

Abstract
Background.
Autophagy is a catabolic pathway that permits cells to recycle intracellular macromolecules, and its inhibition reduces pancreatic cancer growth in model systems. We evaluated hydoxychloroquine (HCQ), an inhibitor of autophagy, in patients with pancreatic cancer and analyzed pharmacodynamic markers in treated patients and mice.
Methods.
Patients with previously treated metastatic pancreatic cancer were administered HCQ at 400 mg (n = 10) or 600 mg (n = 10) twice daily. The primary endpoint was 2-month progression-free survival (PFS). We analyzed peripheral lymphocytes from treated mice to identify pharmacodynamic markers of autophagy inhibition that were then assessed in peripheral lymphocytes from patients.
Results.
Among 20 patients enrolled, 2 (10%) were without progressive disease at 2 months. Median PFS and overall survival were 46.5 and 69.0 days, respectively. Treatment-related grade 3/4 adverse events were lymphopenia (n = 1) and elevated alanine aminotransferase (n = 1). Tolerability and efficacy were similar at the two dose levels. Analysis of treated murine lymphocytes suggested that LC3-II expression by Western blot is a reliable marker for autophagy inhibition. Analysis of LC3-II in patient lymphocytes demonstrated inconsistent autophagy inhibition.
Conclusion.
Mouse studies identified LC3-II levels in peripheral lymphocytes as a potential pharmacodynamic marker of autophagy inhibition. In patients with previously treated metastatic pancreatic cancer, HCQ monotherapy achieved inconsistent autophagy inhibition and demonstrated negligible therapeutic efficacy.
Abstract
摘要
背景. 自噬是一种分解代谢过程,使得细胞内大分子得以回收再利用,自噬抑制在模型系统研究中显示能够阻碍胰腺癌生长。我们在胰腺癌患者中评估了自噬抑制剂羟氯喹(HCQ),并在经治患者与小鼠中分析了药效动力学标记物。
方法.入组经治的转移性胰腺癌患者,给予HCQ 400 mg(n = 10)或600 mg(n = 10),每日2次。主要终点为2个月无疾病进展生存期(PFS)。我们针对经治小鼠开展了外周血淋巴细胞分析,以识别自噬抑制的药效动力学标记物,随后在患者外周血淋巴细胞中进行评估。
结果.共入组20例患者,2例(10%)在2个月时未出现疾病进展。中位PFS以及总生存期分别为46.5、69.0天。 治疗相关3/4级不良事件为淋巴细胞减少(n = 1),丙氨酸氨基 转移酶升高(n = 1)。两个剂量水平的耐受性和疗效相似。经治小鼠淋巴细胞分析表明,经蛋白印迹法检测的LC3-II表达可作为自噬抑制的可靠标记物。患者淋巴细胞LC3-II分析则显示出自噬抑制的不一致性。
结论.小鼠研究证实,外周血淋巴细胞LC3-II水平是自噬抑制的潜在药效动力学标记物。在经治的转移性胰腺癌患者中,HCQ单药治疗并不能普遍达到自噬抑制效果,这提示疗效甚微。Oncologist 2014; 19:637–638
Author Summary
Discussion
Autophagy is a catabolic pathway that permits cells to recycle intracellular macromolecules and organelles [1, 2]. The role of autophagy in cancer is complex and likely is dependent on tumor type, genetic landscape, and phase of tumorigenesis [2–4]. Nevertheless, a subset of malignancies require autophagy for growth and survival [1]. Pancreatic cancers have high basal levels of autophagy, and inhibition of autophagy impeded their growth in vitro and in mouse models [5]. Chloroquine (CQ) and hydroxychloroquine (HCQ) inhibit autophagy in vitro [5–7]. We conducted a phase II clinical trial and translational study of HCQ in patients with previously treated metastatic pancreatic cancer. Concurrently, we examined peripheral lymphocytes from CQ-treated mice to identify pharmacodynamic markers of autophagy inhibition. With more than 35 trials assessing HCQ as cancer therapy, it is paramount to identify reliable pharmacodynamic markers in humans.
In mice receiving CQ at doses sufficient to inhibit autophagy in tumors and to cause tumor regression, we noted increased levels of LC3-II, but not p62, in peripheral lymphocytes and hepatocytes (Fig. 1). This suggests that monitoring LC3-II levels in human peripheral lymphocytes may provide a useful pharmacodynamic marker for monitoring autophagy inhibition. In our patients, HCQ at 800 mg or 1,200 mg daily resulted in inconsistent autophagy inhibition, as measured by LC3-II in peripheral lymphocytes. Furthermore, the 2-month PFS rate of 10% was inadequate to justify further studies of single-agent HCQ in this patient population.
Figure 1.
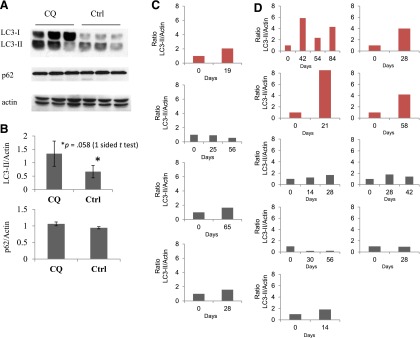
Ratio of LC3-II to actin as a biomarker for autophagy inhibition. (A): Autophagy inhibition in mouse lymphocytes. A Western blot probed for LC3, p62, and β-actin in the presence (first three lanes) or absence (second three lanes) of CQ treatment. Each lane comprised pooled lymphocyte samples from two to three individual mice treated with drug or control. (B): A bar graph displays the relative quantity of LC3-II (upper graph) and p62 (lower graph) as a ratio to β-actin as assessed by densitometry. Autophagy inhibition in circulating lymphocytes from patients receiving hydroxychloroquine (HCQ) at either 400 mg b.i.d. (C) or 600 mg b.i.d. (D). Each bar graph reflects results from a single patient prior to treatment (day 0), and then at one time point or more while receiving HCQ. For each patient, a baseline ratio of LC3-II to actin was determined based on assessment by densitometry of Western blot prior to starting (day 0) and then at one time point or more following initiation of HCQ. Graphs in red depict patients with a more than twofold increase in relative LC3-II levels on serial blood draws.
Abbreviations: CQ, chloroquine; Ctrl, control.
Several mechanisms may explain the lack of efficacy for HCQ. First, autophagy inhibition alone in metastatic human pancreatic cancer may not be sufficient to affect tumor growth. Indeed, studies have suggested that autophagy inhibition can act synergistically with cytotoxic chemotherapy [6, 8]. Second, autophagy inhibition at the HCQ doses tested appeared inconsistent when assessed in circulating lymphocytes. Consequently, the doses tested may not adequately inhibit autophagy within tumors. The use of HCQ with concurrent chemotherapy may obviate the need for complete autophagy inhibition in tumors, and such trials are ongoing. Optimization of HCQ dosing or administration of more potent inhibitors may also be necessary in future studies. Third, this study was conducted in patients who received multiple lines of prior chemotherapy. Given the short survival of patients with previously treated pancreatic cancer, patients may not have received sufficient HCQ to manifest a tumor response. Alternatively, chemotherapy may promote the upregulation of autophagy as a survival mechanism [9], making autophagy inhibition in future lines of therapy more difficult.
Despite our negative efficacy results for HCQ in patients with previously treated metastatic pancreatic cancer, inhibition of autophagy remains an intriguing therapeutic strategy for pancreatic cancer and other tumor types. Successful implementation of this therapeutic approach will require reliable markers of autophagy inhibition, and our data suggest LC3-II as a candidate pharmacodynamic marker for use in clinical trials.
Supplementary Material
Acknowledgments
Brian M. Wolpin is supported by NIH Grant K07 CA140790 from the National Cancer Institute, an American Society of Clinical Oncology Career Development Award, a Howard Hughes Medical Institute Early Career Physician-Scientist Award, the Lustgarten Foundation, and Promises for Purple. Alec C. Kimmelman is supported by NIH Grant 1R01CA157490 from the National Cancer Institute, American Cancer Society Grant RSG-13-298-01-TBG, a Department of Defense Discovery Award (W81XWH-12-1-0459), and the Lustgarten Foundation for Pancreatic Cancer Research. Charles S. Fuchs is supported by NIH Grant 1R01CA124908 and P50127003 from the National Cancer Institute and the Robert T. and Judith B. Hale Fund for Pancreatic Cancer Research.
Footnotes
Brian M. Wolpin and Douglas A. Rubinson contributed equally as joint first authors.
Access the full results at: Kimmelman_Wolpin-14-86.theoncologist.com
ClinicalTrials.gov Identifier: NCT01273805
Sponsor(s): Dana-Farber Cancer Institute
Principal Investigator: Brian M. Wolpin
IRB Approved: Yes
Author disclosures and references available online.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.