

Abstract
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease characterized by autoantibodies targeting nucleic acid-associated antigens. The endosomal toll-like receptors TLR7 and TLR9 are critical for generation of Abs targeting RNA- or DNA-associated antigens, respectively. In murine lupus models, deletion of TLR7 limits autoimmune inflammation, while deletion of TLR9 exacerbates disease. Whether B cell or myeloid TLR7/TLR9 signaling is responsible for these effects has not been fully addressed. Here, we utilize a chimeric strategy to evaluate the effect of B cell-intrinsic deletion of TLR7 vs. TLR9 in parallel lupus models. We demonstrate that B cell-intrinsic TLR7 deletion prevents RNA-associated Ab formation, decreases production of class-switched Abs targeting non-nuclear antigens and limits systemic autoimmunity. In contrast, B cell-intrinsic TLR9 deletion results in decreased DNA-reactive Ab, but increased Abs targeting a broad range of systemic autoantigens. Further, we demonstrate that B-intrinsic TLR9 deletion results in increased systemic inflammation and immune-complex (IC) glomerulonephritis despite intact TLR signaling within the myeloid compartment. These data stress the critical importance of dysregulated B cell-intrinsic TLR signaling in the pathogenesis of SLE.
INTRODUCTION
Despite numerous potential autoantigen targets, patients with SLE frequently develop a restricted autoantibody repertoire targeting nucleic acid-associated antigens. In addition to exogenous pathogens, the Toll-like receptor (TLR) family of germ-line encoded, pattern-recognition receptors are able to recognize endogenous ligands. Nucleic acid-containing apoptotic particles promote activation of autoreactive B cells via dual B cell receptor (BCR)/Toll-like receptor (TLR)-mediated signals, thereby explaining the prominence of antinuclear Abs in autoimmunity (1–3). The Myd88-dependent, endosomal receptors TLR7 and TLR9 are critical in this context, with TLR7 required for the generation of Abs targeting RNA and RNA-associated proteins, while TLR9 activation promotes production of Abs targeting dsDNA and chromatin (4–7).
Importantly, two alternate, but not mutually-exclusive, mechanisms may explain the role of TLR7 and TLR9 in autoimmune pathogenesis in vivo. Dual BCR/TLR activation may either promote a B cell-intrinsic break in tolerance, or IC-mediated TLR activation of plasmacytoid dendritic cells may promote autoimmunity via increased type 1 IFN production (4, 8). In addition, tlr7-deficient lupus-prone mice are significantly protected from autoimmunity, whereas disease is exacerbated in tlr9−/− autoimmune strains (1–3). The mechanisms underlying accelerated autoimmunity in the absence of TLR9 remain unclear. However, tlr9−/− MRL.Mplpr/lpr mice develop greater plasmacytoid dendritic cell activation and increased serum IFN-α levels suggesting that loss of tlr9 in the myeloid compartment exacerbates autoimmunity on the MRL.Mplpr/lpr background (6).
While several models (9–13) have implicated B cell Myd88 signaling in autoimmune pathogenesis, the B-intrinsic impact of TLR7 and TLR9 has only been addressed in a limited number of studies. The role of B cell-intrinsic TLR7 signaling was evaluated in two studies using the TLR7 transgenic (tlr7Tg) model of spontaneous autoimmunity. First, using a chimeric transplant strategy comparing wild-type (WT) vs TLR7 transgenic (tlr7Tg) hematopoietic cells, Walsh et al. demonstrated that tlr7Tg B cells are preferentially recruited into germinal centers (GC) and generate CD138+ plasmablasts (14). Second, Hwang et al. used a CD19Cre recombinase system to normalize B cell TLR7 expression in a low copy tlr7Tg strain crossed with the Sle1 lupus susceptibility locus. B cell-intrinsic TLR7 normalization decreased RNA-associated anti-SnRNP titers, but did not impact GC and plasma cell formation and only moderately reduced autoimmune glomerulonephritis (15). To address the B cell-intrinsic impact of TLR9, a recent study utilized MRL.Faslpr mixed bone marrow (BM) chimeras in which tlr9-deficiency was primarily limited to the B cell compartment and demonstrated a specific reduction in anti-nucleosome reactivity. Whether B cell-intrinsic tlr9 deletion accelerated systemic autoimmunity, however, was not addressed in that study (16).
We recently developed a murine model of autoimmunity that provides important information regarding how self-reactive B cells are initially activated and can drive generation of pathogenic Ab (12). In this model, B cells, but not other hematopoietic lineages, harbor a mutation that abolishes the expression of Wiskott Aldrich syndrome protein (WASp). In the absence of WASp, peripheral B cells are rendered mildly hyper-responsive to both B cell receptor (BCR) and Toll-like receptor (TLR) ligands. In this setting, Wiskott-Aldrich syndrome (WAS)-null (was−/−) B cells drive the development of humoral autoimmunity that is characterized by spontaneous GCs, class-switched IgG2c autoantibodies and IC glomerulonephritis. Disease development is dependent on wild-type CD4+ T cells and MyD88-dependant B cell-intrinsic TLR signaling. An important advantage of the WASp chimera model is that dysregulated immune responses are limited to the B cell compartment, allowing genetic manipulation in a B cell-intrinsic fashion. In the current study, we detail the relative contributions of B cell TLR signaling in humoral autoimmunity and demonstrate that B cell-intrinsic TLR7 vs TLR9 activation is sufficient to alter the autoantibody repertoire. In addition, we demonstrate that B cell TLR7 and TLR9 signals exert opposing pathogenic and protective effects on systemic inflammation and autoimmune pathology.
MATERIALS AND METHODS
Mice
Ly.5.1+ and Ly5.2+ C57BL/6, μMT, was−/−, tlr7−/− and tlr9−/− mice were bred and maintained in the specific pathogen-free (SPF) animal facility of Seattle Children’s Research Institute (Seattle, WA). All animal studies were conducted in accordance with Seattle Children’s Research Institute IACUC approved protocols.
Bone Marrow Transplantation
WT, was−/−, was−/−.tlr7−/− or was−/−.tlr9−/− donor BM and μMT BM (20:80 ratio, 5 × 106 total BM), was injected into lethally irradiated (450cGy x 2 doses) μMT recipients. Chimeras were sacrificed at 24–36 weeks post-transplant. For CD4-depletion assays, mice were treated weekly with IP injection of 250μg anti-CD4 (GK1.5) or isotype control (rat IgG2b) Ab (UCSF Antibody Core) from 5–24 weeks post-transplant, as described (12). Data are representative of four (BWT, BWAS−/−, and BW/TLR9−/−), three (BW/TLR7−/−) or two (BWAS−/− CD4 depletion) independent experimental cohorts.
Flow Cytometry and Antibodies
Flow cytometry was performed as described (12). Antibodies used: B220 (RA3-6B2), CD4 (RM4-5), Thy1.2 (53-2.1), CD138 (281-2), CXCR5 (2G8) from BD Biosciences; CD62L (MEL-14), CD11c (N418), Gr-1 (RB6-8C5), Ly5.1 (A20), Ly5.2 (104), CD11b (M1/70), GL7 (GL-7), PD-1 (J43) from eBioscience; goat anti-mouse IgM-, IgG-, IgG2c- HRP conjugated, unlabeled, or isotype from Southern Biotechnology; CD19 (ID3), CD44 (IM7) from BioLegend; PNA (Fl-1071) from Vector Labs; and Fas (Jo2) from BD Pharmingen.
Measurement of autoantibodies
For ANA assays and determination of dsDNA reactivity by kinetoplast staining, diluted serum (1:200 for ANA or 1:50 for kinetoplast) was added to fixed Hep-2 ANA slides (Bio-Rad 30472) or Crithidia luciliae slides (Bio-Rad 31069). FITC-conjugated goat anti-mouse IgG served as the detection antibody and slides were counterstained with DAPI. Fluorescence images were obtained using Leica DM6000B microscope, Leica DFL300 FX camera, and Leica Application Suite Advanced Fluorescence software at 40x with a constant 5 second (ANA) or 2 second (kinetoplast) exposure. ANAs were scored as nuclear homogeneous, nucleolar, cytoplasmic or a combination of these patterns. Kinetoplast reactivity was defined by co-localization of DAPI-positive C. luciliae DNA and IgG FITC staining; with staining intensity scored from 0–3. ANA patterns and kinetoplast intensity were scored by two independent observers blinded to genotype.
For specific Ab ELISAs, 96 well Immuno plates (Nunc) were coated with: calf thymus dsDNA (100ug/ml; Sigma-Aldrich D3664-5X2MG); sm/RNP (5ug/ml; Arotec Diagnostic Limited ATR01-10); PC-BSA (10ug/ml; Biosearch Technologies PC-1011-10); or MDA-LDL (10ug/ml; Academy Bio-medical Co. 20P-MD-L110). Plates were blocked for 1 hour with 1% BSA/PBS prior to addition of diluted serum for 2 hours. Specific Ab were detected using goat anti-mouse IgM-, IgG- or IgG2c-HRP (1:2000 dilution; Southern Biotechnology Associates) and peroxidase reactions developed using OptEIA TMB substrate (BD Biosciences). Absorbance at 450nm was read using a Victor 3 plate reader (PerkinElmer) and data analyzed using GraphPad Prism (GraphPad Software, Inc.). Autoantigen microarrays were performed at the UT Southwestern Medical Center Microarry Core Facility, Dallas, TX (17).
Spleen and kidney immunofluorescence staining
Mouse spleens and kidneys were embedded in OCT compound and snap frozen over liquid nitrogen. 10 μm sections were cut on a cryostat, mounted on Superfrost plus slides, and fixed in −20°C acetone for 20 minutes. After rehydration in staining buffer (PBS, 1% goat serum, 1% BSA, 0.1% Tween-20), slides were stained with: B220-PE, CD4-FITC and GL7-APC (spleen); or, IgG-FITC, IgG2c-FITC, or C3-FITC (kidney). Images were acquired using a Leica DM6000B microscope, Leica DFL300 FX camera, and Leica Application Suite Advanced Fluorescence software. For glomerular immune-complex quantification, images were obtained using a constant exposure and scored from 0–3 by two independent observers blinded to genotype.
Histopathology
Tissues were fixed in 10% neutral buffered formalin, embedded in paraffin and tissues sections stained with Hematoxylin and eosin (H&E) (lung, liver, pancreas) or Jones’ methenamine silver-Periodic acid-Schiff (kidney) according to standard practices. Immunohistochemistry staining was performed using a Leica Bond–automated immunostainer and HRP-conjugated secondary Abs. Histology images were acquired using a Nikon OptiPhot-2 microscope and a Canon Eos 5D Mark II camera. Tissue sections were examined by a board-certified veterinary pathologist (D.L.) who was blinded to study design. Kidney sections were analyzed by two observers blinded to genotype (K.L.H. and C.E.A.) and scored from 0–2 based on degree of mesangial expansion, GBM thickening/reduplication and glomerular hypercellularity. For quantification of MAC-2 area, glomeruli were manually delineated in 4 independent kidney sections and MAC-2+ area as a percentage of glomerular area determined using Image Pro Plus (Media Cybernetics, Inc. Rockville, MD).
Statistical Evaluation
P-values were calculated using one-way ANOVA, followed by Tukey’s multiple comparison test (GraphPad Software, Inc.).
RESULTS
B cell-intrinsic TLR7 and TLR9 signals alter the autoantibody repertoire
To test the impact of B cell-intrinsic TLR7 and TLR9 deletion in humoral autoimmunity, we generated mixed bone marrow (BM) chimeras by transplanting a mix of 20% WT, Wiskott-Aldrich syndrome (WAS)-null (was−/−), double-deficient was−/−.tlr7−/−, or was−/−.tlr9−/− BM with 80% B cell-deficient μMT BM into lethally irradiated (450cGy x 2 doses) μMT recipients. After reconstitution, all B cells were donor-derived (either WT, was−/−, was−/−.tlr7−/−, or was−/−.tlr9−/−), while ~80% of myeloid cells and >97% CD4+ T cells were wild-type (not shown). Utilizing this strategy, in the TLR-deficient cohorts, all B cells, but only ~20% of myeloid cells, lacked TLR7 or TLR9. Respective B cell chimeras will henceforth be referred to as BWT, BWAS−/−, BW/TLR7−/− and BW/TLR9−/−.
We initially screened for autoimmunity using fluorescent antinuclear antibody (ANA) assays. The majority of BWAS−/− were ANA positive with mixed homogenous and nucleolar staining patterns, consistent with both DNA and RNA Ab reactivity. In contrast, BW/TLR7−/− ANA staining was homogenous with no nucleolar reactivity, while BW/TLR9−/− showed nucleolar and cytoplasmic, but not homogenous, ANA reactivity (Fig. 1A). These altered ANA staining patterns suggested a B-intrinsic impact of TLR7 vs. TLR9 on RNA- and DNA-associated Abs, which we evaluated using specific autoantigen ELISAs. Prior reports have demonstrated persistent anti-dsDNA Ab in tlr9-deficient murine lupus models; an effect attributed to a lack of DNA specificity of standard anti-dsDNA ELISA assays (6, 18). Although we noted anti-dsDNA Ab by ELISA in BW/TLR9−/−, DNA-reactivity by highly-specific Crithidia luciliae kinetoplast staining was abrogated in BW/TLR9−/−, but unaffected in BW/TLR7−/− (Fig. 1B, Supplemental Fig. 1A). In contrast, RNA-associated anti-sm/RNP Ab were abolished in BW/TLR7−/−. Further, anti-sm/RNP Ab titers of the pathogenic IgG2c subclass were significantly increased in BW/TLR9−/− vs. BWAS−/− chimeras, an observation consistent with elevated anti-RNA titers in tlr9−/− MRL.Mplpr/lpr mice (Fig. 1C). Therefore, the requirement for TLR7 in anti-RNA and TLR9 in anti-DNA Ab production is B cell-intrinsic; and lack of TLR9 enhances anti-RNA Ab formation in a B-intrinsic manner.
Figure 1. B cell-intrinsic TLR7 and TLR9 signals promote anti-nuclear Ab production.

(A) Upper: ANA staining patterns were scored as homogeneous, nucleolar, or cytoplasmic by two independent, blinded observers. Unfilled portion of circle represents ANA-negative animals. The number in the circle represents the total number of mice analyzed. Lower: Representative ANA images showing: combined homogeneous/nucleolar staining in BWAS−/−; homogenous pattern with nucleolar sparing in BW/TLR7−/−; nucleolar and cytoplasmic staining in BW/TLR9−/−. Bars, 50μm. (B) Anti-dsDNA IgG Ab by ELISA and kinetoplast staining. (C) Anti-sm/RNP IgG and IgG2c Ab by ELISA. (B, C) Mean ± s.e.m. *, P<0.05; **, P<0.01; ***, P<0.001. (A–C) Total mice analyzed: BWT (n=6), BWAS−/− (n=18) BW/TLR7−/− (n=11), and BW/TLR9−/− (n=16), pooled from 4 independent experimental cohorts.
Generation of Ab targeting non-nuclear antigens was recently shown to be Myd88 dependent (11, 19), but the specific TLRs responsible for these Ab specificities have not been determined. For this reason, we measured IgG reactivity to apoptotic cell epitopes malondialdehyde conjugated with low-density lipoprotein (MDA-LDL) and phosphoryl choline (PC). In contrast to abrogation of anti-RNA responses, anti-apoptotic IgG responses were decreased in BW/TLR7−/−; but were not absent. Rather, we noted a specific decrease in pathogenic IgG2c subclass titers. Since we previously observed a lack of anti-dsDNA IgG2c in the absence of cognate T cell help (12), we measured anti-PC and anti-MDA-LDL IgG2c subclass titers in BWAS−/− treated weekly with monoclonal CD4-depleting antibody. In the absence of CD4+ T cells, BWAS−/− chimeras developed markedly reduced IgG2c Ab targeting PC and MDA-LDL; findings that mirrored BW/TLR7−/− (Fig. 2A, 2B). Therefore, in addition to the B cell-intrinsic requirement for TLR7 in generating Abs targeting RNA and RNA-associated antigens (Fig. 1C, and (15)), our data demonstrate that B cell-intrinsic TLR7 signals are required for generation of class-switched Abs targeting non-nuclear, apoptotic antigens.
Figure 2. B cell-intrinsic TLR7 and TLR9 signals impact autoantibody reactivity to apoptotic and disease-associated antigens.

(A) Left: Anti-MDA-LDL IgG. Right: Anti-MDA-LDL IgG2c in BWAS−/−, BW/TLR7−/−, BW/TLR9−/− and CD4-depleted BWAS−/− chimeras (BWAS−/− CD4 DEP). Data normalized to BWT anti-MDA-LDL IgG2c titer. (B) Left: Anti-PC IgG. Right: Anti-PC IgG2c in BWAS−/−, BW/TLR7−/−, BW/TLR9−/− and CD4-depleted BWAS−/− chimeras (BWAS−/− CD4 DEP). Data normalized to BWT anti-PC IgG2c titer. (A, B) Mean ± s.e.m. *, P<0.05; **, P<0.01; ***, P<0.001. Total mice analyzed: BWT (n=7), BWAS−/− (n=13) BW/TLR7−/− (n=13), BW/TLR9−/− (n=14), and BWAS−/− CD4 DEP (n=10). (C) Serum IgG Abs from BWT (n=4), BWAS−/− (N=6), BW/TLR7−/− (n=4) and BW/TLR9−/− (n=8) chimeras determined using an autoantibody array chip containing 88 specific autoantigens. Data are represented as a heat map of Z-scores ranging from −1 (blue) to 3 (red). Representative of two independent microarray analyses.
Finally, to quantify the impact of B cell tlr7 vs. tlr9 deficiency on the diversity of the autoantibody repertoire of BWAS−/− chimeras, sera was evaluated for reactivity to 88 distinct autoantigens by microarray. While BW/TLR7−/− demonstrated a broad reduction in serum Ab titers, the reactivity of BW/TLR9−/− sera was increased relative to BWAS−/− to a wide-range of different autoantigens (Fig. 2C). Consistent with our ELISA data, these Abs were predominantly of the pathogenic IgG2c subclass (Supplemental Fig. 1B). While absolutely required for the generation of anti-dsDNA Abs, TLR9 thus acts B-intrinsically to limit Ab responses to RNA-associated autoantigens, apoptotic cell epitopes and a broad range of disease-associated autoantigens.
B cell-intrinsic TLR7 and TLR9 exert opposing signals on immune activation
We previously demonstrated that BWAS−/− chimeras develop systemic inflammation that is dependent on B cell Myd88 signaling (12). B cell TLR7 and TLR9 signaling exerted opposing impacts on immune activation in this model, as splenic inflammation is exacerbated in BW/TLR9−/−, but abrogated in BW/TLR7−/−. Relative to BWAS−/−, BW/TLR9−/− chimeras developed increased splenomegaly, without marked alterations in total B cell numbers (Fig. 3A, 3B). Notably, splenomegaly in BWAS−/− and BW/TLR9−/− resulted from a significant expansion of CD4+ T cells, CD11b+GR1LO monocyte/macrophages and CD11b+GR1+ neutrophils. Consistent with a role for TLR9 in limiting autoimmune responses, B cell-intrinsic TLR9 deletion resulted in statistically greater numbers of CD4+ T cells and monocyte/macrophages relative to BWAS−/−, with a trend towards more neutrophils (Fig. 3C, 3D).
Figure 3. B cell-intrinsic TLR7 and TLR9 exert opposing signals on immune activation.
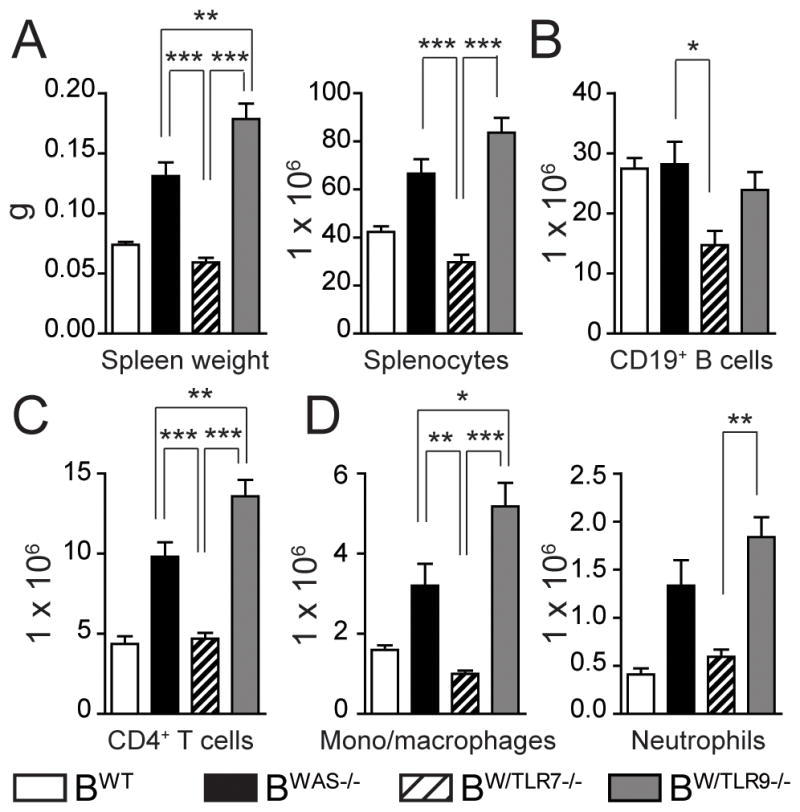
Splenic monocyte subsets were analyzed in BWT, BWAS−/−, BW/TLR7−/− and BW/TLR9−/− chimeras at sacrifice. (A) Spleen weight and total splenocyte numbers. (B–D) Number of splenic: (B) CD19+ B cells; (C) CD4+ T cells; (D) CD11b+GR1LO monocyte/macrophages and CD11b+GR1+ neutrophils. (A-D) Mean ± s.e.m. *, P<0.05; **, P<0.01; ***, P<0.001. Total mice analyzed: BWT (n=5), BWAS−/− (n=12) BW/TLR7−/− (n=12), and BW/TLR9−/− (n=13), pooled from 4 independent experimental cohorts.
Although total B cell numbers were similar, activated B cell subsets including PNA+FAS+GL7+ GC B cells and B220LOCD138+ plasma cells/plasmablasts were markedly expanded in spleens of BWAS−/− and BW/TLR9−/− chimeras (Fig. 4A, 4C). In addition, splenic immunofluorescence staining revealed spontaneous GL7+ GCs in BWAS−/− and BW/TLR9−/− mice, which were absent in BW/TLR7−/− (Fig. 4D). Relative to BWAS−/−, BW/TLR9−/− animals exhibited a higher percentage of GC B cells with greater numbers of splenic CD138+ plasma cells/plasmablasts, suggesting that B cell-intrinsic TLR9 signals serve to limit B cell activation. Consistent with this, total serum IgM, IgG and IgG2c Ab titers were markedly elevated in BW/TLR9−/− mice relative to BW/TLR7−/− mice, with a trend towards greater Ab titers than BWAS−/− chimeras (Fig 4E).
Figure 4. Divergent impact of B cell-intrinsic TLR7 and TLR9 on B cell activation.

(A, B) Representative FACS plots (left), total number (middle) and percentage (right) of: (A) PNA+ FAS+ GL7+ germinal center B cells (gated on CD19+ B cells); and (B) CD11b+ CD11c+ “age-associated B cells” (gated on CD19+ B cells). (C) Representative FACS plots (left) and total number (right) of B220LO CD138+ plasmablasts/plasma cells (gated on total splenocytes). (A–C) Number in FACS plot represents percentage within gated population. (D) Representative examples of splenic sections stained with B220 (red), CD4 (green) and GL7 (blue). Stars denote spontaneous GCs in BWAS−/− and BW/TLR9−/− chimeras (*). Images were taken with 10X objective: bars, 100μm. (E) Total IgM, IgG and IgG2c serum titers in BWT, BWAS−/−, BW/TLR7−/− and BW/TLR9−/− chimeras. (A–C, E) Mean ± s.e.m. *, P<0.05; **, P<0.01; ***, P<0.001. Total mice analyzed: BWT (n=5), BWAS−/− (n=12) BW/TLR7−/− (n=12), and BW/TLR9−/− (n=13), pooled from 4 independent experimental cohorts.
BWAS−/− and BW/TLR9−/− also developed significant expansion of autoimmune disease-associated CD11b+CD11c+ “age-associated B cells” (ABCs), with up to 75% of total B cells expressing CD11b and CD11c in BW/TLR9−/−, exceeding the percentage ABCs previously reported in aged NZB/WF1 and Mer−/− autoimmune strains (20, 21). In keeping with importance of chronic TLR7 activation in ABC generation, this population was absent BW/TLR7−/− (Fig. 4B).
In addition to B cell activation, CD4+ effector/memory (EM) subsets (CD44HICD62LLO and CD44HICD62LHI) were increased in BWAS−/− and BW/TLR9−/− (with relatively greater T cell activation in BW/TLR9−/−), while the number of CD44LOCD62LHI naïve CD4 T cells did not differ between experimental groups (Fig. 5A). Consistent with greater GC B cell numbers, PD1+CXCR5+ T follicular helper cells were also expanded in BWAS−/− and BW/TLR9−/− (Fig. 5B). B cell-intrinsic TLR7 activation appears critical for T cell activation in this model, as CD4+ effector/memory (EM) and T follicular helper cell subset expansion is abrogated in BW/TLR7−/−.
Figure 5. B cell-intrinsic TLR7 and TLR9 exert opposing impacts on T cell activation.

(A, B) Representative FACS plots (left) and total number (right) of splenic: (A) naïve (CD44LOCD62LHI) and effector/memory (EM; CD44HICD62LLO and CD44HICD62LHI) CD4+ T cells; and, (B) PD1+CXCR5+ T follicular helper (TFH) cells. Number represents percentage within gated population (gated on splenic CD4+ T cells). Mean ± s.e.m. ***, P<0.001. Total mice analyzed: BWT (n=5), BWAS−/−, (n=12) BW/TLR7−/− (n=12), and BW/TLR9−/− (n=13), pooled from 4 independent experimental cohorts.
In conclusion, B cell-intrinsic activation of autoreactive was−/− B cells via TLR7 promotes global immune activation and expansion of activated B and T cell populations; while B cell TLR9 signaling serves to limit these processes.
B cell intrinsic TLR7 signaling promotes systemic autoimmunity
Aged BWAS−/− and BW/TLR9−/− chimeras demonstrated diffuse inflammation and lymphoid infiltrates involving multiple organs, in particular the lungs and liver; findings that were absent in all BWT and BW/TLR7−/− animals evaluated (Fig. 6A). Immunohistochemistry staining demonstrated that these lymphoid infiltrates were consistent with ectopic lymphoid follicles comprising separate B220+ B cell and CD3+ T cells zones (Fig. 6B). The presence of ectopic lymphoid tissues within target organs has been reported in several human autoimmune diseases, including rheumatoid arthritis and type 1 diabetes, and is correlated with disease severity in autoimmunity (22). We demonstrate that hyper-responsive was−/− B cells are sufficient to recruit WT T cells to ectopic lymphoid follicles in a TLR7-dependent manner, reinforcing the critical role of B cell-intrinsic TLR7 signals in the pathogenesis of systemic autoimmunity.
Figure 6. B cell-intrinsic TLR7 signals promote systemic inflammation and ectopic lymphoid follicles.
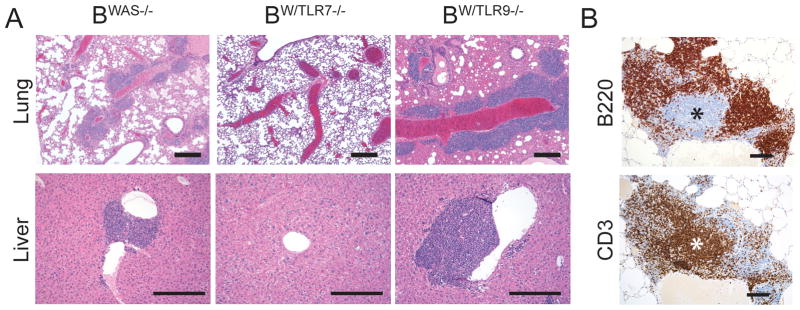
(A) Hematoxylin and eosin (H&E) stained tissues sections showing prominent lymphoid aggregates surrounding pulmonary vasculature in the lungs (upper panels, 4X) and portal tracts of the liver (lower panels, 10X) in BWAS−/− and BW/TLR9−/− chimeras. Bars, 100μm. (B) Immunohistochemistry of serial lung sections from a representative BW/TLR9−/− mouse showing distinct B220+ B cell follicle and CD3+ T cell zone (*). Image taken with 10X objective. Bars, 100μm. (A, B) Data are representative of: BWT (n=4), BWAS−/− (n=10), BW/TLR7−/− (n=9), and BW/TLR9−/− (n=11) mice, analyzed from 2 independent experimental cohorts.
B cell TLR7 deletion eliminates, while TLR9 loss exacerbates, IC glomerulonephritis
Given divergent impacts on Ab production and systemic inflammation, we evaluated the role of B cell-intrinsic TLR signaling on autoimmune glomerulonephritis. BWAS−/− chimeras develop inflammatory glomerulonephritis characterized by deposition of class-switched Ab, activated complement and recruitment of MAC-2+ macrophages (12). In keeping with an overall decrease in Ab titer and systemic inflammation, BW/TLR7−/− chimeras did not develop glomerulonephritis (Fig. 7A), an observation consistent with decreased glomerulonephritis in tlr7−/− MRL/Mplpr/lpr mice (6). Despite absent histologic glomerulonephritis, evaluation of glomerular Ig deposition by immunofluorescence staining showed persistent glomerular IgG in BW/TLR7−/− chimeras (albeit at lower levels than BWAS−/− and BW/TLR9−/−) (Fig. 7B). However, deposition of the pathogenic IgG2c subclass was absent in BW/TLR7−/− (Fig. 7C). In keeping with the role for IgG2c in complement and Fc-receptor activation (23), C3 complement deposition was absent in BW/TLR7−/− and glomerular MAC-2+ macrophages were not increased over BWT controls (Fig. 7D,E).
Figure 7. B cell-intrinsic TLR7 and TLR9 exert opposing effects on IC glomerulonephritis.

(A) Left panels: Representative examples of glomeruli from BWAS−/−, BW/TLR7−/− and BW/TLR9−/− chimeras stained with Jones’ methenamine silver-Periodic acid-Schiff stain. Right panel: Glomerular inflammation scored as: (0+) minimal mesangial expansion consistent with radiation injury; (1+) focal glomerular changes with moderate mesangial expansion, GBM thickening/reduplication and glomerular hypercellularity; or, (2+) diffuse glomerular changes with severe mesangial expansion, GBM thickening/reduplication and glomerular hypercellularity. Pathology was scored by two observers blinded to genotype. (B–D) Glomerular immune-complex deposits were determined by immunofluorescence staining for: (B) IgG (C) IgG2c, and (D) complement C3. Representative images are shown (left), together with intensity of glomerular fluorescent staining (right) scored from 0–3+ by two independent, blinded observers. (E) Left panels: Representative images of immunohistochemistry staining for glomerular MAC-2+ macrophages. Right panel: MAC-2+ area as a percentage of total glomerular area determined using Image Pro Plus (Media Cybernetics, Inc.). 4 independent kidney sections were examined per mouse. Bars, 50μm. (A–E) Results are mean ± s.e.m. *, P<0.05; **, P<0.01; ***, P<0.001. Total mice analyzed (A–D, E): BWT (n=6, 4), BWAS−/− (n=14, 6), BW/TLR7−/− (n=11, 9), and BW/TLR9−/− (n=15, 8), from 3 (A–D) and 2 (E) independent experimental cohorts.
In contrast to the protective effect of B cell TLR7 deletion, BWAS−/− and BW/TLR9−/− chimeras developed significant glomerulonephritis characterized by mesangial expansion, glomerular basement membrane thickening and glomerular hypercellularity (Fig. 7A). In keeping with increased serum IgG2c Ab, BW/TLR9−/− mice demonstrated statistically greater glomerular IgG2c deposition than BWAS−/−, with a trend towards greater C3 complement deposition and abundant infiltration by MAC-2+ macrophages, likely recruited by glomerular IgG2c ICs binding activating Fc-receptors (Fig. 7B-E). Increased IC glomerulonephritis in BW/TLR9−/− parallels accelerated autoimmune pathology in tlr9−/− MRL/Mplpr/lpr mice, but demonstrates that the disease exacerbating effects of TLR9 deletion can occur independently of the lack of TLR9 expression in the majority of the myeloid compartment.
DISCUSSION
The importance of the Myd88-dependant toll-like receptors TLR7 and TLR9 in lupus pathogenesis has been clearly established (1–3). In this context, global deletion of TLR7 in murine lupus models protects against autoimmunity, while TLR9 deficiency paradoxically exacerbates disease (4–7). The mechanisms underlying these divergent effects of TLR7 and TLR9 remain unclear, in part because studies involving globally gene deficient autoimmune strains are unable to address the differential impacts of TLR signaling on B cells vs. type 1 IFN-producing plasmacytoid dendritic cells. Our study provides the novel observation that B cell-intrinsic deletion of TLR7 or TLR9 recapitulates several of the key features of global TLR7/TLR9 deficiency in murine lupus models, despite intact TLR signaling within the myeloid compartment.
In this context, our data provide several important new insights into autoimmune pathogenesis. First, this study is the first to compare the B cell-intrinsic impact of TLR7 vs. TLR9 in parallel models of murine autoimmunity. Similar to global TLR7/TLR9 deletion in murine lupus models, we demonstrate that B cell-intrinsic TLR7 and TLR9 signals are required for in vivo generation of RNA- and DNA-reactive Ab, respectively. In addition, we also establish TLR7 as the major Myd88-dependent receptor responsible for B intrinsic breaks in tolerance to a broad range of non-nuclear, tissue-specific autoantigens.
Second, we identify a critical role for B cell-intrinsic TLR7 signals in sustaining spontaneous autoimmune GC responses. While the absence of anti-sm/RNP antibodies in BW/TLR7−/− may have been predicted from in vitro studies (24), the observation that antibodies targeting apoptotic and phospholipid epitopes develop in BW/TLR7−/−, but fail to undergo efficient CSR suggests an additional role for B cell TLR7 signaling in enhancing GC responses during autoimmune disease; a finding that parallels the requirement for TLR7 in Ab responses to chronic viral infection (25).
Third, we document a striking expansion of effector and TFH CD4+ T cells in BWAS−/− and BW/TLR9−/− animals. Similar expansion of the effector/memory T cell compartment has been documented on the lupus-prone MRL/Mplpr/lpr background (6), although it should be noted that CD4+ T cells in our model are genetically WT and not Fas-deficient. Our data, therefore, suggests that BCR/TLR-mediated activation of autoreactive B cells directly promotes T cell proliferation and activation during autoimmunity. These data are consistent with recent reports describing decreased T effector/memory expansion in the setting of B cell-intrinsic Myd88 deletion in the MRL.Faslpr and Lyn deficient models of autoimmunity (11, 13). Our data suggests that B cell TLR7 is likely the major Myd88-dependent receptor driving this CD4+ T cell activation. Surprisingly, dual BCR/TLR7 B cell activation was also sufficient for the establishment of ectopic lymphoid follicles within target organs, as have previously been described in several human autoimmune diseases (22). Although not formally demonstrated, we predict that these expanded effector/memory T cell compartments are enriched for autoreactive T cell clones, supporting the emerging hypothesis that activated autoreactive B cells can initiate breaks in T cell tolerance.
Forth, we demonstrate the importance of autoantibody isotype (in particular IgG2a/c) in promoting autoimmune nephritis. Despite producing anti-dsDNA Ab, BW/TLR7−/− fail to develop IC glomerulonephritis. These renal protective effects of TLR7 deletion despite persistent DNA-reactive Ab are surprising given clinical data implicating anti-dsDNA Abs in lupus nephritis (26). In keeping with this paradox, we observed glomerular IgG deposition by immunofluorescence in BW/TLR7−/−, in the absence of histologic glomerulonephritis. However, deposition of the pathogenic Ig subclass IgG2c was abrogated in the absence of B cell TLR7. In keeping with the role for IgG2c in promoting complement activation and activation of myeloid Fc-receptors (23), glomerular C3 deposition and recruitment of inflammatory macrophages was absent in BW/TLR7−/−. These data emphasize the need to consider IgG subclass in addition to Ab specificity in mediating end-organ damage in SLE
Finally, we show that B cell-intrinsic TLR9 deletion: enhances class-switched Abs targeting RNA-associated antigens and broadens the autoantibody repertoire; exacerbates systemic inflammation and expansion of activated CD4+ T cell and myeloid populations; and promotes glomerular deposition of pathogenic IgG2c IC. Although protective roles for TLR9 in autoimmunity have been described in several murine autoimmune models, how TLR9 deletion promotes autoimmunity has not yet been determined. Potential underlying mechanisms include: enhanced interaction of TLR7 with the shared endoplasmic reticulum trafficking protein Unc93b1 in the setting of TLR9-deletion (27); facilitation of entry of autoreactive cells into the mature B cell repertoire (16); or decreased generation of TLR9-dependent regulatory B cells (2). Our study makes the important observation that B cell-intrinsic TLR9 deletion can accelerate autoimmunity despite largely intact myeloid TLR signaling.
Importantly, however, our data does not exclude the possibility that myeloid TLR7 and TLR9 signals may exert additional impacts on lupus pathogenesis. Two recent studies, utilizing a cre-recombinase strategy to delete the TLR adaptor Myd88 in CD11c+ dendritic cells, demonstrated that myeloid TLR activation accelerates autoimmunity, likely via enhanced type 1 IFN production by IC-mediated activation of plasmacytoid dendritic cells (11, 13). Given this observation, it will be important to evaluate whether deletion of myeloid TLR7 vs. TLR9 exerts similar divergent effects on autoimmune pathogenesis. In this regard, a potential caveat of our study is that reconstitution with an 80:20 ratio of μMT:donor (was−/−, was−/−.tlr7−/−, or was−/−.tlr9−/−) BM, will result in ~20% myeloid cells also lacking TLR7 or TLR9. However, since the majority of myeloid cells (~80%) in our model are TLR sufficient, it is highly likely that our data reflect the B cell-intrinsic effects of TLR7 vs TLR9 in autoimmune pathogenesis.
In conclusion, our study provides critical new insights into how dual BCR and TLR signals promote B cell activation resulting in class-switched antibody production, CD4+ T cell activation and the development of IC glomerulonephritis. B cell TLR7 and TLR9 exert opposing impacts in this process, reinforcing the importance of dysregulated B cell TLR signaling in autoimmunity and informing efforts to therapeutically target TLR signals in SLE and other disorders characterized by autoantibody production.
Supplementary Material
Abbreviations used
- ABC
age-associated B cells
- ANA
antinuclear antibody
- BM
bone marrow
- CSR
class switch recombination
- EM
effector/memory T cells
- GC
germinal center
- IC
immune complex
- MDA-LDL
malondialdehyde conjugated with low-density lipoprotein
- PC
phosphoryl choline
- SLE
systemic lupus erythematosus
- TFH
T follicular helper cells
- μMT
B cell-deficient
- WAS
Wiskott-Aldrich syndrome
- WASp
Wiskott-Aldrich syndrome protein
References
- 1.Shlomchik MJ. Activating systemic autoimmunity: B’s, T’s, and tolls. Curr Opin Immunol. 2009;21:626–633. doi: 10.1016/j.coi.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green NM, Marshak-Rothstein A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Semin Immunol. 2011;23:106–112. doi: 10.1016/j.smim.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat Rev Immunol. 2012;12:282–294. doi: 10.1038/nri3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, Wortis HH, Kearney JF, Ucci AA, Imanishi-Kari T. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. 2006;25:429–440. doi: 10.1016/j.immuni.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Lartigue A, Courville P, Auquit I, Francois A, Arnoult C, Tron F, Gilbert D, Musette P. Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J Immunol. 2006;177:1349–1354. doi: 10.4049/jimmunol.177.2.1349. [DOI] [PubMed] [Google Scholar]
- 8.Yasuda K, Richez C, Maciaszek JW, Agrawal N, Akira S, Marshak-Rothstein A, Rifkin IR. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol. 2007;178:6876–6885. doi: 10.4049/jimmunol.178.11.6876. [DOI] [PubMed] [Google Scholar]
- 9.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, Mackay F. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med. 2007;204:1959–1971. doi: 10.1084/jem.20062567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med. 2006;203:553–561. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38:528–540. doi: 10.1016/j.immuni.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becker-Herman S, Meyer-Bahlburg A, Schwartz MA, Jackson SW, Hudkins KL, Liu C, Sather BD, Khim S, Liggitt D, Song W, Silverman GJ, Alpers CE, Rawlings DJ. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J Exp Med. 2011;208:2033–2042. doi: 10.1084/jem.20110200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hua Z, Gross AJ, Lamagna C, Ramos-Hernandez N, Scapini P, Ji M, Shao H, Lowell CA, Hou B, Defranco AL. Requirement for MyD88 Signaling in B Cells and Dendritic Cells for Germinal Center Anti-Nuclear Antibody Production in Lyn-Deficient Mice. J Immunol. 2013 doi: 10.4049/jimmunol.1300683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh ER, Pisitkun P, Voynova E, Deane JA, Scott BL, Caspi RR, Bolland S. Dual signaling by innate and adaptive immune receptors is required for TLR7-induced B-cell-mediated autoimmunity. Proc Natl Acad Sci U S A. 2012;109:16276–16281. doi: 10.1073/pnas.1209372109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang SH, Lee H, Yamamoto M, Jones LA, Dayalan J, Hopkins R, Zhou XJ, Yarovinsky F, Connolly JE, Curotto de Lafaille MA, Wakeland EK, Fairhurst AM. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol. 2012;189:5786–5796. doi: 10.4049/jimmunol.1202195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nickerson KM, Christensen SR, Cullen JL, Meng W, Luning Prak ET, Shlomchik MJ. TLR9 promotes tolerance by restricting survival of anergic anti-DNA B cells, yet is also required for their activation. J Immunol. 2013;190:1447–1456. doi: 10.4049/jimmunol.1202115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li QZ, Zhou J, Wandstrat AE, Carr-Johnson F, Branch V, Karp DR, Mohan C, Wakeland EK, Olsen NJ. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin Exp Immunol. 2007;147:60–70. doi: 10.1111/j.1365-2249.2006.03251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isenberg DA, Dudeney C, Williams W, Addison I, Charles S, Clarke J, Todd-Pokropek A. Measurement of anti-DNA antibodies: a reappraisal using five different methods. Ann Rheum Dis. 1987;46:448–456. doi: 10.1136/ard.46.6.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh YT, Scatizzi JC, Gahan JD, Lawson BR, Baccala R, Pollard KM, Beutler BA, Theofilopoulos AN, Kono DH. Role of nucleic acid-sensing TLRs in diverse autoantibody specificities and anti-nuclear antibody-producing B cells. J Immunol. 2013;190:4982–4990. doi: 10.4049/jimmunol.1202986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, Marrack P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood. 2011;118:1305–1315. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118:1294–1304. doi: 10.1182/blood-2011-01-330530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carragher DM, Rangel-Moreno J, Randall TD. Ectopic lymphoid tissues and local immunity. Semin Immunol. 2008;20:26–42. doi: 10.1016/j.smim.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 24.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clingan JM, Matloubian M. B Cell-Intrinsic TLR7 Signaling Is Required for Optimal B Cell Responses during Chronic Viral Infection. J Immunol. 2013 doi: 10.4049/jimmunol.1300244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isenberg DA, Manson JJ, Ehrenstein MR, Rahman A. Fifty years of anti-ds DNA antibodies: are we approaching journey’s end? Rheumatology (Oxford) 2007;46:1052–1056. doi: 10.1093/rheumatology/kem112. [DOI] [PubMed] [Google Scholar]
- 27.Fukui R, Saitoh S, Kanno A, Onji M, Shibata T, Ito A, Onji M, Matsumoto M, Akira S, Yoshida N, Miyake K. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity. 2011;35:69–81. doi: 10.1016/j.immuni.2011.05.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.