome 4). The values on the y axis for ChIP-Seq data are input normalized intensities. kb, kilobases. b, An overview of the predicted regulatory elements in the 19 tissue and cell types. E14.5, embryonic day 14.5; MEF, murine embryonic fibroblast. c, Percentages of known cis-regulatory elements recovered in this study.
To determine the accuracy and completeness of our cis-regulatory sequence mapping, we first compared the identified promoters with knownpromoters.Werecovered 79% of RefSeq-annotated promoters15 (Fig. 1c and Supplementary Fig. 2a) and confirmed an additional 62% of University of California, Santa Cruz (UCSC)-annotated promoters (13,205 out of 21,433) that are not annotated in RefSeq. As expected, annotated promoters not recovered by our study are generally expressed in tissues that were not investigated in this work (Supplementary Table 3). In addition to the annotated promoters, we also identified 13,438 novel promoters. When tested with a luciferase reporter, 85% of 65 randomly selected novel promoters showed significant promoter activity in at least one orientation (P < 0.01, Student's t-test) (Supplementary Fig. 3a, b), supporting their function as promoters. Next we compared the predicted enhancers with a list of 726 experimentally validated enhancers16 and found that 82% of them were correctly identified in this study (Fig. 1c and Supplementary Fig. 2b). We also randomly selected eight predicted murine embryonic fibroblast (MEF) enhancers for validation and found that six of them (75%) gave positive results (Supplementary Fig. 4) (P < 0.01, Student's t-test), supporting the reliability of our enhancer identification method. In addition, we recovered 94.5% of previously reported CTCF-binding sites in mouse embryonic stem cells (mESCs)17 (Fig. 1c), demonstrating the high sensitivity of our detection method for CTCF binding. Further, we detected 77,236 novel CTCF-binding sites, 87.5% of which contained the canonical CTCF motifs (P < 2.23 × 10−16, binomial distribution). The novel CTCF-binding sites tend to be more tissue-specific than the sites identified previously (Supplementary Fig. 5). The above evidence indicates that we have correctly identified most known cis-regulatory sequences and have uncovered many novel ones.
Functional elements are often under negative selection during evolution, so a high level of sequence conservation is frequently used as evidence of function. However, there are also reports showing that transcription factor binding may be rapidly lost or gained during evolution18,19, arguing that the usage of cis-elements may evolve more quickly. We examined the sequence conservation of different classes of the cis-regulatory sequences identified in this study, and found that promoters are characterized by the highest degree of sequence conservation (Fig. 2a). In contrast, CTCF-binding sites and enhancers have a much lower but still significant level of sequence conservation. We next assessed the level of conservation of cis-regulatory element usage between the mouse and human genomes in embryonic stem cells (ESCs)10 (Fig. 2b). More than 70% of homologous promoters are associated with H3K4me3 in both species, confirming a high degree of conservation in promoter usage (Fig. 2c, d). However, only 25.7% and 24.8% of enhancers and CTCF-binding sites, respectively, found in human ESCs are still associated with H3K4me1 or CTCF binding in mESCs, despite a high degree of sequence conservation (Fig. 2c). These results suggest that the cis-regulatory elements identified in the mouse genome are under different selective pressure during evolution, with promoters being most conserved in both sequence and usage, whereas enhancers and CTCF-binding sites are undergoing a considerable degree of evolution. This result agrees well with the recent findings of large interspecies differences and divergence of transcriptional regulation18.
Figure 2. Evolutionary conservation of the identified cis-regulatory elements.

a, Evolutionary conservation of cis-regulatory elements, in comparison with exons and random genomic sequences. Asterisk, P < 0.001, Fisher's exact test. b, UCSC genome browser views of chromatin state and CTCF-binding sites at Sox2 loci for mESCs and human ESCs (hESCs) on chromosome 3. DNA sequences, chromatin states and CTCF binding are all conserved in this region. c, Number of hESC regulatory elements that are conserved and predicted as regulatory elements in mESCs. d, Number of mESC regulatory elements that are conserved and predicted as regulatory elements in hESCs. e, Functional annotation of the conserved non-coding sequences based on the cis-regulatory elements identified in this study. The asterisk in c, d and e indicates CTCF-binding sites that do not overlap with either promoters or enhancers.
Comparative genomic methods have identified a significant number of mammalian sequences as non-protein coding but undergoing negative selection during evolution, commonly referred to as conserved non-protein-coding sequences (CNSs). These sequences are suspected to have important biological roles, yet their precise function remains to be defined.Wecompared our map of cis-regulatory elements with a list of CNSs20 and found that 70% of them fall into one of the three classes of predicted cis-elements: 15%as promoters, 53%as enhancers and 2% as CTCF-binding sequences. Additionally, 1% of the CNSs seem to be non-coding RNA sequences as supported by the RNA-Seq data (Fig. 2e and Supplementary Fig. 2c). Most CNSs therefore seem to function in regulating transcription.
We previously showed that enhancers in the human genome are associated with active chromatin marks in a cell-type-specific manner, whereas promoter and insulator elements tend to be ubiquitously occupied in multiple cell lines10. Here we found that the occupancy of enhancers by H3K4me1 in the mouse genome is still the most tissue-specific (Fig. 3a). In contrast, we observed that whereas H3K4me3 occupies most RefSeq promoters in multiple tissues, a significant number of promoters, especially the novel promoters discovered in this study, show tissue-specific occupancies by H3Kme3 or polII (Fig. 3a) (Supplementary Fig. 3d), with many of them corresponding to alternatively used promoters (Supplementary Table 4 and Supplementary Fig. 6). We also found that most CTCF-binding sites are occupied in multiple tissues (Fig. 3a). The tissue-specific CTCF-binding sites showed significant overlap with enhancers (P < 1.83 × 10−143, binomial distribution), whereas the ubiquitous CTCF-binding sites overlapped significantly with promoters (P < 9.0 × 10−43, binomial distribution) (Supplementary Fig. 5b, c), suggesting that a fraction of the CTCF-binding sites may function through promoters and enhancers, although the exact role of CTCF at these regions remains unclear. These results indicate that a large fraction of cis-regulatory elements are active in a tissue-specific manner and are most probably involved in regulating tissue-specific gene expression.
Figure 3. Genomic organization of co-regulated promoters and enhancers.
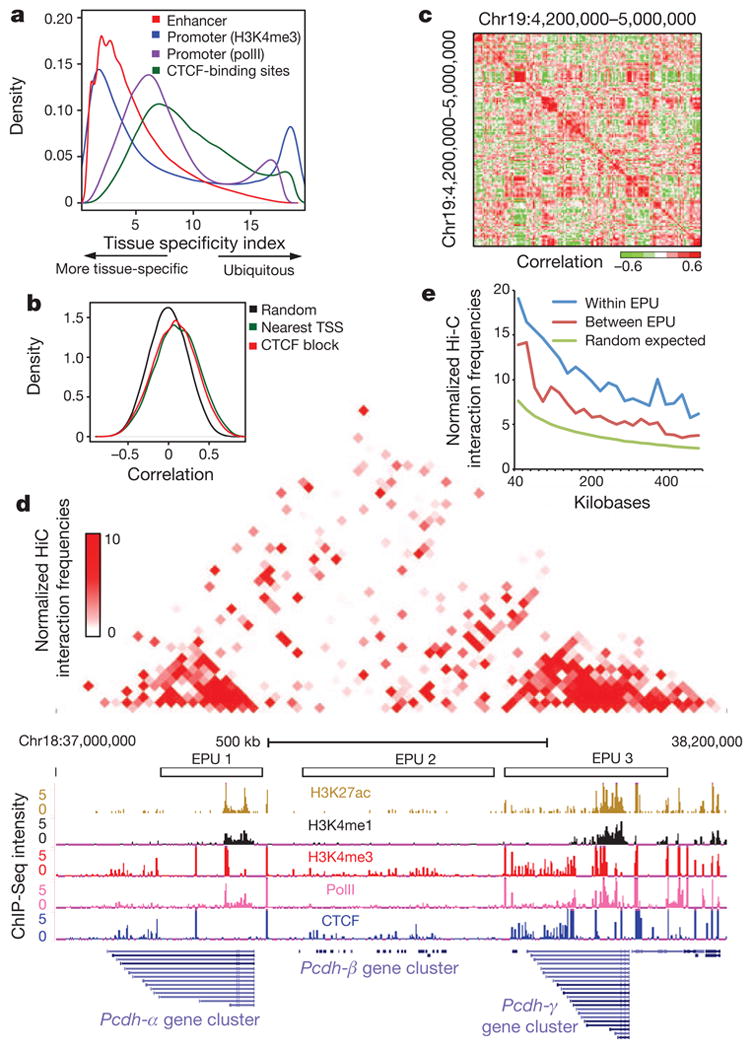
a, Tissue specificity of the usages of promoters (H3K4me3 and polII), enhancers (H3K4me1) and CTCF-binding sites. b, Distribution of the Spearman correlation coefficient of H3K4me1 at enhancers and polII at promoters of random permutation, the nearest TSS model, and the CTCF block model. c, Enhancers and promoters form co-regulated clusters of different sizes, as shown by the Spearman correlation coefficient of H3K4me1 at enhancers and polII at promoters on chromosome 19. d, Hi-C interaction heatmap showing that the physical partitioning of the genome is highly correlated with the EPUs that encompass Pcdhα, Pcdhβ and Pcdhγ gene clusters on chromosome 18. Top: normalized Hi-C interaction frequencies in mouse cortex as a two-dimensional heatmap. Bottom: UCSC genome browser views of the same regions, including the identified EPUs and the ChIP-Seq data (H3K27ac, H3K4me1, H3K4me3, polII and CTCF) in cortex. e, The average normalized Hi-C interaction frequencies for enhancer–promoter pairs within EPUs, between EPUs, and expected by random chance.
Enhancers are important in regulating tissue-specific expression patterns during mammalian development. However, finding target genes for enhancers is not straightforward because they are frequently distal from the genes they control. Assigning enhancers to the nearest transcription start sites is the most widely used method. A recently published strategy associates enhancers and promoters located within the same domain defined by the CTCF-binding sites, assuming that insulators can block promoter–enhancer interactions10. We evaluated these two methods by assessing the Spearman correlation coefficients (SCCs) between H3K4me1 signals at enhancers and the polII intensities at target promoters (Supplementary Methods).As a control, we observed that the SCCs from the randomly paired enhancers and promoters have a bell-shaped distribution with a median of 0 (Fig. 3b). The distribution of the SCCs from enhancer–promoter pairs identified by the nearest transcription start site (TSS) model and CTCF block model are only slightly better than the random control, with medians at 0.11 and 0.08, respectively (Fig. 3b). In addition, 34% and 38% of the enhancer/promoter pairs in the nearest TSS model and the CTCF block model, respectively, are negatively correlated, indicating potentially incorrect promoter assignment. To improve the linking of enhancers to their targets, a logistic regression classifier was recently introduced and shown toper form better than the nearest TSS model21. However, this model is still based on the one-to-one relationship between an enhancer and a gene, with a bias towards the nearby genes. It has been reported that a significant fraction of enhancers may not target the nearest promoters22. Therefore, to gain a better understanding of enhancer/promoter organization we assessed the correlation of the chromatin state at enhancers and polII occupancy at promoters for each possible pair of elements along a chromosome. We observed that co-regulated promoters and enhancers tend to form clusters with variable sizes (Fig. 3c). We developed an algorithm to detect these local clusters, defined as enhancer–promoter units (EPUs) (Supplementary Methods). Performing this analysis genome-wide, we defined 8,792 EPUs that contained at least one promoter and one enhancer (Supplementary Table 5), encompassing 1,258 million base pairs, or nearly half of the mouse genome. The median enhancer-to-promoter ratio per EPU was 5.67 (Supplementary Table 6), which is consistent with the idea that multiple enhancers may be used to regulate a gene23. We confirmed that previously defined enhancer–promoter pairs are frequently located within the same EPU. For example, out of the 2,605 putative enhancer–promoter pairs recently defined in the human genome21, most of their mouse homologues are found within the same EPU (83.8% observed versus 43% expected; P < 2.2 × 10−16, Fisher's exact test). In addition, each of the four linked enhancer–promoter pairs reported by a recent study24 was found within the same EPU. Finally, seven locus-control regions for Hbb genes were all identified within the same EPU25.
The discovery of EPUs provides strong evidence that the genome is partitioned into functional domains in which cis-regulatory elements are coordinately regulated, whereas elements located in different domains are relatively insulated from each other. This organization is reminiscent of recently identified topological domains, defined by chromatin interactions, in the mammalian genome26,27. Indeed, comparison of the EPUs with the higher order chromatin organization shows that physical partitioning of the genome is highly correlated with functional partitioning on the basis of the coordinated activities of cis-regulatory sequences (Fig. 3d and Supplementary Fig. 7).
EPUs provide a new approach for associating enhancers with their target genes. Instead of being linked to the nearest genes, an enhancer could be assigned to one or more promoters within an EPU that show significant correlation. To validate the enhancer–promoter relationship predicted by this approach (Supplementary Table 7), we examined long-range looping interactions between the enhancers and promoters, reasoning that true enhancer–promoter target pairs should have higher interaction frequencies than neighbouring non-target sites. We performed chromosome confirmation capture (3C) experiments for five enhancer–promoter pairs predicted to be linked in the cortex but not in mouse ES cells, and two enhancer–promoter pairs predicted not to be linked in either tissue or cell type. The five linked pairs showed enrichment of 3C signals, whereas the two non-linked pairs did not, indicating that the EPU analysis can accurately reveal a enhancer– promoter targeting relationship (Supplementary Fig. 8 and Supplementary Table 8). For a systematic evaluation of the enhancer– promoter pairing relationships as defined by this approach, we examined long-range looping interactions in adult mouse cortex genome-wide by using the Hi-C method28. We observed that interactions between predicted enhancer–promoter pairs within the same EPUs occured significantly more frequently than interactions between enhancer–promoter pairs of the same genomic distance but across different EPUs or by random chance (Fig. 3e; P < 2.2 × 10−16, Wilcoxon test). These results suggest that EPUs may help in assigning enhancers to their target promoters.
Mammalian development requires a precise temporal gene expression program that is tightly controlled by transcription factors and cis-regulatory elements. The map of cis-regulatory sequences now provides a chance for us to analyse the potential mechanisms involved in temporal regulation of gene expression. First, we identified enhancers specific to embryonic and adult brain on the basis of H3K4me1 intensities (Fig. 4a). We observed that the former class was associated with genes expressed in neuron differentiation and neuron development, whereas the latter was associated with genes important for adult brain functions, for example the transmission of nerve impulses (Fig. 4b, c and Supplementary Fig. 9). We made similar observations for stage-specific enhancers in liver and heart (Supplementary Figs 9 and 10).
Figure 4. Motif analysis of tissue-specific enhancers.

a, Classification of development stage-specific enhancers based on their chromatin state (H3K4me1) between embryonic (embryonic day 14.5; E14.5) and adult brain. b and c, Gene Ontology analysis for the genes associated with embryonic brain-specific enhancers and adult cortex-specific enhancers. d, Classification of tissue-specific enhancers on the basis of their chromatin state (H3K4me1) among different tissue and cell types. The first 19 tissue-specific clusters were used for further motif analysis. The last cluster contains enhancers enriched in multiple tissues with no clear patterns. e, Enrichment of three transcription factor recognition motifs in the predicted enhancers. REST, RE1-silencing transcription factor. f, Heatmap showing the clustering of 270 transcription factor motifs on the basis of their enrichment in the various groups of enhancers as identified in e.g, Boxplot showing that the de novo motifs found in tissue-specific enhancers are evolutionarily conserved. h–k, Examples of motifs that show high sequence conservation: h, REST motif in cortex-specific enhancers; i, Hnf1 motif in kidney-specific enhancers; j, Oct4 motif in mESC-specific enhancers; k, Atoh1 motif in cerebellum-specific enhancers.
We also systematically identified potential transcriptional regulators acting on tissue-specific gene expression programs. We first defined 19 groups of tissue-specific enhancers on the basis of H3K4me1 occupancy (Fig. 4d). Gene Ontology term analysis confirmed that the enhancers in each group are linked to genes specifically expressed in the corresponding tissue or developmental stage (Supplementary Fig. 11). We also observed that the known motifs of transcription factors that have been reported to function in certain tissues are enriched in the tissue-specific enhancers from the same tissue (Fig. 4e). To identify new transcription factors involved in each group of tissue-specific enhancers, we performed de novo motif analysis and identified 206 motifs with a very stringent cutoff (P,< 10−20; Supplementary Tables 9 and 10). We found that 91% of them (188 out of 206) showed significant levels of evolutionary conservation among the vertebrate species (Fig. 4g, h–k). We annotated the most likely transcription factor for each motif by comparing it with public transcription factor databases and verified that the matching transcription factor was expressed in the corresponding tissue. A total of 62% of the conserved de novo motifs (117 out of 188) were associated with a known transcription factor, and 75% of them (88 out of 117) have previously been implicated in the regulation of gene expression in specific tissues (Supplementary Tables 9 and 11). We performed a similar motif analysis for promoters, and compared the top motifs enriched in promoter and enhancer sequences in the same tissue (Supplementary Table 12). Only 11 motifs were shared between the two groups of motifs, whereas 93% of transcription factor motifs enriched in the tissue-specific enhancer were unique only to enhancers, confirming that enhancers and promoters contain different regulatory sequences, as we reported previously10.
Here we have described an initial survey and a draft annotation of the cis-regulatory sequences in the mouse genome. The wide range of tissue and cell types examined in this study provides an unprecedented opportunity to detect tissue-specific and development-specific promoters and enhancers, analyses of which have yielded potential clues to transcription regulators of tissue-specific gene expression programs. We show that nearly half of the mouse genome is organized into EPUs containing enhancers and promoters with correlated activities. These EPUs overlap significantly with recently discovered topological domains, defined by chromatin interactions, thus linking physical partitioning of the genome with transcriptional regulation. Such multigene structures22,29 probably represent a general feature of genome organization in mammals.
Methods Summary
Mouse tissues were harvested from eight-week-old male C57Bl/6 mice (Charles River). The murine embryonic fibroblasts were isolated from C57Bl/6 embryos at embryonic day 14.5. ChIP-Seq and RNA-Seq experiments were performed as described14,30, with the use of Illumina GAIIx and HiSequation (2000) instruments (details are provided in Supplementary Information). Hi-C experiments in adult cortex were conducted as described28. A software pipeline to process ChIP-Seq data and predict enhancers is described in Supplementary Methods. Highly correlated biological replicates for ChIP-Seq experiments were pooled for all subsequent data analyses. An algorithm to define the enhancer–promoter unit is given in Supplementary Methods.
Supplementary Material
Acknowledgments
We thank F. Jin, Y. Luu, S. Klugman, A. Y.-J. Kim, Q.-M. Ngo, B. A. Gomez and S. Selvaraj for consultation. The mESC line Bruce4 was a gift from UCSD Transgenic Core. Research funding was provided by the National Human Genome Research Institute (R01HG003991) and the Ludwig Institute for Cancer Research to B.R. Y.S. is supported by a postdoctoral fellowship from the International Rett Syndrome Foundation. J.D. is supported by a pre-doctoral fellowship from the California Institute for Regenerative Medicine.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions Y.S., F.Y. and B.R. designed the experiments. Y.S., D.M., Z.Y. and L.L. conducted experiments. F.Y. performed computational analysis. U.W. contributed to RNA-Seq data analysis. J.D. contributed to Hi-C data analysis. S.K. and L.E. performed DNA sequencing and initial data processing. V.L. provided CTCF monoclonal antibodies. Y.S., F.Y. and B.R. prepared the manuscript.
Author Information Data sets are available from the ENCODE website (http://genome.ucsc.edu/ENCODE), the supporting website for this paper (http://chromosome.sdsc.edu/mouse/index.html) and the Gene Expression Omnibus (GSE29184)
Reprints and permissions information is available at www.nature.com/reprints
The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at www.nature.com/nature.
References
- 1.Waterston RH, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 2.Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461:199–205. doi: 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The ENCODE Project Consortium. A user's guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011:9–e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerstein MB, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roy S, et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787–1797. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim TH, et al. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rada-Iglesias A, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 12.Kim TH, et al. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkhomchuk D, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence data base of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt D, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li G, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nature Rev Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kagey MH, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Splinter E, et al. CTCF mediates long-range chromatin looping and local histone modification in the β-globin locus. Genes Dev. 2006;20:2349–2354. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nora EP, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dixon JR, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chepelev I, Wei G, Wangsa D, Tang Q, Zhao K. Characterization of genomewide enhancer–promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 2012;22:490–503. doi: 10.1038/cr.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hawkins RD, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.