

Abstract
Variants that disrupt the translation initiation sequences in cancer predisposition genes are generally assumed to be deleterious. However few studies have validated these assumptions with functional and clinical data. Two cancer syndrome gene variants likely to affect native translation initiation were identified by clinical genetic testing: MLH1:c.1A>G p.(Met1?) and BRCA2:c.67+3A>G. In vitro GFP-reporter assays were conducted to assess the consequences of translation initiation disruption on alternative downstream initiation codon usage. Analysis of MLH1:c.1A>G p.(Met1?) showed that translation was mostly initiated at an in-frame position 103 nucleotides downstream, but also at two ATG sequences downstream. The protein product encoded by the in-frame transcript initiating from position c.103 showed loss of in vitro mismatch repair activity comparable to known pathogenic mutations. BRCA2:c.67+3A>G was shown by mRNA analysis to result in an aberrantly spliced transcript deleting exon 2 and the consensus ATG site. In the absence of exon 2, translation initiated mostly at an out-of-frame ATG 323 nucleotides downstream, and to a lesser extent at an in-frame ATG 370 nucleotides downstream. Initiation from any of the downstream alternative sites tested in both genes would lead to loss of protein function, but further clinical data is required to confirm if these variants are associated with a high cancer risk. Importantly, our results highlight the need for caution in interpreting the functional and clinical consequences of variation that leads to disruption of the initiation codon, since translation may not necessarily occur from the first downstream alternative start site, or from a single alternative start site.
Keywords: Unclassified Variant, In vitro assay, Cancer syndrome genes
Introduction
Identification of germline mutations in cancer syndrome genes is important to ensure most appropriate medical management for high-risk carrier individuals, namely genetic counselling, pre-symptomatic screening, and risk-reducing medication or surgery. However, ambiguity arises for clinicians when gene sequencing identifies variations of unknown clinical significance, such as missense, small in-frame insertions/deletions, intronic and regulatory region variants. Unclassified variants may be evaluated using multifactorial likelihood modelling that combines bioinformatic, genetic and clinical information to provide a probability that a variant has the characteristics of a high-risk pathogenic mutation, and models have been developed and improved for the BRCA1 (MIM# 113705), BRCA2 (MIM# 600185) and DNA mismatch repair (MMR) genes commonly tested in the familial cancer setting, including MLH1 (MIM# 120436), MSH2 (MIM# 609309), MSH6 (MIM# 609309) and PMS2 (MIM# 600259) [1–6]. However, such analysis is often hampered by paucity of relevant information for example, segregation and tumour pathology data. Laboratory assays are an alternative approach to assess the biological effect of these variants, and include assays of protein function for missense or small in-frame insertions/deletion variants [7–10], mRNA splicing for variants that are predicted to create aberrant splicing events [11, 12], and regulation of gene expression for variants in the 3′UTR or 5′UTR [13–15].
Variants in the translation start site are a particular class of unclassified variant that should be amenable to bioinformatic prediction of their effect on translation. Previous reports have demonstrated that alternative downstream and upstream ATG sequences may encode translation start sites even under normal physiological splicing conditions [16, 17]. Thus, genetic variation that directly or indirectly disrupts the initiation codon may lead to use of alternative start site(s) upstream or downstream, determined by the presence of an ATG codon and nearby Kozak sequence recognised by translational machinery [18, 19].
Genetic testing of cancer patients for mutations in BRCA1, BRCA2 and the mismatch repair genes are commonly undertaken. There are a limited number of reports relating to variation that disrupts the translation initiation codon. The start codon variant BRCA2:c.3G>A p.(Met1?) was recently reported as being likely pathogenic using a multifactorial analysis approach that was largely influenced by the position/conservation-based prior probability, however, no functional implications were investigated [20]. Variation at the start codon of the MMR gene MSH2 has also been reported, with the MSH2:c.1A>G p.(Met1?) variant identified in trans with a large deletion in MSH2 in two siblings demonstrating a less severe clinical phenotype than is observed for individuals with classical constitutional mismatch repair deficiency [21]. A subsequent study investigated use of an in-frame methionine, 25 amino acids downstream as the alternative translation initiation codon, and functional analysis of this possible alternative protein product showed slightly reduced function of MSH2 [22].
In this study we investigated the functional consequences of two cancer syndrome gene variants predicted to disrupt the translation initiation codon directly (MLH1:c.1A>G p.(Met1?)), or indirectly via aberrant splicing resulting in loss of exon 2 (BRCA2:c.67+3A>G). Using construct assays that comprehensively assessed use of multiple potential alternative start sites, we showed that alternative start site usage is complex and further studies are justified to investigate the functional and clinical consequences of all variants likely to affect constitutional translation initiation.
Methods
Patient Identification and Assay Design
The genetic variants studied were predicted to have direct or indirect effects on constitutional translation initiation, and were identified in two cancer-affected individuals who underwent clinical genetic testing. Nucleotide numbering is relative to cDNA numbering where +1 is the A in the ATG of the start codon. Codon number 1 is the methionine wild-type translation initiation codon. The Human Genome Variation Society (HGVS) rules for mutation nomenclature were followed, using the following cDNA reference sequences: NM_000249.3 for MLH1 and NM_000059.3 for BRCA2.
MLH1:c.1A>G p.(Met1?)
The family containing the MLH1:c.1A>G p.(Met1?) variant was identified in a woman diagnosed with endometrial cancer at age 56 by an Australian family cancer clinic. MLH1 and MSH2 gene testing was performed based on a reported family history that met the Amsterdam II criteria [23]. The family were participants in the Australasian Colorectal Cancer Family Registry (ACCFR [24]). The variant-carrying proband reported endometrial cancer at age 56, two siblings with colorectal cancer (ages 36, untested and 71, obligate carrier based on carriage of the variant by unaffected child), and a nephew with colorectal cancer (age 46, untested). Potential alternative translation start sites are shown in Figure 1A. The first three possible alternate translation initiation codons, all located in the ATP-binding hydrolysis domain, were selected for construct-based assays to assess alternative translation initiation site usage in the presence of the variant allele.
Figure 1. Alternative translation initiation codons in MLH1 and BRCA2.

A) Translation from all but one of these potential start codons (c.103) is predicted to result in an out-of-frame protein product. Two of the ATP-binding and hydrolysis domain motifs are shown (bases 91–129 and 187–204). Two further ATP-binding and hydrolysis motifs lie at bases 289–321 and 436–441. B) Alternative translation start sites following the aberrant splicing event as a result of BRCA2:c.67+3A>G are highlighted at position c.323 and c.367. The N-terminal transactivation domain that spans bases 67–315 is lost as a result of translation initiation at c.323 and c.367. In the event that the ATG at c.323 is recognized as the translation initiation signal, an out of frame protein would be synthesized terminating 12 amino acids after initiation. Initiation at c.367 would result in an in-frame protein product, lacking the N-terminal transactivation domain.
BRCA2:c.67+3A>G
The BRCA2:c.67+3A>G variant was identified by an Australian family cancer clinic in a patient with bilateral breast cancer, diagnosed at ages 40 and 49. Sequencing and MLPA analysis did not identify any large genomic rearrangements. Reported cancers for relatives included: a sister with endometrial cancer at age 58, a mother with breast cancer diagnosed over age 70, and maternal grandmother with cancer type unknown prior to her death at age 48. Following bioinformatic predictions that suggested the variant had potential to result in aberrant splicing using Human Splicing Finder version 2.4 (which combines Human Splice Finder matrices and MaxEntScan (www.umd.be/HSF/) [25]), a blood sample was taken from the patient in the clinical setting for further investigation of mRNA defects in vitro. Blood was collected using an RNA stabilising PaxGene tube and RNA extracted within 24hrs using the Qiacube PAXgene Blood RNA Kit (Qiagen, Doncaster, Victoria, Australia). Complementary DNA (cDNA) was synthesised using Superscript III First Strand Synthesis System (Invitrogen, Carlsbad, CA). PCR to assess the predicted splicing aberration was performed using Amplitaq Gold (Applied Biosystems, Mulgrave, Victoria, Australia) under the following conditions: 95°C for 7 minutes followed by 35 cycles of 94°C for 30 seconds, 55°C for 30 seconds and 72°C for 1 minute and a final extension step at 72°C for 7 minutes using a forward primer, 5′ CTCGGGTGTCTTTTGCGGCGGTG 3′ and a reverse primer, 5′ TGAAACAAACTCCCACATACCA 3′. PCR products were purified using QIAquick PCR Purification Kit (Qiagen), and sequenced using Big-Dye Terminator version 3.1 sequencing chemistry and the ABI 377 sequencer (Applied Biosystems) in both directions. The mRNA aberration detected in the variant-carrying patient (exon 2 deletion, see Figure 2 and results text) indicated that the constitutional start codon would not be present in this aberrant mRNA transcript. Potential alternative start sites for the BRCA2 del exon 2 transcript are shown in Figure 1B. Two downstream ATG sites in exon 4 at c.323 and c.367 were selected for functional analysis to determine their potential use as alternate translation initiation sites.
Figure 2. BRCA2:c.67+3A>G splicing products detected by RT-PCR.
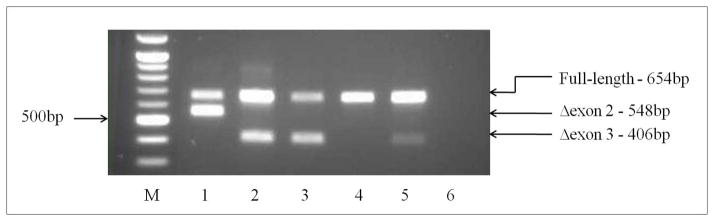
M – 100bp DNA marker (New England Biolabs); Lane 1 variant carrier; Lanes 2–5, normal unaffected female controls; lane 6, no-template control. Controls revealed only expected product sizes of 654bp for full length transcript, and 406bp for common exon 3 deletion isoform previously reported in healthy controls. The variant carrier in Lane 1 shows an aberrant product of 548bp, that was confirmed by sequencing to be an exon 2 deletion. The loss of exon 2 includes the position of the ATG start site, complicating prediction of the molecular consequences of this aberration.
Fusion Constructs
MLH1:c.1A>G p.(Met1?)
To generate cDNA for constructs assessing the functional consequences of the MLH1:c.1A>G p.(Met1?) variant, an Epstein Barr Virus transformed lymphoblastoid cell line was established from patient blood and cultured in RPMI with 10% fetal bovine serum. RNA was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions and treated with rDNase I (Invitrogen). cDNA was synthesised using SuperScript III First-Strand Synthesis kit (Invitrogen) according to manufacturer’s instructions. Four MLH1-GFP fusion constructs were synthesised as shown in figure 3A by fusing 5′ sequence preceding the ATG at nucleotide c.1, c.89, c.103 and c.121 to green fluorescence protein (GFP) coding sequence from the pEGFP-N1 plasmid (Clontech, Mountain View, CA). The MLH1 sequence in each construct begins at c.-56 in the MLH1 5′UTR. For each construct, MLH1 and GFP coding sequences were amplified independently, using primers detailed in Supplementary Table 1. The MLH1 and GFP components for each construct were fused using primers MLH1_NHE_F (containing a Nhe1 site) and GFP_R (containing a Not1-HF site) and cycling conditions, 95°C for 7 minutes followed by 35 cycles of 94°C for 30 seconds, 55°C for 30 seconds and 72°C for 1 minute and a final extension step at 72°C for 7 minutes. Primers were added after 12 cycles. Fused fragments were digested using Nhe1 and Not1-HF to create sticky ends. The pEGFP-N1 vector was digested in an independent reaction to remove the GFP coding sequence from the vector and provide complementary sticky ends. MLH1-GFP fusion fragments and the vector with GFP coding sequence removed were ligated using DNA ligase (Invitrogen) and then cloned using a pGEM-T Vector System (Promega, Auburn, Victoria, Australia). Recombinant clones were selected from a single colony and sequence confirmed. Site-directed mutagenesis was performed using primers MLH1_103G_F and MLH1_103G_R (Supplementary Table 1).
Figure 3. Relative GFP fluorescent level from alternative downstream translation initiation codons in MLH1 and BRCA2.

Schematic representation of each construct is shown. The GFP sequence is in-frame with the most 3′ potential start site. Error bars are based on the standard error between repeat experiments. A) Relative GFP fluorescent level of predicted potential alternate start sites with and without the presence of the variant MLH1c.1A>G(p.Met1Val). The ATG at position c.103 is in-frame with the ATG at c.1, and produced higher GFP protein level than the two initiation sites not in-frame with position c.1 (c.89 and c.122, which are in-frame with each other). B) Effect of exon 2 loss from BRCA2 transcripts on the initiation of translation. An out-of-frame ATG codon at position c.323 in exon 4 is preferred to an in-frame alternative at c.370. Each experiment was conducted in triplicate, and then repeated twice.
BRCA2:c.67+3A>G
Existing cDNA prepared for mRNA transcript analysis, as detailed above, was used to generate constructs to assess the functional consequences of the BRCA2:c.67+3A>G variant. Three BRCA2-GFP fusion constructs were synthesised using primers detailed in Supplementary Table 1 by combining 5′ sequence preceding the ATG at nucleotide c.1, c.323 and c.370 to GFP coding sequence in the pEGFP-N1 plasmid. Each construct contained BRCA2 mRNA sequence beginning at c.-158 in the BRCA2 5′UTR. The constructs fusing the potentially alternative ATG start sites at c.323 and c.370 did not contain exon 2 shown to be deleted in the presence of the variant. Fused BRCA2-GFP fragments and vector were ligated, cloned, selected and sequence confirmed as noted above for MLH1-GFP constructs.
GFP-Fluorescence Assay
The colorectal cancer cell line HT29 was used for MLH1 construct assays. HT29 cells were maintained using RPMI (Invitrogen), supplemented with 10% foetal bovine serum (Lonza, Portsmouth, NH) and 1% penicillin/streptomycin (Invitrogen). The breast cancer cell line MDA-MB-231 was used for the BRCA2 construct assays. MDA-MB-231 cells were maintained using DMEM (Invitrogen), supplemented with 10% foetal bovine serum and 1% penicillin/streptomycin. Sixty thousand cells were pre-plated 24 hours prior to transfection in antibiotic free media with 10% foetal bovine serum and transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were then cultured for 24 hours before harvesting for flow cytometry analysis using a FACS Canto (BD Biosciences, North Ryde, NSW, Australia) for MLH1 analysis and an LSR Fortessa (BD Biosciences) for BRCA2 analysis. All constructs were transfected in triplicate within each experiment. The geometric mean intensity of GFP protein level in transfected cells was measured in each sample and normalised to pEGFP-N1 control. To account for transfection efficiency, analysis was performed on GFP positive cells only.
Mismatch Repair Functional Analysis
The in vitro cell-free assay described by Drost et al. [7] was used to assess the functional consequences of the MLH1:c.1A>G p.(Met1?) variant allele, using two different templates: (i) the entire MLH1 cDNA containing the variant allele; (ii) a manufactured MLH1 cDNA lacking nucleotides c.1 to c.102, to force translation of an in-frame MLH1 protein lacking the first 34 amino acids and so mimic the protein product determined from construct assays to be the predominant transcript produced by the MLH1:c.1A>G p.(Met1?) variant allele. Templates for full-length wildtype MLH1 protein, a known MMR function abrogating mutation (MLH1:c.199G>A(p.Gly67Arg)) and a known benign polymorphism (MLH1:c.655A>G(p.Ile219Val) were used as controls. The templates were generated by PCR (Figure 4A) and sequence confirmed (Figure 4B). In vitro protein synthesis was carried out in the presence of radioactive methionine, and protein expression was measured by gel electrophoresis and autoradiography. In vitro mismatch repair activity of this N-terminal truncated protein was measured and compared to the controls described above.
Figure 4. In vitro cell-free mismatch repair assay confirms loss of function proteins translated from MLH1c.1A>G p.(Met1?) alleles.

A) Alleles generated by PCR including the full-length variant allele and an allele carrying a deletion of 34 amino acids as anticipated from MLH1-GFP fusion construct assays. B) Sequence confirmed alleles. C) 35S-Methionine labelled proteins derived for the cell-free assay. The p.Met1Val allele was unable to produce a detectable protein. D) A protein lacking amino acids 1–34 reduces MMR activity to a level equivalent to known pathogenic missense mutation, MLH1:p.Gly67Arg.
Statistical Analysis
Statistical analyses of the GFP-Fluorescence assay were performed on the normalised geometric mean expression relative to geometric mean peGFP-n1 control for each experiment (3 per variant) and replicate (3 per experiment). General linear models (GLM) were used to assess for differences in GFP between groups. This model allowed adjustment for experimental variability and design covariates, with further analyses using contrasts to test specific hypotheses.
To determine differences in GFP between wildtype and variant alleles, a GLM adjusted for experiment, construct and a construct by wildtype/variant interaction was used. Appropriate contrasts were set up to assess differences in GFP between wildtype and variant at each construct, and, also to determine if wildtype and variant constructs differ from wildtype allele construct at c.1.
GLM was used to assess for differences in GFP between mutagenesis and non-mutagenesis at c.103. The model adjusted for experiment, wildtype/variant and an experiment by wildtype/variant interaction. Contrasts were set up to assess whether differences between mutagenesis and non-mutagenesis occurred in the wildtype only or variant only, and whether there were differences between wildtype and variant within mutagenesis.
For the BRCA2 analysis GLM was used to determine differences between the alternative translation initiation constructs and the c.1ATG construct, and were adjusted for experiment and replicates. Contrasts were set up to assess pairwise differences between the constructs and the c.1ATG.
All statistical analyses were performed in IBM SPSS Statistics 19.0 and p-values <0.05 were considered significant.
Results
MLH1:c.1A>G p.(Met1?)
GFP protein level assays were conducted using MLH1-GFP fusion constructs synthesized by combining MLH1 sequence 5′ to alternative downstream ATG sequences to determine the capacity for these sites to act as Kozak sequences and initiate translation in the presence of the variant allele. Results are presented in Figure 3A.
Comparison of the wildtype allele construct (c.1ATG) to the variant allele construct (c.1GTG) showed a significant reduction in GFP protein level at the consensus site (p<0.001). This suggests that the c.1A>G substitution is likely to disrupt translation initiation of full-length MLH1 in vivo, possibly leading to use of alternative initiation codons downstream. Initiation of translation at the first downstream alternative ATG motif at c.89 is unlikely given the minimal GFP protein level observed from the c.1GTG_c.89ATG construct. The second downstream alternative ATG motif is located at c.103 and maintains the reading frame. GFP protein level comparable to the wildtype allele (c.1ATG) was observed in constructs containing an ATG at c.103 with either an ATG at c.1 (c.1ATG_c.103ATG) or GTG at c.1 (c.1GTG_c.103ATG). Considering the significant reduction in GFP protein level from the c.1GTG construct compared with the c.1ATG construct (p<0.001) but similar GFP protein level in the constructs assessing the second alternative downstream ATG, it seemed likely that the GFP protein level from the c.1GTG_c.103ATG construct was due to translation from position c.103 rather than c.1. It was also likely that GFP protein level observed for the c.1ATG_c.103ATG construct was attributable to initiation at the consensus start site (c.1). To investigate this, constructs c.1ATG_c.103AGG and c.1GTG_c.103AGG were synthesised to disrupt the c.103ATG while maintaining an ATG at c.1 or GTG at c.1, testing the wildtype and variant alleles, respectively. The c.1ATG_c.103AGG construct contains the wild-type start site and produced GFP protein level that was not significantly different to the c.1ATG_c.103ATG construct, indicating that translation for both of these c.103 constructs actually initiated at the c.1ATG position. However, GFP protein level of the c.1GTG_c.103AGG construct was markedly reduced in comparison to the c.1GTG_c.103ATG (p<0.001) construct, suggesting that the in-frame c.103ATG site is functional from the variant allele but not from the wildtype allele.
Two further constructs representing initiation at a third downstream, alternative ATG motif at c.122 were synthesised. The assay results suggest that some translation initiation occurs at c.122. However reading frame would be altered, with a stop codon encoded eight amino acids downstream that is expected to result in transcript nonsense mediated decay (NMD) or a short peptide.
Together, the construct assay results indicate that in vivo translation initiation at the c.103ATG position of MLH1 would result in an MLH1 protein with a 103 nucleotide (34 amino acid) deletion from the 5′ end. This would exclude the first four amino acids of one of four ATP-binding and hydrolysis motifs [26], as shown in Figure 1a. Subsequent to construct assay results, protein expression analysis suggested that, at least in the cell-free in vitro system, the template containing the MLH1 c.1A>G variant allele does not produce a protein (Figure 4C). In addition, MMR activity assays indicated that functional capacity of a protein with loss of the initial 34 amino acids in MLH1:(p.Met1_Glu34del) is reduced to the MMR activity of a known MMR-deficient variant (p.Gly67Arg) (Figure 4D), and would result in MMR deficiency if produced in vivo.
BRCA2:c.67+3A>G
Messenger RNA splicing analysis detected a transcript with deletion of exon 2, visualised by gel electrophoresis (Figure 2), and confirmed by sequencing. Exon 2 includes the consensus ATG start site at c.1. In the absence of exon 2, the next downstream ATG motif that may be recognised as a translation initiation codon in the BRCA2 mRNA begins at nucleotide 323, followed by a second ATG at nucleotide 370 (Figure 1B). Alternatively, the mRNA transcript may be subject to NMD as a result of the aberrant splicing [27–29]. It was not possible to experimentally determine the extent of NMD due to an absence of a polymorphism to be utilised for quantitative assessment. In the event that the second downstream ATG beginning at c.370 is recognised as the translation initiation site, the transcript would remain in-frame and code for a protein lacking the initial 123 amino acids. Several other alternative ATG sites may be recognised in exons 5–8, but these would result in loss of the open reading frame and transcripts would also likely be subject to NMD.
To assess the effect on translation from this allele, BRCA2-GFP fusion constructs were synthesized, combining BRCA2 sequence 5′ to alternative downstream ATG sequences at c.323 and c.370. Results shown in Figure 3B indicated that, in the absence of exon 2, the ATG sequence at c.323 will be preferentially used as an alternative translation initiation site, with less translation initiation from position c.370 (p<0.001). These data suggest that the BRCA2:c.67+3A>G allele causing an exon 2 deletion would primarily lead to translation initiation from position c.323 and an out-of-frame transcript likely subject to NMD. The construct assay results also indicate production of an in-frame transcript initiating at c.370, which if translated, would encode a protein lacking the first 123 amino acids and a portion of the N-terminal transactivation domain. However, we cannot exclude the possibility that the variant allele also produces correctly spliced, full-length BRCA2 mRNA, since, in this instance, it was not possible to use exonic variation in cis to measure the ratio of full-length to aberrant transcription from the variant allele.
Discussion
The data produced in this study suggest that the predominant transcript from a MLH1:c.1A>G p.(Met1?) allele, which if translated in vivo, will encode an in-frame protein lacking the first 34 amino acids that is deficient in MMR activity. This may involve retention of binding with MMR proteins XO1, MLH3, PMS1 and PMS2 but compromise DNA repair function, acting as a dominant-negative mutant. While these results indicate abrogated function for MLH1:c.1A>G p.(Met1?), following standards for MMR variant classification put forward by the InSiGHT Variant Interpretation Committee (www.insight-group.org/criteria), multiple points of evidence that include supporting clinical data would be necessary to confirm that this variant should be considered pathogenic. Such data could include segregation data, colorectal or endometrial tumor MSI or MMR IHC characteristics of variant carriers, and should they exist, clinical features of individuals who carry the variant of interest in trans with a confirmed pathogenic MLH1 mutation.
We also showed that the BRCA2 c.67+3A>G allele results in an aberrant mRNA transcript lacking exon 2, which in vitro causes translation initiation predominantly at position c.323, producing an mRNA transcript predicted to encode a truncated protein. Together, these findings suggest that BRCA2:c.67+3A>G will encode protein with abrogated BRCA2 function. However, confirmation of the pathogenicity of this variant requires additional quantitative mRNA studies, and clinical information such as segregation or breast tumor pathology data to enable multifactorial analysis. The findings also have implications for other variant carriers reported to lose this exon as a result of a splicing defect, and for carriers of variants that occur in the ATG at position c.1–3 or other sequences that disrupt the initiation of translation.
GFP was chosen as the reporter gene for this in vitro functional assay because it is amenable to flow cytometry analysis methods, providing a straight-forward and economical method of GFP protein level quantification [17, 30, 31]. Compared to luciferase assays, flow cytometric detection of GFP is cheaper and less laborious, as no processing or additional reagents are required for reporter protein level measurement. Luciferase assays could be considered complementary to such GFP-based assays, since the shorter half-life of the luciferase protein may provide a more accurate measure of steady-state protein levels dependent on translation regulatory mechanisms. A potential limitation of the GFP reporter assay is the creation of target gene-GFP fusion proteins. Differences in protein folding may alter the stability of fusion proteins produced compared to GFP [32]. The constructs were designed to test the most 3′ potential translation initiation site of the MLH1 or BRCA2 sequence by ensuring GFP was in-frame with that site. Thus fusion proteins would only occur when another potential translation initiation site was in-frame with the site being tested. In our study, the BRCA2 constructs did not have the potential to create fusion proteins as c.323 and c.360 are not in-frame with each other, but fusion proteins may have occurred in constructs with MLH1 up to ATG at c.103 (in-frame with c.1), and an ATG at c.122 (in-frame with c.89). However, results for the MLH1 constructs mutated to AGG at the c.103 potential translation initiation site suggested that protein stability was not altered for a fusion protein of GFP with 34 amino acids of MLH1 N-terminal sequence, and there was no evidence for a fusion protein initiating at c.89 for the c.122_ATG constructs since there was negligible GFP protein level observed from both c.89 constructs (c.1ATG_c.89ATG and c.1GTG_c.89ATG).
The results from our extensive in vitro assays have shown that the translation does not necessarily occur from the first downstream alternative start site, or from only a single alternative start site. Given that multiple alternative start sites may differ in their predicted effect on transcript/protein sequence, we recommend that further studies should be undertaken for all such variants in order to better inform variant classification. Given the unanticipated and complex translational profiles identified in our study for specific gene variants, we also suggest that in vitro and in vivo studies be used to assay endpoint activity or function of all transcripts produced by a variant allele disrupting native translation initiation. Examples include BRCA1 and BRCA2 mouse embryonic stem assays [33, 34] or MMR activity assays [22, 35]. However, all functional assays require calibration against or clinical data to allow interpretation of findings for clinical use [9, 36–38]. Importantly, our results have highlighted the need to be cautious in interpreting the functional and clinical consequences of variation that leads to disruption of the initiation codon. When investigating the potential pathogenicity of such germline variation, primary bioinformatic analysis may help to identify potential alternative translation start sites and the functional domains that may be impacted.
Supplementary Material
Acknowledgments
Financial support: This work was supported by funding from The National Health and Medical Research Council (NHMRC ID1010719), Cancer Australia (Grant ID#1010859), the National Cancer Institute, National Institutes of Health under RFA # CA-95-011 and through the Australasian Colorectal Cancer Family Registry (U01 CA097735). Amanda B. Spurdle and Mark A. Jenkins are supported by NHMRC Senior Research Fellowships. John L. Hopper is an NHMRC Senior Principal Research Fellow and Distinguished Visiting Professor at Seoul National University, Korea.
The authors thank the patients, and Australasian Colon Cancer Family Registry staff for their contributions to this project. Samples for study of the MLH1 variant were obtained from the Jeremy Jass Memorial Tissue Pathology Bank.
Abbreviations
- ACCFR
Australasian Colorectal Cancer Family Registry
- cDNA
complementary DNA
- GFP
green fluorescence protein
- MMR
mismatch repair
- NMD
nonsense mediated decay
Footnotes
Disclaimer: The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centres in the Cancer Family Registries, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the Cancer Family Registry. Authors had full responsibility for the design of the study, the collection of the data, the analysis and interpretation of the data, the decision to submit the manuscript for publication, and the writing of the manuscript.
Ethics approval: Written informed consent was obtained from all study participants and the study protocol was approved by the QIMR HREC under protocol P628.
References
- 1.Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J Med Genet. 2012;49:151–7. doi: 10.1136/jmedgenet-2011-100714. [DOI] [PubMed] [Google Scholar]
- 2.Thompson BA, Goldgar DE, Paterson C, et al. A Multifactorial Likelihood Model for MMR Gene Variant Classification Incorporating Probabilities Based on Sequence Bioinformatics and Tumor Characteristics: A Report from the Colon Cancer Family Registry. Hum Mutat. 2013;34:200–9. doi: 10.1002/humu.22213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson BA, Greenblatt MS, Vallee MP, et al. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat. 2013;34:255–65. doi: 10.1002/humu.22214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldgar DE, Easton DF, Byrnes GB, Spurdle AB, Iversen ES, Greenblatt MS. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum Mutat. 2008;29:1265–72. doi: 10.1002/humu.20897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plon SE, Eccles DM, Easton D, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–91. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spurdle AB. Clinical relevance of rare germline sequence variants in cancer genes: evolution and application of classification models. Curr Opin Genet Dev. 2010;20:315–23. doi: 10.1016/j.gde.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 7.Drost M, Zonneveld JB, van Dijk L, et al. A cell-free assay for the functional analysis of variants of the mismatch repair protein MLH1. Hum Mutat. 2010;31:247–53. doi: 10.1002/humu.21180. [DOI] [PubMed] [Google Scholar]
- 8.Farrugia DJ, Agarwal MK, Pankratz VS, et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res. 2008;68:3523–31. doi: 10.1158/0008-5472.CAN-07-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millot GA, Carvalho MA, Caputo SM, et al. A guide for functional analysis of BRCA1 variants of uncertain significance. Hum Mutat. 2012;33:1526–37. doi: 10.1002/humu.22150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raevaara TE, Korhonen MK, Lohi H, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–49. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Spurdle AB, Couch FJ, Hogervorst FB, Radice P, Sinilnikova OM. Prediction and assessment of splicing alterations: implications for clinical testing. Hum Mutat. 2008;29:1304–13. doi: 10.1002/humu.20901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houdayer C, Caux-Moncoutier V, Krieger S, et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum Mutat. 2012;33:1228–38. doi: 10.1002/humu.22101. [DOI] [PubMed] [Google Scholar]
- 13.Brewster BL, Rossiello F, French JD, et al. Identification of fifteen novel germline variants in the BRCA1 3′UTR reveals a variant in a breast cancer case that introduces a functional miR-103 target site. Hum Mutat. 2012;33:1665–75. doi: 10.1002/humu.22159. [DOI] [PubMed] [Google Scholar]
- 14.Nicoloso MS, Sun H, Spizzo R, et al. Single-nucleotide polymorphisms inside microRNA target sites influence tumor susceptibility. Cancer Res. 2010;70:2789–98. doi: 10.1158/0008-5472.CAN-09-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pamula J, Krzesniak M, Zientek H, Pekala W, Rusin M, Grzybowska E. Functional Impact of Sequence Alterations Found in BRCA1 Promoter/5′UTR Region in Breast/Ovarian Cancer Families from Upper Silesia, Poland. Hered Cancer Clin Pract. 2006;4:20–4. doi: 10.1186/1897-4287-4-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Prolla G, Rostagno A, Chiarle R, Feiner H, Inghirami G. Initiation of translation from a downstream in-frame AUG codon on BRCA1 can generate the novel isoform protein DeltaBRCA1(17aa) Oncogene. 2000;19:2767–73. doi: 10.1038/sj.onc.1203599. [DOI] [PubMed] [Google Scholar]
- 17.Wang XQ, Rothnagel JA. 5′-untranslated regions with multiple upstream AUG codons can support low-level translation via leaky scanning and reinitiation. Nucleic Acids Res. 2004;32:1382–91. doi: 10.1093/nar/gkh305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kochetov AV. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays. 2008;30:683–91. doi: 10.1002/bies.20771. [DOI] [PubMed] [Google Scholar]
- 19.Lukowski SW, Bombieri C, Trezise AE. Disrupted post-transcriptional regulation of the cystic fibrosis transmembrane conductance regulator (CFTR) by a 5′UTR mutation is associated with a CFTR-related disease. Hum Mutat. 2011;32:E2266–82. doi: 10.1002/humu.21545. [DOI] [PubMed] [Google Scholar]
- 20.Thomassen M, Blanco A, Montagna M, et al. Characterization of BRCA1 and BRCA2 splicing variants: a collaborative report by ENIGMA consortium members. Breast Cancer Res Treat. 2012;132:1009–23. doi: 10.1007/s10549-011-1674-0. [DOI] [PubMed] [Google Scholar]
- 21.Kets CM, Hoogerbrugge N, van Krieken JH, Goossens M, Brunner HG, Ligtenberg MJ. Compound heterozygosity for two MSH2 mutations suggests mild consequences of the initiation codon variant c.1A>G of MSH2. Eur J Hum Genet. 2009;17:159–64. doi: 10.1038/ejhg.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cyr JL, Brown GD, Stroop J, Heinen CD. The predicted truncation from a cancer-associated variant of the MSH2 initiation codon alters activity of the MSH2-MSH6 mismatch repair complex. Mol Carcinog. 2012;51:647–58. doi: 10.1002/mc.20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–6. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 24.Winship I, Win AK. The Australasian Colorectal Cancer Family Registry. Med J Aust. 2012;197:480–1. doi: 10.5694/mja12.11395. [DOI] [PubMed] [Google Scholar]
- 25.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ban C, Junop M, Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell. 1999;97:85–97. doi: 10.1016/s0092-8674(00)80717-5. [DOI] [PubMed] [Google Scholar]
- 27.Senapathy P. Origin of eukaryotic introns: a hypothesis, based on codon distribution statistics in genes, and its implications. Proc Natl Acad Sci U S A. 1986;83:2133–7. doi: 10.1073/pnas.83.7.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senapathy P, Shapiro MB, Harris NL. Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol. 1990;183:252–78. doi: 10.1016/0076-6879(90)83018-5. [DOI] [PubMed] [Google Scholar]
- 29.Stover ML, Primorac D, Liu SC, McKinstry MB, Rowe DW. Defective splicing of mRNA from one COL1A1 allele of type I collagen in nondeforming (type I) osteogenesis imperfecta. J Clin Invest. 1993;92:1994–2002. doi: 10.1172/JCI116794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang XQ, Hadwen T, Rothnagel JA. Green fluorescent protein as a reporter in translational assays. Anal Biochem. 2005;336:135–7. doi: 10.1016/j.ab.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 31.Wang XQ, Rothnagel JA. Post-transcriptional regulation of the gli1 oncogene by the expression of alternative 5′ untranslated regions. J Biol Chem. 2001;276:1311–6. doi: 10.1074/jbc.M005191200. [DOI] [PubMed] [Google Scholar]
- 32.Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17:691–5. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 33.Bouwman P, van der Gulden H, van der Heijden I, et al. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 2013 doi: 10.1158/2159-8290.CD-13-0094. [DOI] [PubMed] [Google Scholar]
- 34.Kuznetsov SG, Liu P, Sharan SK. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat Med. 2008;14:875–81. doi: 10.1038/nm.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinrichsen I, Brieger A, Trojan J, Zeuzem S, Nilbert M, Plotz G. Expression defect size among unclassified MLH1 variants determines pathogenicity in Lynch syndrome diagnosis. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-12-3299. [DOI] [PubMed] [Google Scholar]
- 36.Iversen ES, Jr, Couch FJ, Goldgar DE, Tavtigian SV, Monteiro AN. A computational method to classify variants of uncertain significance using functional assay data with application to BRCA1. Cancer Epidemiol Biomarkers Prev. 2011;20:1078–88. doi: 10.1158/1055-9965.EPI-10-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rasmussen LJ, Heinen CD, Royer-Pokora B, et al. Pathological assessment of mismatch repair gene variants in lynch syndrome: past, present and future. Hum Mutat. 2012 doi: 10.1002/humu.22168. [DOI] [PubMed] [Google Scholar]
- 38.Heinen CD, Juel Rasmussen L. Determining the functional significance of mismatch repair gene missense variants using biochemical and cellular assays. Hered Cancer Clin Pract. 2012;10:9. doi: 10.1186/1897-4287-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.