

Abstract
Recombinant chimeras of small heat shock proteins (sHsp) HspB1, HspB5, and HspB6 containing enhanced yellow fluorescent protein (EYFP) attached to their C-terminal ends were constructed and purified. Some properties of these chimeras were compared with the corresponding properties of the same chimeras containing EYFP attached to the N-terminal end of sHsp. The C-terminal fluorescent chimeras of HspB1 and HspB5 tend to aggregate and form a heterogeneous mixture of oligomers. The apparent molecular weight of the largest C-terminal chimeric oligomers was higher than that of the corresponding N-terminal chimeras or of the wild-type proteins; however, both homooligomers of N-terminal chimeras and homooligomers of C-terminal chimeras contained fewer subunits than the wild-type HspB1 or HspB5. Both N-terminal and C-terminal chimeras of HspB6 form small oligomers with an apparent molecular weight of 73–84 kDa. The C-terminal chimeras exchange their subunits with homologous wild-type proteins. Heterooligomers formed by the wild-type HspB1 (or HspB5) and the C-terminal chimeras of HspB6 differ in size and composition from heterooligomers formed by the corresponding wild-type proteins. As a rule, the N-terminal chimeras possess similar or slightly higher chaperone-like activity than the corresponding wild-type proteins, whereas the C-terminal chimeras always have a lower chaperone-like activity than the wild-type proteins. It is concluded that attachment of EYFP to either N-terminal or C-terminal ends of sHsp affects their oligomeric structure, their ability to form heterooligomers, and their chaperone-like activity. Therefore, the data obtained with fluorescent chimeras of sHsp expressed in the cell should be interpreted with caution.
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-013-0477-0) contains supplementary material, which is available to authorized users.
Introduction
Small heat shock proteins (sHsp) form a large family of ubiquitously expressed proteins having important housekeeping roles in the cell (Basha et al. 2012; Hilton et al. 2012; Mymrikov et al. 2011). These proteins participate in regulation of the redox state of the cell and cytoskeleton, in proliferation and apoptosis, as well as in proteostasis that prevents aggregation of improperly folded proteins (Arrigo 2007; Ciocca et al. 2013; Boncoraglio et al. 2012). The human genome contains 10 genes encoding sHsp differently expressed in practically all tissues (Garrido et al. 2012). The monomers of sHsp have a small molecular weight (12–43 kDa) and contain a conservative α-crystallin domain, considered as a hallmark of the family of sHsp (Kriehuber et al. 2010). The α-crystallin domain seems to be responsible for the formation of stable dimers of sHsp (Clark et al. 2012). This domain is flanked by variable N-terminal and C-terminal regions that have important roles in the interaction of sHsp with protein targets, as well as in the formation of sHsp oligomers (Hilton et al. 2012; Delbecq et al. 2012). As a rule, sHsp form homooligomers or heterooligomers with a very flexible structure (Hilton et al. 2012; Mymrikov et al. 2012; Arrigo 2013). These oligomers can undergo rapid association and dissociation by exchanging their subunits and can change their intracellular location upon different stimuli (Bryantsev et al. 2002; Clarke and Mearow 2013). In order to analyze the mechanism of sHsp functioning, it is desirable to develop a method that can follow the formation of different oligomers of sHsp and their translocation in the cell. Utilization of fluorescent proteins seems to be a very promising tool for this purpose (Chudakov et al. 2010). A number of different fluorescent chimeras of sHsp have been designed and used for investigation of the intracellular location (Doshi et al. 2009; Qian et al. 2006; Borrelli et al. 2002; Shelden et al. 2002; Raju and Abraham 2011, 2013; Irobi et al. 2004; Sun et al. 2007) and formation of homooligomeric and heterooligomeric complexes of sHsp (Fontaine et al. 2005; Fontaine et al. 2006; Sun et al. 2004; Datskevich et al. 2012a). Utilization of fluorescent chimeras of sHsp has provided important and interesting information. However, the molecular weight of fluorescent proteins is comparable with that of sHsp, and therefore, attachment of fluorescent protein to sHsp can significantly affect their structure and properties. Indeed, we found that fusion of fluorescent proteins to the N-terminal end of sHsp affects some of their properties (Datskevich et al. 2012b). Therefore, it seems reasonable to change the location of the fluorescent protein inside fluorescent chimeras and to obtain chimeras containing fluorescent protein attached to the C-terminal end of sHsp. This paper deals with the isolation and characterization of chimeras of sHsp containing enhanced yellow fluorescent protein (EYFP) on their C-terminal ends and comparison of some properties of the N-terminal and C-terminal chimeras of three sHsp, HspB1 (Hsp27), HspB5 (αB-crystallin), and HspB6 (Hsp20).
Materials and methods
Cloning of fluorescent chimeras of sHsp
EYFP was fused to the C-terminal ends of one of four human sHsp (HspB1, HspB5, HspB6, HspB8) through a flexible linker (GGGSGGGTGGG) coded by the sequence carrying the AgeI restriction site. The properties of these chimeras were compared with the corresponding properties of chimeras carrying EYFP at their N-terminal ends. These N-terminal chimeras were expressed and purified as described earlier (Datskevich et al. 2012b). cDNA of EYFP and the corresponding sHsp with parts of the linker was amplified by Pfu DNA polymerase (Thermo Scientific) utilizing as a matrix the pET23b(+) plasmid carrying the correspondent sequences cloned at the NdeI and XhoI restriction sites (Datskevich et al. 2012b). sHsp fragments were amplified using a pair of gene-specific primers: sHsp-NdeI forward and sHsp-AgeI reverse primers, which also carried part of the linker. To obtain cDNA of EYFP, we used gene-specific EYFP-AgeI forward primer carrying a second part of the linker and T7 reverse primer. Primer sequences are presented in Online Resource 1. Amplified fragments of sHsp and EYFP were restricted and ligated at the AgeI restriction site to obtain full-length fusion protein inserts, which were amplified by PCR using the corresponding forward primer and a T7 reverse primer. Inserts were cloned into the pET23b(+) vector at the NdeI and XhoI restriction sites. All plasmids were verified by sequencing.
Expression of fluorescent chimeras
Developing the method of expression of fluorescent chimeras in a soluble state, we used different expression strains of Escherichia coli (BL 21 (DE3), C41 (DE3), and C43 (DE3)) (Miroux and Walker 1996) and various expression conditions.
Seed cultures were prepared as follows: Competent cells (Chung et al. 1989) were transformed with the pET23b(+) plasmid carrying full-size fluorescent chimeras cDNA, sowed on Petri dishes (Luria–Bertani (LB) agar, 0.1 mg/ml ampicillin (Amp)), and grown overnight at 37 °C. One colony from the dish was inoculated to 20 ml of LB broth with 0.1 mg/ml Amp and grown overnight (37 °C, 230 rpm). For test expression, 1 ml of seed culture was transferred to 19 ml of medium and grown under selected conditions. For preparative expression, 40 ml of seed culture was added to 800 ml of medium and grown under selected conditions in two 2-l flasks.
Several sets of conditions for fusion protein expression have been tested. In the first case, bacteria were grown in 1× LB broth with 0.1 mg/ml Amp at 37 °C up to OD600 equal to 0.6. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to the final concentration of 1 mM and cultivation was continued either at 37, 30, or 20 °C for 5–7 or 20 h. In the second case, we used the so-called autoinduction method (Studier 2005). In this case, 1 ml of seed culture was transferred to 19 ml of 3× LB broth with 0.1 mg/ml Amp and the cells were grown for 7 h at 37 °C on a shaker. Afterwards, the temperature was changed to 20, 25, or 30 °C or was left unchanged and culturing was continued for an additional 15–18 h.
In the course of test expression, 1 ml of bacterial culture was collected and subjected to centrifugation (5,000×g, 5 min). The pellet collected was resuspended in 200 μl of lysis buffer (50 mM Tris/HCl pH 8.0, 100 mM NaCl, 15 mM mercaptoethanol (ME), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), and 1 mM ethylenediaminetetraacetic acid (EDTA)). Suspension was sonicated two times for 20 s on ice and centrifuged (14,000×g, 10 min). The pellet was suspended in 200 μl of lysis buffer, and the protein composition of the pellet and supernatant was determined by sodium dodecyl sulfate (SDS) gel electrophoresis (Laemmli 1970). Conditions providing the most efficient accumulation of fluorescent chimeras in supernatant were used for preparative expression. HspB1-EYFP and HspB6-EYFP were expressed in E. coli BL21 strain using the autoinduction method with the temperature of the second stage of expression kept at 30 °C, i.e., under conditions described earlier for the expression of N-terminal chimeras (Datskevich et al. 2012b). Expression of HspB5-EYFP was performed in an E. coli C43 strain. In this case, expression was performed in the 1× LB medium and lasted for 21 h at 30 °C after the addition of IPTG. Although we varied the bacterial strains and the conditions of expression, we failed to obtain recombinant C-terminal chimera of HspB8 in a soluble state.
Isolation of fluorescent chimeras
Purification of soluble recombinant fluorescent chimeras was performed as described earlier (Datskevich et al. 2012b). Briefly, bacterial cells in the lysis buffer (50 mM Tris/HCl pH 8.0, 100 mM NaCl, 15 mM ME, 0.5 mM PMSF, and 1 mM EDTA) were sonicated and subjected to centrifugation (25,000×g, 20 min). Extraction with a lysis buffer was repeated two more times and supernatants collected were subjected to ammonium sulfate fractionation. Protein fractions obtained in the ranges of 0–20, 20–40, and 40–60 % ammonium sulfate saturation were collected and analyzed by SDS gel electrophoresis (Laemmli 1970). Fractions enriched by fluorescent chimeras were dialyzed against buffer B (20 mM Tris/acetate pH 7.6, 10 mM NaCl, 15 mM ME, 0.1 mM PMSF, and 0.1 mM EDTA), subjected to ultracentrifugation (105,000×g, 30 min), and loaded onto a 5 ml HiTrapQ column equilibrated with buffer B. Proteins were eluted by linear gradient (12 column volumes) of NaCl (10–510 mM). The samples containing fluorescent chimeras were collected, concentrated, and loaded onto a HiLoad 26/60 Superdex 200 column equilibrated with buffer B containing 150 mM NaCl. The fractions containing fluorescent chimeras were combined, dialyzed against buffer B containing 2 mM dithiothreitol (DTT) instead of 15 mM ME, and stored at −20 °C.
Different methods were used to isolate fluorescent chimeras from inclusion bodies. The inclusion bodies, containing fluorescent chimeras, were purified by the method of Yang et al. (2011). The inclusion bodies were first dissolved in the buffer containing 7 M guanidine chloride (50 mM Tris/HCl, 50 mM NaCl, 10 mM ME, and 7 M guanidine chloride pH 8.0), and then rapidly precipitated by dilution in the buffer without guanidine chloride (50 mM Tris/HCl, 50 mM NaCl, 1 mM EDTA, and 10 mM ME pH 8.0) (Yang et al. 2011). The pellet of denatured protein was dissolved in the buffer containing 8 M urea (50 mM Tris/HCl, 50 mM NaCl, 10 mM ME, and 8 M urea pH 8.0) and dropwise added to the large volume of renaturation buffer (20 mM Tris/HCl pH 8.0, 1 mM EDTA, and 15 mM ME) and stirred slowly at 4 °C for 2 days. The obtained sample was subjected to centrifugation followed by ion-exchange chromatography on a HiTrapQ column and size-exclusion chromatography (SEC) as described previously. In this case, the yield of fluorescent chimeras was either very low or the spectral properties of fluorescent chimera were different from those of intact fluorescent protein, thus indicating improper folding.
Alternatively, inclusion bodies were dissolved either in 2 M arginine (pH 8.0) or buffer B, containing 8 M urea. After centrifugation, the samples were dialyzed for 2 days against buffer B. In the course of dialysis, the largest part of the protein was precipitated, making further purification impossible.
Size-exclusion chromatography
The quaternary structure of the wild-type sHsp and their fluorescent chimeras was analyzed by means of SEC performed on a Superdex 200 HR 10/30 column equilibrated with buffer B containing 150 mM NaCl. All experiments were performed at room temperature (20–25 °C), i.e., under conditions where the rate of sHsp subunits exchange is rather low (Bukach et al. 2009). Samples (150 μl) containing 12–130 μg of protein were loaded on the column and eluted at a rate of 0.5 ml/min. The apparent molecular weight was determined by using thyroglobulin (669 kDa), ferritin (440 kDa), catalase (240 kDa), glyceraldehyde-3-phosphate dehydrogenase (144 kDa), bovine serum albumin (68 kDa), and ovalbumin (43 kDa) as molecular weight standards.
Dynamic light scattering
All measurements were performed at 25 °C on Zetasizer Nano (Malvern) in buffer B containing 150 mM NaCl at protein concentrations equal to 0.3 mg/ml. Each measurement lasted 15 s and was repeated 10 times. This cycle of measurements was repeated 10 times, and thus, 100 measurements were accumulated for each sample. The data were evaluated by a built-in program, and the number distribution was used to estimate the particle size.
Formation of homooligomeric and heterooligomeric complexes of sHsp
Different sHsp (1 mg/ml) in buffer B containing 150 mM NaCl were incubated at 37 °C for 1 h in the presence of 15 mM DTT. To obtain mixed homooligomers of the wild-type sHsp and their chimeras, we mixed equal volumes of two reduced proteins and incubated this mixture for 1 h either at 4 or 42 °C. Heterooligomers of the wild-type sHsp and fluorescent chimeras of another sHsp were obtained in a similar manner. Thus, obtained samples were analyzed by means of SEC on a Superdex 200 HR 10/30 column, loading on the column either isolated proteins or their mixtures incubated at two different temperatures. The column was operated at a rate of 0.5 ml/min and fractions (300 μl) were collected. The protein compositions of these fractions were analyzed by means of SDS gel electrophoresis (Laemmli 1970).
Chaperone-like activity
The chaperone-like activity of sHsp and their fluorescent chimeras was determined by using two model protein substrates, namely, lysozyme and insulin. In the first case, all experiments were performed in 50 mM potassium phosphate buffer (pH 7.4) containing 50 mM NaCl at 37 °C at lysozyme concentration equal to 10 μM and variable concentrations of sHsp (5–10 μM per monomer). After preincubation for 5 min at 37 °C, reaction was started by the addition of DTT up to a final concentration of 20 mM and lysozyme aggregation was followed by an increase of the optical density at 340 nm. In the second case, reaction was performed in the buffer containing 100 μl of aggregation buffer (100 mM potassium phosphate (pH 8.5), 100 mM NaCl), 10–15 μl of insulin (dissolved in 2.5 % acetic acid) so that the final concentration of insulin was equal to 50 μM, variable quantities of buffer B and sHsp or their fluorescent chimeras (2–5 μM per monomer). The samples were preincubated for 5 min at 37 °C and reaction was started by the addition of DTT up to the final concentration 20 mM. Aggregation of the B-chain of insulin was followed by an increase of optical density at 340 nm.
Spectroscopic methods
The absorbance spectra of proteins were recorded on either Ultrospec 3100 (Amersham-Pharmacia) or NanoVue (GE Healthcare) spectrophotometers. The protein concentration was determined spectrophotometrically, taking A2800.1 % equal to 1.775 for HspB1 (UniProt P04792), 0.693 for HspB5 (UniProt P02511), and 0.582 for HspB6 (UniProt O14558). The concentration of chimeras of EYFP was determined by ε514 equal to 83,400.
Results
Oligomeric structure of sHsp chimeras containing fluorescent protein attached to their C-terminal end
SEC and dynamic light scattering (DLS) were used for the analysis of the oligomeric state of the wild-type sHsp and their fluorescent chimeras. As reported earlier (Datskevich et al. 2012b), the wild-type HspB1 forms large oligomers with an apparent molecular weight of 650–700 kDa and was eluted as a peak with an elution volume ∼10.3 ml, the position of which was independent of the quantity of protein loaded on the column (Fig. 1d). The elution profile of the C-terminal fluorescent chimera of HspB1 was strongly dependent on the quantity of protein loaded on the column (Fig. 1a). At high load, HspB1-EYFP was eluted as a main peak with an elution volume of 9.4 ml (∼800 kDa), whereas at low load, an additional peak eluted at ∼13.0 ml was detected on the elution profile (Fig. 1a). In addition to these two peaks, we also detected a small peak that was eluted close to the exclusion volume of the column (∼7.8 ml).
Fig. 1.

Analysis of the oligomeric structure of sHsp and their fluorescent chimeras by SEC. Normalized elution profiles of the C-terminal fluorescent chimeras of HspB1 (a), HspB5 (b), and HspB6 (c). In each case, the column was loaded with 12, 34, 68, or 136 μg of proteins (curves 1–4, respectively). Dependence of the elution volume of the main protein peak on the quantity of protein loaded on the column for the wild-type protein (1) and its N-terminal (2) or C-terminal (3) fluorescent chimeras for HspB1 (d), HspB5 (e), or HspB6 (f)
Both the wild-type HspB5 and its N-terminal fluorescent chimeras formed large stable oligomers, which, independent of protein concentration, were eluted as more or less symmetrical peaks with elution volumes of ∼10.4 ml corresponding to Mr ∼650 kDa (Fig. 1e). HspB5-EYFP formed very large oligomers with Mr ∼1,000 kDa, which, independent of the quantity of protein loaded on the column, were eluted at ∼8.5 ml (Fig. 1b).
The wild-type HspB6 formed small oligomers (probably dimers) with Mr ∼43–45 kDa with an elution volume (∼14.8–14.9 ml) that was practically independent of the quantity of protein loaded on the column (Fig. 1f). The chromatographic behavior of both N-terminal and C-terminal fluorescent chimeras was very similar, and both proteins formed stable small-sized oligomers with Mr 78–82 kDa and an elution volume ∼13.4 ml (Fig. 1c, f).
The data of the SEC were confirmed by means of DLS (Table 1). These experiments were performed at a fixed protein concentration equal to 0.3 mg/ml. Under these conditions, the size of the oligomers of the wild-type HspB1 was slightly larger than that of its N-terminal chimeras (Table 1) and, at the same time, it was significantly smaller than that of its C-terminal chimeras. It is worth mentioning that the dispersion of the size of the wild-type HspB1 oligomers and their N-terminal fluorescent chimeras was similar and significantly smaller than that of the C-terminal fluorescent chimeras. These results completely agree with the data of SEC and indicate that the C-terminal chimeras of HspB1 form a heterogeneous mixture of different sizes of oligomers (Fig. 1a). The oligomers of the wild-type HspB5 and its N-terminal chimeras have similar size and dispersion (Table 1), whereas the C-terminal chimeras of HspB5 form larger oligomers and have wide size dispersion (Table 1). Finally, both N-terminal and C-terminal chimeras of HspB6 form similar-sized small oligomers (Table 1).
Table 1.
Determination of the size (particle diameter D and polydispersity P di) of the wild-type human sHsp and their N-terminal and C-terminal fluorescent chimeras
| Sample | D int. distrib. ± SD, nm | P di ± SD |
|---|---|---|
| HspB1 WT | 18.55 ± 0.73 | 0.28 ± 0.04 |
| EYFP-HspB1 | 16.94 ± 0.54 | 0.24 ± 0.06 |
| HspB1-EYFP | 28.50 ± 1.67 | 0.72 ± 0.16 |
| HspB5-WT | 19.50 ± 0.77 | 0.23 ± 0.06 |
| EYFP-HspB5 | 18.16 ± 0.62 | 0.28 ± 0.07 |
| HspB5-EYFP | 29.14 ± 0.77 | 0.68 ± 0.13 |
| HspB6 WT | 8.98 ± 0.88 | 0.34 ± 0.11 |
| EYFP-HspB6 | 10.04 ± 0.16 | 0.52 ± 0.14 |
| HspB6-EYFP | 10.28 ± 1.94 | 0.49 ± 0.13 |
Comparing the data obtained on the N-terminal fluorescent chimeras (Datskevich et al. 2012b) with those obtained on the C-terminal chimeras, we can conclude that, in the case of HspB1 and HspB5, the molecular weight of their N-terminal chimeras was either slightly lower or equal, whereas the molecular weight of their C-terminal chimeras was significantly higher than that of oligomers formed by the corresponding wild-type proteins (Table 2). However, in all cases, oligomers formed by fluorescent chimeras of HspB1 and HspB5 contained a smaller number of subunits than oligomers formed by the wild-type proteins (Table 2). In the case of sHsp tending to form only small oligomers (HspB6), attachment of yellow fluorescent protein either to the N-terminal ends or to the C-terminal ends does not affect the oligomeric state and both the wild-type protein and its chimeras form only small oligomers (probably dimers). Similar results were obtained earlier with the N-terminal chimeras of HspB8 (Datskevich et al. 2012b) (Table 2).
Table 2.
Apparent molecular weight of the main largest oligomers (M r in kilodaltons) determined by SEC and calculated approximate number of subunits (N) in homooligomers of sHsp and their fluorescent chimeras
| Samples | Wild type | N-terminal chimeras | C-terminal chimeras | |||
|---|---|---|---|---|---|---|
| M r | N | M r | N | M r | N | |
| HspB1 | ∼650–700 | 24–28 | ∼480–500 | 8–10 | ∼790–850 | 14–16 |
| HspB5 | ∼650 | 26–30 | ∼650–670 | 15–17 | ∼1,000 | 20–23 |
| HspB6 | ∼43–45 | 2 | ∼78–82 | 2 | ∼78–82 | 2 |
| HspB8 | ∼36–43 | 1–2 | ∼74–78 | 2 | ND | ND |
ND not determined
Formation of mixed oligomers of the wild-type sHsp and their fluorescent chimeras
As already mentioned, the wild-type sHsp and their fluorescent chimeras form different-sized oligomers. Therefore, the question arises whether oligomers formed by the wild-type proteins and their fluorescent chimeras can exchange their subunits. To answer this question, the mixture of the wild-type protein and its chimeras preincubated at 4 or 42 °C were subjected to SEC.
Elution volume of the wild-type HspB1 (∼10.3 ml) is intermediate between that of the N-terminal (∼11.4 ml) and the C-terminal (∼9.4 ml) chimeras (see Fig. 1d). Preincubation of the mixture of the wild-type HspB1 with its N-terminal chimeras was accompanied by formation of a new peak with an elution volume of about 10.8 ml and a decrease of the amplitude of the peaks corresponding to isolated wild-type protein and isolated N-terminal fluorescent chimera (Online Resource 2). This effect was more pronounced if preincubation was performed at 42 °C. In this case, the peak of mixed oligomers was more sharp and symmetric and it was shifted to lower elution volumes. Similar results were obtained with the C-terminal chimera (Online Resource 2).
Elution volumes of the wild-type HspB5 (∼10.4 ml) and its N-terminal chimera (∼10.6 ml) are similar and are larger than those of the C-terminal chimera of HspB5 (∼8.5 ml). When the mixture of the wild-type HspB5 and its N-terminal chimera was preincubated at 4 °C, the elution profile was exactly equal to the sum of the elution profiles of two isolated proteins, thus indicating that, at low temperature, the exchange of subunits is very improbable (Fig. 2a). When preincubation was performed at 42 °C, the elution profile was changed and a peak corresponding to the mixed oligomers with an elution volume ∼10.0 ml appeared on the elution profile (Fig. 2b). Similar results were obtained in the case of the C-terminal chimera of HspB5. At low temperature, exchange of subunits of the wild-type protein and its fluorescent chimera was practically completely prevented (Fig. 2c). At the same time at elevated temperature, the subunits were easily exchanged, and this was accompanied by formation of a new sharp peak with an elution volume of ∼9.6 ml (Fig. 2d).
Fig. 2.

Formation of mixed oligomers of the wild-type HspB5 and its N-terminal (a, b) or C-terminal (c, d) chimeras. Each panel presents elution profiles of isolated wild-type HspB5 (1, dotted line), isolated fluorescent chimera (2, dashed line), the algebraic sum of these two profiles (3, dash-dotted line), and real elution profile of the mixture of the wild-type protein and its fluorescent chimera (4, solid line) preincubated either at 4 °C (a, c) or 42 °C (b, d)
As already mentioned, both N-terminal and C-terminal chimeras of HspB6 have similar elution volumes (∼13.4 ml), whereas the wild-type protein is eluted at ∼14.8 ml. Preincubation of the mixture of the wild-type HspB6 and its fluorescent chimeras both at low and high temperatures was accompanied by a decrease in the amplitude of the peaks corresponding to isolated proteins, by a shifting of the elution volumes of the peaks corresponding to isolated proteins, and by an increase of absorbency in between these peaks (Online Resource 3). These data indicate that the subunits of the wild-type HspB6 and its fluorescent chimeras either form weak temporal complexes with a high rate of dissociation or can exchange their subunits forming mixed oligomers.
Heterooligomers formed by the C-terminal fluorescent chimera of HspB6 and the wild-type HspB1 and HspB5
It is well known that the sHsp are able to form heterooligomers (Mymrikov et al. 2012; Datskevich et al. 2012a; Arrigo 2013). Therefore, it seems reasonable to analyze the effect of the yellow fluorescent protein attached to the C-terminal end of HspB6 on its ability to interact with the wild-type HspB1 and HspB5.
The wild-type HspB1 and HspB5 form large oligomers having elution volumes much smaller than that of the C-terminal fluorescent chimera of HspB6. This makes it acceptable to use SEC to investigate the interaction of the C-terminal fluorescent chimera of HspB6 with two other sHsp. Mixed together and preincubated at an elevated temperature, the wild-type HspB1 and HspB6 are able to form two types of heterooligomeric complexes with Mr 100 and 300 kDa (Bukach et al. 2009; Mymrikov et al. 2012; Datskevich et al. 2012a) (Fig. 3a). Both these complexes contain roughly equal quantities of both proteins (Bukach et al. 2009) (Fig. 3c). The C-terminal chimera of HspB6 was also able to form heterooligomeric complexes with the wild-type HspB1. However, in this case, heterooligomeric complexes had apparent molecular weights of ∼670 and ∼250 kDa (Fig. 3d). The large heterooligomeric complex contained only trace amounts of fluorescent chimera of HspB6. It is possible that this peak predominantly contains unexchanged wild-type HspB1 with a small population of exchanged heterooligomers containing a few subunits of fluorescent chimeras of HspB6. At the same time, according to the SDS gel electrophoresis data, the small heterooligomeric complex (Mr 250 kDa) contained roughly equal quantities of both proteins (Fig. 3f). Thus, the heterooligomeric complexes formed by HspB1 and by wild-type HspB6 or its C-terminal chimera are different in size and subunit stoichiometry.
Fig. 3.

Formation of heterooligomeric complexes of the wild-type HspB1 and either the wild-type HspB6 (a–c) or HspB6-EYFP (d–f). a, d Elution profiles of isolated HspB6 or its fluorescent chimera (1, dashed line), isolated wild-type HspB1 (2, dotted line), and the mixture of two proteins preincubated either at 4 °C (3, dash-dotted line) or 42 °C (4, solid line). b, c, e, f SDS gel electrophoresis of fractions (indicated above each track) collected in the course of SEC. The temperature of preincubation is indicated below each panel. Positions of the wild-type HspB6 (B6), HspB6-EYFP (B6-Y), and the wild-type HspB1 (B1) and molecular weight standards (in kilodaltons) are marked by arrows
The wild-type HspB6 exchanges its subunits with the wild-type HspB5, and this is accompanied by formation of a heterooligomeric complex with Mr ∼480 kDa. This complex was formed only if the mixture of two proteins was preincubated at an elevated temperature (Fig. 4a–c) (Mymrikov et al. 2012; Datskevich et al. 2012a). We measured the area of the peak of the wild-type HspB6 in the absence and in the presence of the wild-type HspB5 and, by this means, tried to estimate the approximate stoichiometry of the complex formed by these two proteins. By using this approach, we found that the stoichiometry of the heterooligomer HspB5/HspB6 was close to 1/0.5. The C-terminal chimera of HspB6 was also able to form a heterooligomeric complex with the wild-type HspB5. However, in this case, the heterooligomeric complex had Mr indistinguishable from that of the isolated wild-type HspB5, which was close to 680 kDa (Fig. 4d–f), and the approximate stoichiometry of HspB5/HspB6-YFP was equal to 1/0.35. Thus, the heterooligomeric complexes formed by HspB5 and the wild-type HspB6 or its C-terminal fluorescent chimera are different in size.
Fig. 4.
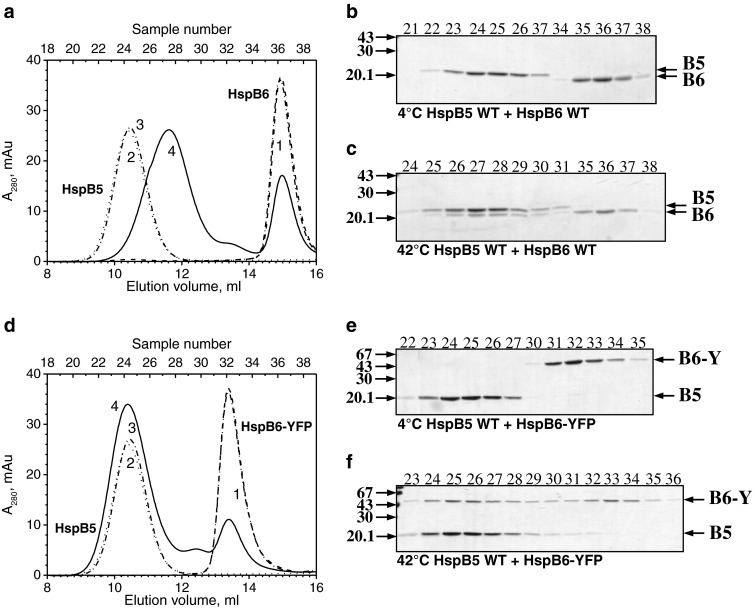
Formation of heterooligomeric complexes of the wild-type HspB5 and either the wild-type HspB6 (a–c) or HspB6-EYFP (d–f). a, d Elution profiles of isolated HspB6 or its fluorescent chimera (1, dashed line), isolated wild-type HspB5 (2, dotted line), and the mixture of two proteins preincubated either at 4 °C (3, dash-dotted line) or 42 °C (4, solid line). b, c, e, f SDS gel electrophoresis of fractions (indicated above each track) collected in the course of SEC. The temperature of preincubation is indicated below each panel. Positions of the wild-type HspB6 (B6), HspB6-EYFP (B6-Y), and the wild-type HspB5 (B5) and molecular weight standards (in kilodaltons) are marked by arrows
Chaperone-like activity of sHsp and their fluorescent chimeras
Two different model substrates were used for analyzing the chaperone-like activity of HspB1. At low concentrations, the wild-type HspB1 only slightly inhibited the reduction-induced aggregation of insulin (Fig. 5a). Under these conditions, the N-terminal chimera effectively retarded insulin aggregation, whereas the C-terminal chimera promoted insulin aggregation (Fig. 5a). Qualitatively similar results were obtained with another model substrate, lysozyme. In this case, at low sHsp concentrations, the wild-type protein slightly inhibited lysozyme aggregation initially (Fig. 5b). Under these conditions, the N-terminal chimera effectively inhibited lysozyme aggregation, whereas the C-terminal chimera promoted aggregation of this substrate (Fig. 5b). Similar results were obtained at high concentrations of sHsp (Fig. 5c). Indeed, under these conditions, the N-terminal chimera completely prevented aggregation of lysozyme, whereas the wild-type protein was less effective and the C-terminal chimera was very ineffective in inhibiting lysozyme aggregation (Fig. 5c).
Fig. 5.

Chaperone-like activity of HspB1 and its fluorescent chimeras. Reduction-induced aggregation of insulin (a) or lysozyme (b, c). Aggregation of isolated model substrate (1) or model substrate in the presence of the wild-type HspB1 (2) or its N-terminal (3) or C-terminal (4) fluorescent chimera was followed by an increase in optical density at 340 nm. The concentration of insulin was equal to 50 μM per monomer, and the concentration of sHsp was equal to 2 μM per monomer. The concentration of lysozyme was equal to 10 μM and the concentration of sHsp was equal to 5 μM (b) and 10 (c) per monomer, respectively. Data are representative of several experiments
Chaperone-like activity of HspB5 was analyzed by using lysozyme as a model protein substrate. At low concentrations, the wild-type HspB5 retarded lysozyme aggregation and decreased the amplitude of optical density at 340 nm, reflecting substrate aggregation at the end of incubation (Online Resource 4). At low concentrations, the N-terminal chimera of HspB5 retarded the onset of aggregation, whereas the C-terminal chimera promoted the onset of aggregation, and both chimeras increased the optical density at 340 nm at the end of incubation (Online Resource 4). At high concentrations, both the wild-type HspB5 and its N-terminal chimera effectively prevented reduction-induced aggregation of lysozyme, whereas the C-terminal chimera promoted reduction-induced aggregation of lysozyme (Online Resource 4).
Since HspB6 and its chimeras possessed only very low chaperone-like activity with lysozyme, we used insulin as a model substrate. The wild-type HspB6 effectively prevented the aggregation of insulin. In agreement with our earlier published data (Datskevich et al. 2012b), we found that the N-terminal chimera of HspB6 was less effective than the wild-type protein; however, it was also able to inhibit insulin aggregation. At the same time, the C-terminal chimera was rather ineffective in preventing insulin aggregation (Fig. 6).
Fig. 6.

Chaperone-like activity of HspB6 and its fluorescent chimeras using insulin as a model substrate. Reduction-induced aggregation of isolated insulin (1) or insulin in the presence of the wild-type HspB6 (2) or its N-terminal (3) or C-terminal (4) fluorescent chimeras. The concentration of insulin was equal to 50 μM per monomer, and the concentration of sHsp was equal to 5 μM per monomer. Data are representative of three experiments
Discussion
Fluorescent chimeric proteins are widely used for the investigation of intracellular localization and protein–protein interaction (Chudakov et al. 2010). Chimeras of HspB1, HspB5, HspB6, and HspB8 containing different fluorescent proteins (green, cyan, yellow, and citrine) fused to the N-terminal ends or to the C-terminal ends of sHsp were designed and successfully used in a number of investigations (Raju and Abraham 2011, 2013; Borrelli et al. 2002; Shelden et al. 2002; Qian et al. 2006; Fontaine et al. 2005, 2006; Sun et al. 2004, 2007; Doshi et al. 2009).
The main part of these investigations was performed at the cell level and it has been shown that some properties of fluorescent chimeras are similar to those of the wild-type proteins. For instance, chimeric HspB1 conferred stress protection in certain cell lines and this protection was comparable with that conferred by the wild-type HspB1 (Borrelli et al. 2002). Under stress conditions, both the wild-type HspB1 and its fluorescent chimeras migrate from cytosol to actin filaments and seem to be involved in protection of the cytoskeleton (Doshi et al. 2009; Clarke and Mearow 2013). Different stimuli can induce translocation of both the wild-type HspB1 and its fluorescent chimeras from the cytoplasm to the nuclei (Qian et al. 2006). However, this similarity was detected only in certain cell lines, whereas in other cell lines, fluorescent chimeras were not translocated to the nuclei (Qian et al. 2006; Borrelli et al. 2002).
At the same time, there are significant differences in the behavior of the wild-type sHsp and their fluorescent chimeras. For instance, heat shock and arsenite induced phosphorylation and dissociation of large oligomers of HspB1 in human A549 lung carcinoma and L929 murine cells, whereas the same stimuli induced only a very moderate dissociation of fluorescent chimera of HspB1 (Borrelli et al. 2002). Unmodified sHsp are highly soluble and do not form any aggregates in the cell, whereas expression of fluorescent chimeras of different wild-type sHsp is accompanied by accumulation of protein aggregates in 5–20 % of cells (Raju and Abraham 2011; Fontaine et al. 2006).
All these data indicate that not all properties of fluorescent chimeras are completely identical to those of unmodified sHsp. Indeed, our results indicate that the C-terminal chimeras of all analyzed sHsp tend to aggregate and require special conditions of expression and special bacterial strains for expression in the soluble state. The C-terminal chimeras of HspB1 and HspB5 form very large oligomers having a molecular weight larger than that of unmodified proteins (Fig. 1d, e). The data of DLS indicate that the C-terminal chimeras of HspB1 and HspB5 form oligomers, which are more heterogeneous and larger than the corresponding oligomers of the wild-type proteins (Table 1). However, although the molecular weight of the C-terminal chimeras of HspB1 and HspB5 is larger than that of the wild-type proteins, large homooligomers of fluorescent chimeras contain a smaller number of subunits than the corresponding homooligomers of the wild-type proteins (Table 2). It is also worth mentioning that the oligomeric structure of the C-terminal chimeras of both HspB1 and HspB5 seems to be significantly different from the oligomeric structure of the N-terminal chimeras of the same proteins (see Fig. 1). This means that, for sHsp tending to form large oligomers (like HspB1 and HspB5), the position of the fluorescent tag might affect the size (and probably the stability) of oligomers formed by these proteins. On the other hand, for HspB6 forming only small oligomers, attachment of fluorescent protein to either side of sHsp had no effect on the oligomeric structure and both the N-terminal and C-terminal fluorescent chimeras, as well as the wild-type protein, form only small oligomers (probably dimers).
Both the N-terminal and the C-terminal fluorescent chimeras may interact with corresponding wild-type proteins and seem to be able to exchange their subunits with the subunits of the wild-type protein (Online Resources 2 and 3; Fig. 2). The mixed homooligomers formed by the C-terminal fluorescent chimeras of HspB1 and HspB5 and the corresponding wild-type proteins were larger than the corresponding homooligomers formed by the wild-type proteins.
The wild-type sHsp can form heterooligomers (Sugiyama et al. 2000; Mymrikov et al. 2012), and the properties of these heterooligomers can be different from those of corresponding homooligomers (Arrigo 2013). Therefore, it seems reasonable to analyze the ability of the C-terminal fluorescent chimeras to form heterooligomeric complexes with other sHsp. The C-terminal fluorescent chimeras of HspB6 formed two types of heterooligomeric complexes with the wild-type HspB1 having Mr ∼250 and ∼670 kDa (Fig. 3). These heterooligomers were different from heterooligomers formed by the wild-type HspB1 and HspB6 and have Mr ∼100 and ∼300 kDa (Bukach et al. 2009; Mymrikov et al. 2012; Datskevich et al. 2012a). Similar results were obtained with two other sHsp, namely, HspB5 and HspB6. The C-terminal chimeras of HspB6 formed with the wild-type HspB5 only one type of heterooligomeric complex with Mr ∼680 kDa, identical to the apparent molecular weight of isolated HspB5 (Fig. 4). The size of this heterooligomeric complex was different from that of heterooligomers formed by the wild-type HspB5 and HspB6 and have Mr ∼480 kDa (Mymrikov et al. 2012). Thus, fluorescent chimeras of HspB6 can form heterooligomers with HspB1 and HspB5; however, the size of these heterooligomeric complexes is different from those formed by the wild-type HspB6.
The question arises, why does the position of a fluorescent protein affect the properties of chimeras? Both the N-terminal and C-terminal ends of sHsp are highly flexible and mobile and have an important role in the formation of sHsp oligomers (Hilton et al. 2012; Jehle et al. 2011; Delbecq et al. 2012; Delbecq and Klevit 2013). The N-terminal domains can be differently oriented in different monomers of the same oligomer; however, at least part of the N-terminal domains is located in the center of oligomers and interact with each other (Jehle et al. 2011; Hilton et al. 2012). Fluorescent protein attached to the N-terminal domain will occupy a rather large volume inside sHsp cages and will probably destabilize interaction of the N-terminal domains. It is also possible that the crowding induced by attachment of the fluorescent protein to the N-terminal domain will also decrease the number of subunits in sHsp oligomers. As a result, the oligomers of N-terminal chimeras of HspB1 and HspB5 contain a smaller number of subunits than the corresponding oligomers of the wild-type proteins (Table 2). The C-terminal ends are also very flexible and are exposed on the surface of oligomers. In addition, they can occupy different positions and interact in at least two different modes with the hydrophobic groove formed by β4 and β8 strands of the same or neighbor dimers inside oligomers (Hilton et al. 2012; Baldwin et al. 2011; Delbecq et al. 2012; Delbecq and Klevit 2013). Attachment of fluorescent protein to the C-terminal end of sHsp tending to form large oligomers (HspB1 and HspB5) might enhance hydrophobic interaction between subunits within the same oligomer or hydrophobic interactions between oligomers, thus leading to the formation of a very large and heterogeneous mixture of different-sized oligomers (Fig. 1). This is probably the reason for the poor solubility of practically all C-terminal chimeras and for the complications that arise in the course of their expression and purification. Thus, in the case of sHsp tending to form large oligomers, attachment of fluorescent proteins to either N-terminal or C-terminal ends can induce different changes in their quaternary structure. In the case of HspB6 tending to form only small oligomers (probably dimers) (Bukach et al. 2004), attachment of fluorescent protein to either N-terminal or C-terminal ends did not induce large changes of the quaternary structure, and both fluorescent chimeras form similar small-sized oligomers with Mr ∼78–82 kDa (Fig. 1c, f).
Having different properties, the N-terminal and C-terminal chimeras form heterooligomeric complexes which are distinct from each other and from those formed by the wild-type proteins (Figs. 3 and 4). These complexes differ both in their size and in subunit composition and can have different properties. It is difficult to determine exactly the chaperone-like activity of heterooligomeric complexes of sHsp. However, we were able to compare the chaperone-like activity of the wild-type proteins and their fluorescent chimeras (Figs. 5 and 6; Online Resource 4). As a rule, the chaperone-like activity of the N-terminal chimeras was comparable or even slightly higher than that of the wild-type proteins, although with certain substrates some N-terminal chimeras possessed slightly lower chaperone-like activity than the wild-type protein. In contrast, the chaperone-like activity of the C-terminal chimeras was always lower than that of the wild-type proteins. Moreover, in certain cases, the C-terminal chimeras promoted rather than prevented substrate aggregation (Fig. 5). It is known that the C-terminal ends have an important role in the chaperone-like activity of sHsp and replacement of charged residues located in the C-terminal end of sHsp by hydrophobic residues decreases their chaperone-like activity (Morris et al. 2008). Attachment of fluorescent protein to the C-terminal end increases hydrophobicity and the probability of sHsp aggregation. All these factors can lead to a decrease in the chaperone-like activity of the C-terminal chimeras of sHsp.
Summing up, we might conclude that attachment of fluorescent proteins to either N-terminal or C-terminal ends can differently affect the oligomeric structure of sHsp, their chaperone-like activity, and their ability to form heterooligomers. This means that the data obtained with fluorescent chimeras of sHsp expressed in the cell should be interpreted with caution, especially in the case of sHsp tending to form large oligomers. We conclude that successful utilization of fluorescent chimeras demands detailed investigation and optimization of their structure and properties.
Electronic supplementary material
(DOC 33 kb)
(DOC 490 kb)
(DOC 489 kb)
(DOC 129 kb)
Acknowledgments
This investigation was supported by the Russian Foundation for Basic Research (grant nos. 13-0400015 to NBG and 14-04-31197 to PND).
Abbreviations
- Amp
Ampicillin
- DLS
Dynamic light scattering
- DTT
Dithiothreitol
- IPTG
Isopropyl β-d-1-thiogalactopyranoside
- ME
Mercaptoethanol
- SEC
Size-exclusion chromatography
- sHsp
Small heat shock proteins
References
- Arrigo AP. The cellular “networking” of mammalian Hsp27 and its functions in the control of protein folding, redox state and apoptosis. Adv Exp Med Biol. 2007;594:14–26. doi: 10.1007/978-0-387-39975-1_2. [DOI] [PubMed] [Google Scholar]
- Arrigo AP. Human small heat shock proteins: protein interactomes of homo- and hetero-oligomeric complexes: an update. FEBS Lett. 2013;587(13):1959–1969. doi: 10.1016/j.febslet.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Baldwin AJ, Hilton GR, Lioe H, Bagneris C, Benesch JL, Kay LE. Quaternary dynamics of alphaB-crystallin as a direct consequence of localised tertiary fluctuations in the C-terminus. J Mol Biol. 2011;413(2):310–320. doi: 10.1016/j.jmb.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Basha E, O’Neill H, Vierling E. Small heat shock proteins and alpha-crystallins: dynamic proteins with flexible functions. Trends Biochem Sci. 2012;37(3):106–117. doi: 10.1016/j.tibs.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncoraglio A, Minoia M, Carra S. The family of mammalian small heat shock proteins (HSPBs): implications in protein deposit diseases and motor neuropathies. Int J Biochem Cell Biol. 2012;44(10):1657–1669. doi: 10.1016/j.biocel.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Borrelli MJ, Bernock LJ, Landry J, Spitz DR, Weber LA, Hickey E, Freeman ML, Corry PM. Stress protection by a fluorescent Hsp27 chimera that is independent of nuclear translocation or multimeric dissociation. Cell Stress Chaperones. 2002;7(3):281–296. doi: 10.1379/1466-1268(2002)007<0281:SPBAFH>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryantsev AL, Loktionova SA, Ilyinskaya OP, Tararak EM, Kampinga HH, Kabakov AE. Distribution, phosphorylation, and activities of Hsp25 in heat-stressed H9c2 myoblasts: a functional link to cytoprotection. Cell Stress Chaperones. 2002;7(2):146–155. doi: 10.1379/1466-1268(2002)007<0146:DPAAOH>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukach OV, Seit-Nebi AS, Marston SB, Gusev NB. Some properties of human small heat shock protein Hsp20 (HspB6) Eur J Biochem. 2004;271(2):291–302. doi: 10.1046/j.1432-1033.2003.03928.x. [DOI] [PubMed] [Google Scholar]
- Bukach OV, Glukhova AE, Seit-Nebi AS, Gusev NB. Heterooligomeric complexes formed by human small heat shock proteins HspB1 (Hsp27) and HspB6 (Hsp20) Biochim Biophys Acta. 2009;1794(3):486–495. doi: 10.1016/j.bbapap.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90(3):1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- Chung CT, Niemela SL, Miller RH. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci U S A. 1989;86(7):2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocca DR, Arrigo AP, Calderwood SK. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol. 2013;87(1):19–48. doi: 10.1007/s00204-012-0918-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AR, Lubsen NH, Slingsby C. sHSP in the eye lens: crystallin mutations, cataract and proteostasis. Int J Biochem Cell Biol. 2012;44(10):1687–1697. doi: 10.1016/j.biocel.2012.02.015. [DOI] [PubMed] [Google Scholar]
- Clarke JP, Mearow KM. Cell stress promotes the association of phosphorylated HspB1 with F-actin. PLoS ONE. 2013;8(7):e68978. doi: 10.1371/journal.pone.0068978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datskevich PN, Mymrikov EV, Gusev NB. Utilization of fluorescent chimeras for investigation of heterooligomeric complexes formed by human small heat shock proteins. Biochimie. 2012 doi: 10.1016/j.biochi.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Datskevich PN, Mymrikov EV, Sluchanko NN, Shemetov AA, Sudnitsyna MV, Gusev NB. Expression, purification and some properties of fluorescent chimeras of human small heat shock proteins. Protein Expr Purif. 2012;82(1):45–54. doi: 10.1016/j.pep.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Delbecq SP, Klevit RE. One size does not fit all: the oligomeric states of alphaB crystallin. FEBS Lett. 2013;587(8):1073–1080. doi: 10.1016/j.febslet.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbecq SP, Jehle S, Klevit R. Binding determinants of the small heat shock protein, alphaB-crystallin: recognition of the ‘IxI’ motif. EMBO J. 2012;31(24):4587–4594. doi: 10.1038/emboj.2012.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi BM, Hightower LE, Lee J. The role of Hsp27 and actin in the regulation of movement in human cancer cells responding to heat shock. Cell Stress Chaperones. 2009;14(5):445–457. doi: 10.1007/s12192-008-0098-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine JM, Sun X, Benndorf R, Welsh MJ. Interactions of HSP22 (HSPB8) with HSP20, alphaB-crystallin, and HSPB3. Biochem Biophys Res Commun. 2005;337(3):1006–1011. doi: 10.1016/j.bbrc.2005.09.148. [DOI] [PubMed] [Google Scholar]
- Fontaine JM, Sun X, Hoppe AD, Simon S, Vicart P, Welsh MJ, Benndorf R. Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. FASEB J. 2006;20(12):2168–2170. doi: 10.1096/fj.06-5911fje. [DOI] [PubMed] [Google Scholar]
- Garrido C, Paul C, Seigneuric R, Kampinga HH. The small heat shock proteins family: the long forgotten chaperones. Int J Biochem Cell Biol. 2012;44(10):1588–1592. doi: 10.1016/j.biocel.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Hilton GR, Lioe H, Stengel F, Baldwin AJ, Benesch JL. Small heat-shock proteins: paramedics of the cell. Top Curr Chem. 2012 doi: 10.1007/128_2012_324. [DOI] [PubMed] [Google Scholar]
- Irobi J, Van Impe K, Seeman P, Jordanova A, Dierick I, Verpoorten N, Michalik A, De Vriendt E, Jacobs A, Van Gerwen V, Vennekens K, Mazanec R, Tournev I, Hilton-Jones D, Talbot K, Kremensky I, Van Den Bosch L, Robberecht W, Van Vandekerckhove J, Van Broeckhoven C, Gettemans J, De Jonghe P, Timmerman V. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004;36(6):597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- Jehle S, Vollmar BS, Bardiaux B, Dove KK, Rajagopal P, Gonen T, Oschkinat H, Klevit RE. N-terminal domain of alphaB-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proc Natl Acad Sci U S A. 2011;108(16):6409–6414. doi: 10.1073/pnas.1014656108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriehuber T, Rattei T, Weinmaier T, Bepperling A, Haslbeck M, Buchner J. Independent evolution of the core domain and its flanking sequences in small heat shock proteins. FASEB J. 2010;24(10):3633–3642. doi: 10.1096/fj.10-156992. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260(3):289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Morris AM, Treweek TM, Aquilina JA, Carver JA, Walker MJ. Glutamic acid residues in the C-terminal extension of small heat shock protein 25 are critical for structural and functional integrity. FEBS J. 2008;275(23):5885–5898. doi: 10.1111/j.1742-4658.2008.06719.x. [DOI] [PubMed] [Google Scholar]
- Mymrikov EV, Seit-Nebi AS, Gusev NB. Large potentials of small heat shock proteins. Physiol Rev. 2011;91(4):1123–1159. doi: 10.1152/physrev.00023.2010. [DOI] [PubMed] [Google Scholar]
- Mymrikov EV, Seit-Nebi AS, Gusev NB. Heterooligomeric complexes of human small heat shock proteins. Cell Stress Chaperones. 2012;17(2):157–169. doi: 10.1007/s12192-011-0296-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Zhang Z, Shi M, Yu M, Hu M, Xia Q, Shen B, Guo N. Expression and distribution of HSP27 in response to G418 in different human breast cancer cell lines. Histochem Cell Biol. 2006;126(5):593–601. doi: 10.1007/s00418-006-0195-0. [DOI] [PubMed] [Google Scholar]
- Raju I, Abraham EC. Congenital cataract causing mutants of alphaA-crystallin/sHSP form aggregates and aggresomes degraded through ubiquitin-proteasome pathway. PLoS ONE. 2011;6(11):e28085. doi: 10.1371/journal.pone.0028085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju I, Abraham EC. Mutants of human alphaB-crystallin cause enhanced protein aggregation and apoptosis in mammalian cells: influence of co-expression of HspB1. Biochem Biophys Res Commun. 2013;430(1):107–112. doi: 10.1016/j.bbrc.2012.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelden EA, Borrelli MJ, Pollock FM, Bonham R. Heat shock protein 27 associates with basolateral cell boundaries in heat-shocked and ATP-depleted epithelial cells. J Am Soc Nephrol. 2002;13(2):332–341. doi: 10.1681/ASN.V132332. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41(1):207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Sugiyama Y, Suzuki A, Kishikawa M, Akutsu R, Hirose T, Waye MM, Tsui SK, Yoshida S, Ohno S. Muscle develops a specific form of small heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. J Biol Chem. 2000;275(2):1095–1104. doi: 10.1074/jbc.275.2.1095. [DOI] [PubMed] [Google Scholar]
- Sun X, Fontaine JM, Rest JS, Shelden EA, Welsh MJ, Benndorf R. Interaction of human HSP22 (HSPB8) with other small heat shock proteins. J Biol Chem. 2004;279(4):2394–2402. doi: 10.1074/jbc.M311324200. [DOI] [PubMed] [Google Scholar]
- Sun X, Fontaine JM, Bartl I, Behnam B, Welsh MJ, Benndorf R. Induction of Hsp22 (HspB8) by estrogen and the metalloestrogen cadmium in estrogen receptor-positive breast cancer cells. Cell Stress Chaperones. 2007;12(4):307–319. doi: 10.1379/CSC-276.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zhang L, Zhang Y, Zhang T, Feng Y, Lu X, Lan W, Wang J, Wu H, Cao C, Wang X. Highly efficient production of soluble proteins from insoluble inclusion bodies by a two-step-denaturing and refolding method. PLoS ONE. 2011;6(7):e22981. doi: 10.1371/journal.pone.0022981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 33 kb)
(DOC 490 kb)
(DOC 489 kb)
(DOC 129 kb)