

Abstract
Efforts to model human pancreatic neuroendocrine tumors (PanNET) in animals have been moderately successful, with minimal evidence for glucagonomas or metastatic spread. The renin gene while classically associated with expression in the kidney is also expressed in many other extra-renal tissues including the pancreas. To induce tumorigenesis within renin specific tissues, floxed alleles of p53 and Rb were selectively abrogated using Cre-recombinase driven by the renin promoter. The primary neoplasm generated is a highly metastatic islet cell carcinoma of the pancreas. Lineage tracing identifies descendants of renin-expressing cells as pancreatic alpha cells despite a lack of active renin expression in the mature pancreas. Both primary and metastatic tumors express high levels of glucagon, furthermore an increased level of glucagon is found in the serum identifying the pancreatic cancer as a functional glucagonoma. This new model is highly penetrant and exhibits robust frequency of metastases to lymph nodes and liver, mimicking human disease and provides a useful platform for better understanding pancreatic endocrine differentiation and development, as well as islet cell carcinogenesis. The use of fluorescent reporters for lineage tracing of the cells contributing to disease initiation and progression provides a unique opportunity to dissect the timeline of disease, examining mechanisms of the metastatic process, as well as recovering primary and metastatic cells for identifying co-operating mutations that are necessary for progression of disease.
Keywords: Glucagonoma, Renin, Pancreas, PanNET
Introduction
Classically, the aspartyl protease renin is recognized for its role in the regulation of blood volume and electrolyte balance as the rate limiting step in the renin angiotensin system (RAS) cascade through its synthesis and release at the juxtaglomerular (JG) cell of the kidney. The RAS has more recently been shown to be critical for normal mammalian renal development with transient expression of renin marking the developing renal vasculature (1–3). However, renin expression is not limited to the developing kidney, it is also found in many extra-renal organs (4) including the pancreas reflecting a potential role as a regulatory system in diverse tissue environments (5–7).
Although there are extensive publications describing renin and RAS function in the pancreas, conflicting observations have made it difficult to identify the cellular source of renin expression (7–15). For example multiple lines of evidence have supported renin expression in the rat pancreas (7), and (pro)renin has been localized to both endothelial cells of pancreatic vasculature as well as beta cells of the human pancreas islets (8). More recently, highly intriguing in vitro studies have detected renin and its substrate, angiotenginogen, in early stage differentiating islet cells but not in undifferentiated progenitor cells (6).
There is a large body of data from many groups describing the relation and origin of different lineages of endocrine islet cells of the pancreas. Current understanding suggests that the multiplicity of primary endocrine cell types all differentiate independently from Pdx1 positive progenitor cells that give rise to unipotent precursor cells transiently expressing the Neurogenin3 (Ngn3) transcription factor (16–19). The specificity appears to be conferred by a shifting window of competency on the part of the pancreatic progenitor cells whereby they form different endocrine cell types at different developmental stages (20). Glucagon-expressing cells are the earliest islet cell-type to appear during endocrine pancreatic morphogenesis and the competence for progenitor cells to differentiate to this cell type appears at about 9–10 days in mouse pancreatic rudiment, while the competence to make ontogenetically related insulin and pancreatic polypeptide lineage appears between 10.5–12.5 days. In the mature islet, the mantle region is the site occupied by glucagon- and somatostatin-expressing cells (21), whereas the core of the mature islet in mice is predominantly occupied by the insulin-expressing beta cells (20).
Various endocrine cell types can give rise to pancreatic neuroendocrine tumors (PanNETs) which are characterized as functional or non-functional based on association with syndromes reflecting overproduction of cognate hormones (22). In humans, glucagonomas are observed to arise at a considerably lesser frequency than insulinomas (23). They appear to occur primarily sporadically but are infrequently found in association with the Multiple Endocrine Neoplasia (Men1) and von Hippel-Lindau (Vhl) syndromes where they can be multicentric (22). While the tumors are generally slow growing, 75–80 % of glucagonomas tend to be malignant at discovery with greater than 50% exhibiting metastasis. The primary metastatic sites are regional lymph nodes and liver (22).
While mutation of p53 is observed in human PanNETs, mutation rates for Rb, per se, appear to be very low (24, 25). In general, 80% of cancers do not have mutations in the Rb protein but have genetic alterations in other key components of the Rb regulatory pathway (26). Recent studies strongly support a more wide extant of involvement of the p53 and Rb pathways in PanNETs that is mediated through copy number alteration of negative regulators of these tumor suppressors. In particular a high fraction of tumors exhibit amplification of the p53 negative regulators MDM2, 4 and WIP1, as well as CDK4 which negatively regulates Rb (24, 25). High expression of Cdk4, both through increased copy number and activation of the mTOR pathway, has been shown to be coincident with Rb phosphorylation leading to inactivation in PanNETs (25). Furthermore, hypermethylation of p16Ink4a, a Cdk inhibitor, has also been identified in PanNETs (27). Therefore, while Rb itself may not be mutated, it is clear that both the p53 and Rb pathways are compromised at high frequency in the human disease.
Efforts to model PanNETs in animals have been variously successful. Genetic deficiencies for p53 and Rb cooperatively result in the development of endocrine tumors reminiscent of MEN1 cancer predisposition with the tumor spectrum encompassing numerous tissues but with minimal evidence for glucagonomas specifically, or metastasis (28, 29). Variant targetings to generate general Men1 deficiency have given variable levels of mimicry of the human disease, predominantly insulinomas with low levels of metastases observed (30–38). The RIP-Tag model of Hanahan in which robust and rapid islet cell tumorigenesis is induced has been a highly valuable productive model for studying the tumor progression process (39). It is limited, however, to development of insulinomas and metastasis is relatively infrequent in most genetic backgrounds. A parallel construct in which the glucagon promoter was used to drive SV40 Tag has been reported. It exhibited relatively low penetrance with most animals exhibiting unifocal dysplastic islets with variable functional character and minimal evidence for progression or metastasis (40).
In this paper we characterize a highly metastatic islet cell carcinoma of the pancreas that results from using a renin driven cre recombinase transgene to delete p53 and Rb in renin-expressing cells of the developing mouse. The cancer model developed can be further described as a glucagonoma with metastatic progression to liver and lymph node, which mimics the metastatic spread observed clinically in human disease. With insufficient mouse models currently available, this metastatic pancreatic neuroendocrine tumor model represents an unprecedented tool for studying the cooperating mutations responsible for development of glucagonomas including progression to metastatic disease.
Results
Renin specific deletion of p53 and Rb tumor suppressor genes leads to pancreatic neuroendocrine metastatic disease
In an attempt to phenocopy our earlier Ren-SV40Tag studies which give rise to kidney and subcutaneous tumors at approximately 3–4 months (41, 42) we utilized a BAC-based RenCre transgenic line we developed to selectively delete the floxed tumor suppressor genes p53 and Rb. The RenCre x floxed p53/Rb model gives rise to subcutaneous tumors, albeit infrequently, at approximately 3–4 months of age. However the primary pathology and lethality of this cross results from the development and metastasis of pancreatic neuroendocrine tumors (Figure 1A and 1B). At low power, the nodular and highly vascularized nature of the neuroendocrine tumor can be observed (Figure 1A), at higher magnification the organoid and trabecular growth pattern as well as the stippled chromatin of the primary pancreatic tumor is consistent with pathology characteristic of neuroendocrine disease (Figure 1B). Furthermore, upon performing necropsies of animals from the RenCre x floxed p53/Rb cross, metastatic involvement of the liver and lymph node was observed (Figure 1D–E, 1G–H). Similar to the primary tumor the liver metastases shared the nodular and highly vascularized phenotype (Figure 1D) as well stippled chromatin (Figure 1E) consistent with neuroendocrine tumors. The liver also contains many separate metastatic foci (Figure 1 D) which is congruent with the metastatic proliferation identified in human disease. The lymph node located adjacent to the pancreas is almost entirely replaced by metastatic neuroendocrine tumor, with only a localized area in the lower left corner containing lymphocytes in the lymph node (Figure 1G and 1H). To further confirm the tumor model as being pancreatic neuroendocrine in origin immunohistochemical stains for the neuroendocrine tumor markers, synaptophysin and chromogranin A were performed (Figure 2). As seen in this figure both the primary pancreatic tumor as well as the liver metastases strongly stain for these neuroendocrine markers (brown staining) which is consistent with a primary pancreatic neuroendocrine tumor that forms multiple metastases in the liver.
Figure 1.

Hematoxylin and eosin staining of paraffin embedded tissue identifying pancreas tumor and liver and lymph node metastasis in the RenCre/p53/Rb tumor model. A–B. Primary pancreatic tumor (A. 40x, B. 200x) C. Glucagon staining of primary pancreas tumor (200x). DE. H&E metastatic liver (D. 20x, E. 400x). F. Serial glucagon staining of liver metastasis (400x). G–H. Regional lymph node metastasis (G.100x, H.200x). I. Serial glucagon staining of regional lymph node (200x).
Figure 2.

Immunohistochemical stains for the neuroendocrine tumor markers, synaptophysin and chromogranin A. Left panels. Primary pancreas tumor and liver metastases (lower) stained with Chromogranin A. Right panels. Primary pancreas tumor and liver metastases (lower) stained with Synaptophysin
To further characterize the lethal phenotype of the PanNET model, 10 animals containing the RenCre transgene and the floxed p53/Rb alleles as well as a negative control group of 10 animals, where RenCre was not present in the genetic background, were monitor until animals succumbed or were sacrificed due to burden of disease. As presented in figure 3A, the 10 RenCre positive animals, which expressed the necessary Cre-recombinase to delete p53 and Rb, were euthanized due to morbidity or died by 29 weeks of age where as the RenCre negative littermates exhibited a normal murine life span. At necropsy all RenCre positive animals exhibited a heavy primary tumor burden and evidence of liver metastases.
Figure 3.

Analysis of RenCre x p53/Rb survival A. Survival curve for RenCre x p53/Rb transgenic animals and their RenCre negative littermates. Animals homozygous for both the floxed p53 and Rb alleles that are also RenCre positive succumb to metastatic pancreatic cancer by 29 weeks of age. RenCre negative litter mates show no sign of pathology or morbidity. B. Weightloss in RenCre x p53/Rb animals (17.7+/−2.16g) when compared to Cre− littermates (healthy) littermates (26.2+/− 3.06g).
Continual monitoring of the RenCre x floxed p53/Rb animals as they become ill has led to several observations in their appearance including scruffy, oily skin/fur and weight loss. When measuring the weight of end-stage RenCre x floxed p53/Rb animals (17.7+/−2.16g) and their RenCre negative (healthy) littermates (26.2+/− 3.06g) there is a clear reduction in the mass of the sick animals (Figure 3B). This is a 32% reduction in body weight in the sick animals when compared to healthy age-matched littermates, a symptom also found in human pancreatic disease.
Minor frequency of secondary tumor types in the RenCre x floxed p53/Rb model
Although the primary mortality of the RenCre x floxed p53/Rb animals is from metastatic pancreatic neuroendocrine disease with lethal onset beginning at approximately 22 weeks of age rare instances of subcutaneous tumors have arisen in the colony leading to the need to euthanize animals due to tumor burden at approximately 12–16 weeks of age. Furthermore, even more rare instances of mice dying as early as 8 weeks old from yet undetermined causes have also been observed. A detailed cataloging of the RenCre x floxed p53/Rb mouse colony over one year has identified 14 of 120 (~12%) RenCre x floxed p53/Rb animals that had to be euthanized due to subcutaneous tumor burden or died of unknown causes prior to becoming morbid from pancreatic disease. As seen in Figure 4 the primary pancreatic tumors and liver metastases are nodular and highly vascularized consistent with neuroendocrine disease, however the subcutaneous tumors have a more solid growth pattern, although at higher power vessels and extravagated red blood cells can be seen (Right panel). The subcutaneous tumor is negative for synaptophysin and chromogranin A and positive for the vascular marker CD31 (data not shown). Therefore, the subcutaneous tumor represents a different primary tumor, consistent with a high grade vascular sarcoma.
Figure 4.

Subcutaneous tumors are not metastases of PanNet origin. Left and middle panel. The nodular and highly vascularized nature of the neuroendocrine tumor can be appreciated in the primary pancreatic neuroendocrine tumor and in the liver metastasis. Right panel. The subcutaneous tumor is negative for synaptophysin and chromogranin A and positive for CD31 (a vascular marker). Therefore, the subcutaneous tumor represents a different primary tumor, a high grade vascular sarcoma.
Pancreatic tumors and liver metastasis express high levels of glucagon
The pancreases of animals containing primary tumors were compared to normal pancreas by qPCR. As is evident in figure 5 (supplemental table 1), glucagon transcript levels are highly elevated relative to normal pancreas with occasional evidence for aberrations in levels of other hormones. Immunoflourescent assessment of primary tumors and metastases to liver (Fig 5B&C) suggests that the primary tumors are primarily glucagon-expressing with some scattered pockets of cells expressing insulin or somatostatin, possibly resulting from inclusion of normal islet fragments within tumor region. The liver metastases are strongly glucagon-expressing with variable proportions of coincident somatostatin expression (Fig 5C). Furthermore, immunohistochemical staining clearly identifies glucagon expression in the primary pancreatic neuroendocrine tumor (Figure 1C), liver metastasis (Figure 1F), and lymph node metastasis (Figure1I). However, subcutaneous tumors did not stain for glucagon (data not shown).
Figure 5.

Assessment of endocrine hormone expression within pancreatic tumors from the RenCre/p53/Rb transgenic line. A. Expression of endocrine hormone genes in eight primary pancreatic tumors using qPCR. Log2 fold change of tumor when compared to normal pancreas. Glucagon has highest fold increase in tumor samples. B. and C. Indirect immunoflourescence identifying Glucagon as the primary hormone expressed in primary pancreatic tumors (B) and liver metastases (C) Glucagon is highly expressed in the pancreatic tumors and liver metastasis derived from the RenCre/p53/Rb cross. Low levels of somatastatin and insulin are present. (40x images)
Since the primary tumors of the pancreas as well as the metastatic lesions of the liver predominately express the alpha cell marker glucagon, serum levels of circulating glucagon were analyzed at four different time points during progression of disease (Figure 6). Six RenCre x floxed p53/Rb animals and 3 Cre− littermates were euthanized at each of the four time-points and their serum was analyzed for circulating glucagon levels. As shown in figure 6 the 2 month Cre+ animals did not show an increase in baseline glucagon levels when compared to negative littermates. However, the 4, 5, and 6 month time-points showed a dramatic increase in circulating glucagon levels. Since the primary and metastatic sites of this pancreatic disease not only express glucagon but also secrete glucagon into the blood this PanNET model can therefore be classified as a glucagonoma.
Figure 6.

Measurement of glucagon levels in the blood of RenCre x p53/Rb animals and Cre – littermates. At 2 months of age the RenCre x p53/Rb animals have normal circulating glucagon levels in their blood stream when compared to normal Cre− age-matched littermates. Dramatic increase in 4,5, and 6 month time-points of glucagon levels, characteristic of a glucagonoma.
Progression and metastatic potential of pancreatic neuroendocrine tumor (PanNET) model
Analyzing primary pancreatic tumors collected from different animals at three time points (4, 5, and 6 months) clearly shows substantial growth in the pancreatic tumor from month to month (Figure 7). At 4 months, the tumor forms two relatively small nodules located in the center of the pancreas. The tumors are approximately 1/3 of the pancreas in this image (Figure 7A). At 5 months, the tumor nodules have more than doubled in size, taking up almost the entire microscopic field (Figure 7B). By 6 months, the pancreatic neuroendocrine tumor cannot fit entirely in the microscopic field and has to be moved off center to get some normal pancreas into the image, thus showing that it is a primary pancreatic tumor (Figure 7C). High power (100X) images show that the tumor’s morphology and grade does not differ between 4, 5, and 6 months (Figure 7D–E). Thus, changes in progression seen are explained by the increased growth and size of the tumor over time.
Figure 7.
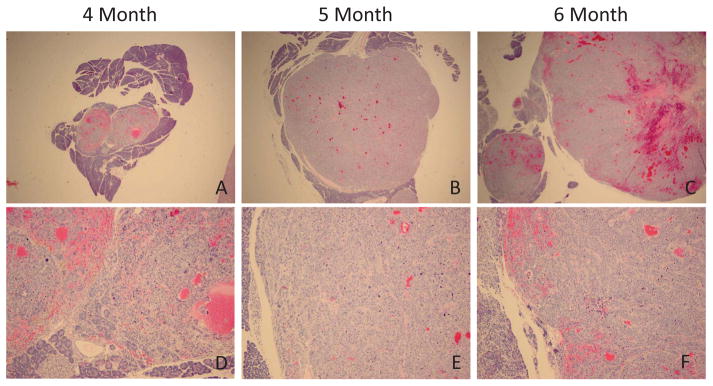
Change in size of the primary pancreatic neuroendocrine tumor among 4, 5, and 6 months. Size of primary pancreatic tumor increases as animals gets older, however the tumor’s morphology and grade does not differ between 4, 5, and 6 months. Upper panels (20x), lower (100x)
To further characterize the progression and metastatic potential of the newly developed pancreatic tumor model ten RenCre x floxed p53/Rb mice were euthanized at each of three time points (4,5, and 6 months of age) and a full necropsy was carried out by RADIL veterinary services including glucagon IHC on pancreas and liver tissue. As detailed in Table 1 all 30 mice (10 from each age group) had primary pancreatic tumors which stained for glucagon. One metastatic lesion (10%) was found in the 4 month old animals whereas at the 5 and 6 month time-points 30% and 40% of the animals had identifiable glucagon positive metastasis to the liver, respectively. All primary tumors of the pancreas and metastasis identified in the liver stained with glucagon, suggesting the alpha cell may be the only source of this metastatic disease. A high level of variation in the severity of the metastatic phenotype was observed in the six month old mice (Figure 8). One animal within the 6 month time-point contained several central veins of the liver filled with large neoplastic glucagon-staining cells without any detectable metastatic nodules (data not shown). Furthermore, as seen in figure 8, the level of metastatic spread identified in different animals varies from a few small aggregates of glucagon staining cells in the sinusoids (upper panels) to larger clusters of glucagon-expressing cells (middle panels) to multiple large metastatic nodules. Within the most advanced liver metastases the glucagon staining is not evident within every metastatic lesion (figure 8, lower panel). Although some of the large advanced foci lack glucagon expression they are composed of neoplastic cells similar to those found in the primary pancreatic nodules (previous IHC) and may represent a non-functional neuroendocrine tumor where no or insignificant amounts of excess hormone are produced by the tumor cells. The varying level of severity of metastatic spread within the disease model may be due to different co-operating mutations responsible for progression of disease where corruption of certain genetic pathways may lead to more progressive metastasis which will allow for the investigation of key genetic alterations responsible for the evolution of the disease.
Table 1.
Primary and Metastatic Frequency of Pancreatic Neuroendocrine Tumor Model
| Primary Tumor Glucagon Positive | Liver Metastases Glucagon Positive | |
|---|---|---|
| 4 Month | 10/10 | 1/10 |
| 5 Month | 10/10 | 3/10 |
| 6 Month | 10/10 | 4/10 |
Figure 8.

Variation in severity of metastatic disease in RenCre/p53/Rb tumor model. Liver sections from six month old end stage RenCre/p53/Rb mice stained with glucagon Ab (left panel, black arrows) and H&E stain (right panels) show varying levels of severity of metastasis to liver.
Identification of lineal descendants of renin expressing cells in the normal adult pancreas and PanNET model
Utilizing the RenCre transgenic line crossed to floxed stop reporter constructs, such as Zsgreen and Confetti, allows for the flourescent tagging of renin-expressing cells even after renin expression is quiescent. Lineage tracing of the renin-expressing cell using RenCre x Zsgreen animals identified GFP expression in the adult pancreas (Figure 9A). To verify faithful reporter expression multiple RenCre founder lines were used to identify lineal descendants of renin expression within the pancreas (supplemental figure 1). The cells of the pancreas that show evidence of having lineally expressed renin are primarily found in the mantle region with few cells evident in the core region (Figure 10). Using immunohistochemistry glucagon and somatostatin-expressing cells located in the mantle region were distributed in a similar pattern as GFP however only a minimal number of GFP expressing cells appear to localize within the insulin-expressing core domain (Figure 10). Using the RenCre x Confetti model, where the Confetti reporter will randomly label each renin-expressing cell with one of four fluorescent reporters allowing for independent lineage tracing of individual cells, further confirmed the distinct labeling of the islet cells of the pancreas (Figure 9B). Moreover, due to the fact that the islets are not all the same color, nor are individual islets monochromatic, the expression of renin in pancreatic development occurs after, or is coincident with, commitment of a pool of cells destined for the alpha cell lineage and that each islet is made up of multiple progenitors.
Figure 9.

Reporter expression tagging lineal descendants of renin in normal mouse pancreas and PanNET model. A. GFP expression identifying cell populations that have once expressed renin in the normal pancreas within the RenCre x Zsgreen model. Whole pancreas tissue visualized using a flourescence dissecting microscope (12x) B. Confetti reporter expression of islet cells of the adult pancreas using confocal microscopy (48x) C &D. Zsgreen was crossed into the RenCre/p53/Rb tumor model and lineal descendants of renin-expressing cells were identified using GFP. The primary tumor (12x)(C) as well as live metastasis(8x)(D) express GFP. Black star represents normal islet in the pancreas. Black arrows identify larger metastases of the liver.
Figure 10.

Co-localization of quiescent renin-expressing cells (GPF) with glucacon and somatostatin in the mantle region of the pancreas. GFP expression which identifies lineal descendants of renin expression co-localizes with glucagon and as well as somatostatin in the mantle region of the endocrine pancreas. Minimal overlap is identified between GFP and insulin expressing cells of the core region. (40x).
To trace the lineal descendant of the renin-expressing cell within the PanNET model the renin-expressing cells were tagged within the RenCre/p53/Rb cross by introducing the Zsgreen reporter into the RenCre/p53/Rb background. This complex genetic cross allows for the cells that have lineally expressed renin to be tagged with GFP even after renin expression is turned off. The primary tumors identified in the pancreas (fig 9C) as well as metastasis within the liver (fig 9D) express GFP further confirming that the cells that propagate these are lineal descendants of renin-expressing cells. Future studies will utilize the confetti reporter crossed into the RenCre/p53/Rb cross in an effort to connect metastases with primary pancreatic tumors responsible for their origin.
Discussion
Conditional deletion of p53 and Rb, specifically within the renin-expressing cells of the developing mouse, has led to the serendipitous development of a highly robust model for metastatic glucagonoma which faithfully mimics the key features of corresponding human disease including a high metastatic potential to clinically relevant sites such as the liver and regional lymph nodes. Initially, in an effort to phenocopy our earlier Ren-SV40TAg studies which were observed to give rise primarily to kidney and subcutaneous tumors at approximately 3–4 months (41, 42), we generated a genetic cross with RenCre and floxed alleles for both p53 and Rb which are the primary targets of SV40 T-antigen. This genetic cross allows for the renin specific deletion of the p53 and Rb alleles within renin-expressing cells of the developing mouse. Although the RenCre/p53/Rb does give rise to infrequent subcutaneous tumors at approximately 3–4 months, the primary pathology and cause of morbidity in this model is metastatic islet cell carcinoma of the pancreas. Interestingly, incidental pancreatic tumors have been noted in the Ren SV40Tag animals at autopsy (data not shown), although morbidity of these animals was due to kidney and subcutaneous tumor burden.
While mutation of p53 is observed in human PanNETs, the mutation rates for Rb appear to be very low (24, 25). However, recent studies strongly support a greater involvement of these pathways in PanNETs which are mediated through copy number alteration of negative regulators of these tumor suppressors. In particular a high fraction of tumors exhibit amplification of the p53 negative regulators MDM2, 4 and WIP1, and CDK4 which is also known to negatively regulate Rb (24, 25). Specifically, increased expression of CDK4 due to elevations in copy number and through the activation of the mTOR pathway via PTEN and TSC2 mutations leads to phosphorylation of Rb in a majority of human PanNETs (25). Rb has also been shown to be inactivated in PanNETs through hypermethylation of the CDK inhibitor p16INK4a (27). Mouse models have further validated the clinical observation that the p53 and Rb pathways are involved in the tumorigenic process in PanNETs. Included in these studies are the development of insulinomas found in the RIP1-Tag2 mouse, which inactivates p53 and Rb (43). Other studies using double knock-out mice for the Cdk inhibitors p18INK4c and p27KIP1 have been shown to develop islet cell hyperplasia (44). Furthermore, mice containing a mutant Cdk4 (R24C) allele, which cannot be down regulated by p16INK4a, produce a large number of tumors including PanNETs (45, 46). Therefore, while Rb itself may not be mutated, it is clear that both the p53 and Rb pathways are compromised at high frequency in pancreatic neuroendocrine tumors.
Renin is not expressed with abundance in mature pancreas, yet as previously discussed several independent transgenic lines report evidence for renin expression in lineal precursors of mature glucagon-expressing islet cells. Notably, in support of this interpretation some highly intriguing results have very recently been reported by Leung and colleagues in their studies of human pancreatic progenitor cells and their differentiation to islet cells during in vitro culture (6). They observe highly regulated expression of various components of the renin-angiotensin system (RAS) throughout the progenitor cell differentiation process. In particular renin and the precursor substrate, angiotensinogen, were detected selectively in early stage differentiating islet cells but not in undifferentiated progenitor cells. These highly provocative results are fully consistent with our observations suggesting transient renin expression during the differentiation process.
We also performed independent lineage tracing analysis of individual islet cells to determine the nature of the progenitor cell differentiation process of the endocrine pancreas. Using the Confetti mice crossed to the RenCre mice (wild type p53 and Rb) identified a distinct labeling pattern of the islet cells of the pancreas (Fig 9B). Moreover, by virtue of the fact that the islets are not all of uniform color, nor are individual islets monochromatic, we can infer that the expression of renin in pancreatic development occurs after, or is coincident with, commitment of a pool of cells destined for the α-cell lineage and that each islet is composed of descendants of multiple progenitors. Such inferences are consistent with recent findings that indicate there is no expansion of the committed α-cell population arising from the unipotent progenitor cells (17).
We believe our data can be coherently unified by a central hypothesis proposing that renin is transiently expressed during early glucagon lineage differentiation. We further argue that the RenCre-driven loss of p53 and Rb in these cells at this early stage predispose them to stochastic acquisition of further genetic and epigenetic damage resulting in carcinogenesis in those rare cells in which the correct cooperating mutations occur. We propose that a specific genomic insult is required for the cells to attain a metastatic phenotype. Whether the high levels of metastasis observed reflects the time-dependent occurrence of additional mutational hits or a different spectrum of co-operating ‘initial’ hits will ultimately need to be examined further. This way include the discovery of novel genes responsible for progression of disease or the identification of already known players in human pancreatic neuroendocrine cancer including Men1 and Vhl, as well as components of the p53 and Rb pathways.
As previously mentioned, efforts to model PanNETs in animals has been moderately successful, with minimal evidence for glucagonomas or metastatic spread (28, 29). In contrast, the model we have created is unique for the glucagonoma subtype of PanNET, highly penetrant, and exhibits robust frequency of metastases to lymph nodes and liver, which are also the major sites of metastatic disease in man. This model will provide a useful platform to better understand pancreatic neuroendocrine differentiation and development, as well as islet cell carcinogenesis. Furthermore, the ability to use fluorescent reporters for lineage tracing of the cells contributing to disease initiation and progression provides a unique opportunity to dissect the timeline of disease pathology. Given the robust metastatic spread, this model also holds promise as a platform for examining mechanisms and features of the metastatic process generally, as well as devising and optimizing therapeutic intervention strategies. Additionally, the increased palette of colors conferred by the multiple flourephores of the Confetti model affords an enhanced ability to address fundamental issues such as assessing hyperplasia within pre-tumorigenic islets, determining clonality of primary tumor foci, and correlating metastases with their primary tumor of origin. This model will allow for dissecting clonal evolution of metastases – will, indeed, allow for the dissection of multiple potential routes of acquisition of the metastatic phenotype - in an experimental format that cannot be matched by studying human tumors.
Materials and Methods
Construction of RenCre BAC Transgenic line
Insertion of the Cre recombinase downstream of the renin promoter region within the RP23-88k07 BAC was performed as previously described and the transgenic line created is termed RenCre within this manuscript (47, 48). Multiple expressing RenCre transgenic lines were developed to ensure faithful transgene expression with natural renin expression without positional effects due to insertion within the BAC construct. Validation of faithful reporting of the multiple RenCre transgenic lines was carried out by crossing RenCre to the floxed-stop-floxed-GFP reporter construct, B6.Cg-Gt(ROSA)26Sortm6(CAG-ZsGreen1)Hze/J (Zsgreen) reporter line. Kidneys from animals positive for both RenCre and Zsgreen were dissected and visualized to confirm accurate GFP reporting of endogenous renin expression (manuscript in submission). Furthermore, to test the recombination efficiency of our RenCre transgenic line we crossed it with both the Zsgreen reporter as well as an floxed-stop-floxed-RFP reporter, B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (Tomato) simultaneously. By assessing the percent of cells that are both green and red compared to the total cells marked with only a single reporter within the kidney of the mouse containing all three transgenes we deduce approximately 75% recombination efficiency of the RenCre transgenic line.
Generation of Pancreatic Tumor Model
To generate the metastatic pancreatic tumor model a RenCre BAC transgenic line was crossed to p53loxP/loxPRbloxP/loxP mice (49, 50). Subsequent backcrosses of RenCre positive, heterozygous p53wt/loxPRbwt/loxP female mice to homozygous p53loxP/loxPRbloxP/loxP male mice yielded the desired RenCre positive homozygous p53loxP/loxPRbloxP/loxP genotype allowing for the excision of all p53 and Rb alleles in the renin expressing cells of the mouse. Females which have both RenCre and the floxed alleles present in their genetic background are always backcrossed to the homozygous p53loxP/loxPRbloxP/loxP stock. Male mice which have RenCre and floxed alleles in the genetic background are not used for backcrossing due to the fact that possible renin expression during spermatogenesis leads to germline deletion of the floxed alleles in subsequent generations. Mice that have germline deletions of one of each of the p53 and Rb alleles have been maintained indefinitely without any change in phenotype. No neonatal or in utero (decreased litter sizes or re-absorption sites) lethality have been identified within this model.
Genotyping
Homozygous p53loxP/loxPRbloxP/loxP animals were maintained as a homozygous stock. Identification of wild-type, un-recombined floxed allele, and recombined floxed allele for both p53 and Rb have been previously described (51). The RenCre transgenic animals were identified using the primers CreF 5′-GAATTGTAATACGACTCACTATAGG-3′ and CreR-5′-ATCGCCATCTTCCAGGCGCACC-3. A 1100bp Cre fragment is generated using the PCR cycling parameters: 94°C 5min initiation, followed by 40 cycles of 94°C 45sec, 55°C for 45sec, 72°C for 90sec, with a final extension of 72°C for 7min. Genotyping guidelines for the floxed-stop-floxed-GFP reporter construct, B6.Cg-Gt(ROSA)26Sortm6(CAG-ZsGreen1)Hze/J are available on the Jackson Laboratory website (http://jaxmice.jax.org/strain/007906.html). This reporter line will be will be designated as Zsgreen through-out the rest of this manuscript. Genotyping guidelines for the floxed-stop-floxed-Confetti reporter construct, B6.Cg-Gt(ROSA)26Sortm1(CAG-Brainbow2.1)Cle/J are available on the Jackson Laboratory website (http://jaxmice.jax.org/strain/013731.html).
Pathology Assessment
Mice were sacrificed using CO2 affixiation followed by cervical dislocation according to guidelines from Roswell Park Cancer Institute’s (RPCI) Animal Care and Use Committee (IACUC). Full necropsies were performed on animals with pancreatic disease. Samples of tissue from multiple organs including pancreas, liver, lung, and heart were fixed in 4% paraformaldehyde. Fixed tissues were sent to IDEXX RADIL (Columbia, MO) and processed for paraffin embedment, blocked in wax and cut in 5-μm sections which were stained with hematoxylin and eosin. Additional sections for immunohistochemistry were probed with an anti-glucagon, anti-synaptophysin, anti-chromogranin A, and anti-CD 31 antibody using standard procedures. Briefly, 5-μm sections were mounted onto ProbeOn Plus microscope slides (Fisher Scientific Inc., Pittsburgh, PA), de-waxed in xylene, and rehydrated through graded concentrations of ethanol, and finally in water. Sections were treated with 3% hydrogen peroxide (to inactivate endogenous peroxidase activity), and rinsed prior to incubation in blocking buffer with 5% bovine serum albumin for 20 min. Sections were then incubated for 60 min at room temperature with anti-glucagon antibody (1:50 dilution of a rabbit polyclonal antibody; 180064, Invitrogen, Grand Island, NY). Sections were then washed, incubated for 30 minutes with a horseradish peroxidase–labeled polymer conjugated to anti-rabbit antibodies (EnVision, DAKO, Carpenteria, CA). Bound antibodies were visualized following incubation with 3,3′-diaminobenzidine solution (0.05% with 0.015% H2O2 in PBS; DAKO) for 3–5 minutes. Sections were counterstained with Mayer’s hematoxylin, dehydrated, and cover-slipped for microscopic examination.
Immunohistochemical Analysis
Tissues for analysis were placed into ice cold 2% buffered formalin-20% sucrose immediately after collection and kept overnight at 4C. These tissues were snap frozen in OCT and 5 um serial sections were collected onto silane treated slides. Slides were stored at −20C until staining when they were dried at room temperature, and briefly heat fixed at 55C. To stain, slides were rehydrated in PBS, blocked in 5% normal donkey serum and 0.05% Tween 20. Slides were stained using antibody to Glucagon (rabbit polyclonal SKU Ab932, Millepore), Somatostatin (rabbit polyclonal SKU NB100-91966, Novus Biologicals) or Insulin (guinea pia polycolonal SKU 4011-01F, Millepore) and incubated for 1 hr at RT. After washing, either donkey anti-rabbit Alexa Fluor 594 (SKU 21207, Molecular Probes) or donkey anti-guinea pig DyLight 594 (SKU 706-515-148, Jackson Immuno) was applied for 30 min at RT. After washing in PBS, slides were mounted with Fluoroshield with DAPI (SKU F6057, Sigma-Aldrich). Slides were viewed on a Nikon Eclipse E600 microscope using an X-Cite 120 fluorescent illumination system. Images were recorded with an RT SPOT digital camera (Diagnostic Instruments).
In Situ Visualization of GFP
To visualize GFP expression in tissues of RenCre x Zsgreen genetic crosses fluorescent stereomicroscopy was performed (model ZX12, Olympus) with mercury bulb illumination through an MGFP excitation/emission filter (Olympus). Images were captured with a Spot RT digital camera and software (Diagnostic Instruments) at identical exposures.
In Situ Visualization of Confetti
Confocal images were acquired with a Zeiss LSM 710 by sequential scanning of individual probes using a 20x, 0.8 NA objective. A 405 nm diode laser was used for CFP excitation (em:465-495), a 488 nm argon laser for GFP (em:499-511), a 514 nm Argon laser for YFP (em 520-560), and the 553nm line from an ‘In Tune’ Laser for RFP (em 589-652). Excitation and collection settings used for each color of the confetti line have been previously described (52).
RNA Isolation and Real-Time PCR
RNA was isolated using Trizol Reagent (Invitrogen, Carlsbad, CA, USA). One microgram of RNA was reverse transcribed in a 100 μl reaction using the high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City CA, USA) according to manufacturer’s recommendation. SYBR Green qPCR primers were designed using Beacon Design Software (Bio-Rad, Hercules, CA, USA) using the default parameters for qPCR primers. When possible, amplicons were designed to span intron-exon boundaries. The qPCR reaction was carried out in 10 μl volume using 2× Bio-Rad iQ SYBR Green Master Mix (Bio-Rad), 1ul cDNA, and 200ng of primer. The qPCR reactions were performed a CFX96 (Bio-Rad) using the manufacturer’s suggested cycling conditions.
Glucagon Serum Measurements
Mice were sacrificed using CO2 affixiation followed by cervical dislocation according to guidelines from Roswell Park Cancer Institute’s (RPCI) Animal Care and Use Committee (IACUC). Cardiac puncture with a 32-gauge needle and 1ml syringe was used to extract blood from mouse. Serum was collected by centrifugation, snap frozen, and subsequently stored at −80C. Once all time-points and samples were collected serum was shipped to Ani Lytics Inc. (Gaithersburg, MD) where glucagon levels were measured in pg/ml and reported back to RPCI.
Supplementary Material
Acknowledgments
This work was funded by the NIH grants 5R01HL048459, R21CA164795 and R21CA169717. We would like to thank the CCSG funded (CA016056) Roswell Park Gene Targeting and Transgenic Resource and the Genomics Shared Resource (GSR). Confocal Microscopy was performed at the University at Buffalo North Campus Imaging Facility using a Zeiss LSM 710 purchased through National Science Foundation Major Research Instrumentation grant DBI 0923133. We would also like to thank MK Ellsworth and Caretta Reese for maintenance of the mouse colonies as well as Dominic Smiraglia and Elizabeth Repasky for critical reading and discussion of work. A special thank you to Dr. George Deeb for his insightful observations on anatomy.
Footnotes
Conflict of Interest
The authors have nothing to disclose.
References
- 1.Gomez RA. Role of angiotensin in renal vascular development. Kidney international Supplement. 1998;67:S12–6. doi: 10.1046/j.1523-1755.1998.06703.x. [DOI] [PubMed] [Google Scholar]
- 2.Nishimura H, Ichikawa I. What have we learned from gene targeting studies for the renin angiotensin system of the kidney? Intern Med. 1999;38:315–23. doi: 10.2169/internalmedicine.38.315. [DOI] [PubMed] [Google Scholar]
- 3.Jones CA, Hurley MI, Black TA, Kane CM, Pan L, Pruitt SC, et al. Expression of a renin/GFP transgene in mouse embryonic, extra-embryonic, and adult tissues. Physiological genomics. 2000;4:75–81. doi: 10.1152/physiolgenomics.2000.4.1.75. [DOI] [PubMed] [Google Scholar]
- 4.Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Developmental cell. 2004;6:719–28. doi: 10.1016/s1534-5807(04)00134-0. [DOI] [PubMed] [Google Scholar]
- 5.Lau T, Carlsson PO, Leung PS. Evidence for a local angiotensin-generating system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia. 2004;47:240–8. doi: 10.1007/s00125-003-1295-1. [DOI] [PubMed] [Google Scholar]
- 6.Leung KK, Liang J, Ma MT, Leung PS. Angiotensin II type 2 receptor is critical for the development of human fetal pancreatic progenitor cells into islet-like cell clusters and their potential for transplantation. Stem Cells. 2012;30:525–36. doi: 10.1002/stem.1008. [DOI] [PubMed] [Google Scholar]
- 7.Leung PS. The physiology of a local renin-angiotensin system in the pancreas. The Journal of physiology. 2007;580:31–7. doi: 10.1113/jphysiol.2006.126193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tahmasebi M, Puddefoot JR, Inwang ER, Vinson GP. The tissue renin-angiotensin system in human pancreas. The Journal of endocrinology. 1999;161:317–22. doi: 10.1677/joe.0.1610317. [DOI] [PubMed] [Google Scholar]
- 9.Lam KY. The pancreatic renin-angiotensin system: does it play a role in endocrine oncology? JOP: Journal of the pancreas. 2001;2:40–2. [PubMed] [Google Scholar]
- 10.Lam KY, Leung PS. Regulation and expression of a renin-angiotensin system in human pancreas and pancreatic endocrine tumours. European journal of endocrinology/European Federation of Endocrine Societies. 2002;146:567–72. doi: 10.1530/eje.0.1460567. [DOI] [PubMed] [Google Scholar]
- 11.Carlsson PO. The renin-angiotensin system in the endocrine pancreas. JOP: Journal of the pancreas. 2001;2:26–32. [PubMed] [Google Scholar]
- 12.Leung PS, Carlsson PO. Tissue renin-angiotensin system: its expression, localization, regulation and potential role in the pancreas. Journal of molecular endocrinology. 2001;26:155–64. doi: 10.1677/jme.0.0260155. [DOI] [PubMed] [Google Scholar]
- 13.Leung PS, Carlsson PO. Pancreatic islet renin angiotensin system: its novel roles in islet function and in diabetes mellitus. Pancreas. 2005;30:293–8. doi: 10.1097/01.mpa.0000158028.76666.76. [DOI] [PubMed] [Google Scholar]
- 14.Leung PS, Chappell MC. A local pancreatic renin-angiotensin system: endocrine and exocrine roles. The international journal of biochemistry & cell biology. 2003;35:838–46. doi: 10.1016/s1357-2725(02)00179-6. [DOI] [PubMed] [Google Scholar]
- 15.Tikellis C, Cooper ME, Thomas MC. Role of the renin-angiotensin system in the endocrine pancreas: implications for the development of diabetes. The international journal of biochemistry & cell biology. 2006;38:737–51. doi: 10.1016/j.biocel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, et al. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127:3533–42. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- 17.Desgraz R, Herrera PL. Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development. 2009;136:3567–74. doi: 10.1242/dev.039214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lammert E, Cleaver O, Melton D. Role of endothelial cells in early pancreas and liver development. Mechanisms of development. 2003;120:59–64. doi: 10.1016/s0925-4773(02)00332-5. [DOI] [PubMed] [Google Scholar]
- 19.Haumaitre C, Lenoir O, Scharfmann R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Molecular and cellular biology. 2008;28:6373–83. doi: 10.1128/MCB.00413-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G, et al. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Developmental cell. 2007;12:457–65. doi: 10.1016/j.devcel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 21.Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocrine reviews. 2007;28:84–116. doi: 10.1210/er.2006-0007. [DOI] [PubMed] [Google Scholar]
- 22.Ehehalt F, Saeger HD, Schmidt CM, Grutzmann R. Neuroendocrine tumors of the pancreas. The oncologist. 2009;14:456–67. doi: 10.1634/theoncologist.2008-0259. [DOI] [PubMed] [Google Scholar]
- 23.Ruttman E, Kloppel G, Bommer G, Kiehn M, Heitz PU. Pancreatic glucagonoma with and without syndrome. Immunocytochemical study of 5 tumour cases and review of the literature. Virchows Archiv A, Pathological anatomy and histology. 1980;388:51–67. doi: 10.1007/BF00430676. [DOI] [PubMed] [Google Scholar]
- 24.Hu W, Feng Z, Modica I, Klimstra DS, Song L, Allen PJ, et al. Gene Amplifications in Well-Differentiated Pancreatic Neuroendocrine Tumors Inactivate the p53 Pathway. Genes & cancer. 2010;1:360–8. doi: 10.1177/1947601910371979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang LH, Contractor T, Clausen R, Klimstra DS, Du YC, Allen PJ, et al. Attenuation of the retinoblastoma pathway in pancreatic neuroendocrine tumors due to increased cdk4/cdk6. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:4612–20. doi: 10.1158/1078-0432.CCR-11-3264. [DOI] [PubMed] [Google Scholar]
- 26.Benson C, Kaye S, Workman P, Garrett M, Walton M, de Bono J. Clinical anticancer drug development: targeting the cyclin-dependent kinases. British journal of cancer. 2005;92:7–12. doi: 10.1038/sj.bjc.6602229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu L, Broaddus RR, Yao JC, Xie S, White JA, Wu TT, et al. Epigenetic alterations in neuroendocrine tumors: methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2005;18:1632–40. doi: 10.1038/modpathol.3800490. [DOI] [PubMed] [Google Scholar]
- 28.Harvey M, Vogel H, Lee EY, Bradley A, Donehower LA. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer research. 1995;55:1146–51. [PubMed] [Google Scholar]
- 29.Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nature genetics. 1994;7:480–4. doi: 10.1038/ng0894-480. [DOI] [PubMed] [Google Scholar]
- 30.Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol Endocrinol. 2003;17:1880–92. doi: 10.1210/me.2003-0154. [DOI] [PubMed] [Google Scholar]
- 31.Loffler KA, Biondi CA, Gartside M, Waring P, Stark M, Serewko-Auret MM, et al. Broad tumor spectrum in a mouse model of multiple endocrine neoplasia type 1. International journal of cancer Journal international du cancer. 2007;120:259–67. doi: 10.1002/ijc.22288. [DOI] [PubMed] [Google Scholar]
- 32.Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1118–23. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crabtree JS, Scacheri PC, Ward JM, McNally SR, Swain GP, Montagna C, et al. Of mice and MEN1: Insulinomas in a conditional mouse knockout. Molecular and cellular biology. 2003;23:6075–85. doi: 10.1128/MCB.23.17.6075-6085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertolino P, Tong WM, Herrera PL, Casse H, Zhang CX, Wang ZQ. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer research. 2003;63:4836–41. [PubMed] [Google Scholar]
- 35.Shen HC, He M, Powell A, Adem A, Lorang D, Heller C, et al. Recapitulation of pancreatic neuroendocrine tumors in human multiple endocrine neoplasia type I syndrome via Pdx1-directed inactivation of Men1. Cancer research. 2009;69:1858–66. doi: 10.1158/0008-5472.CAN-08-3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu J, Herrera PL, Carreira C, Bonnavion R, Seigne C, Calender A, et al. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology. 2010;138:1954–65. doi: 10.1053/j.gastro.2010.01.046. [DOI] [PubMed] [Google Scholar]
- 37.Shen HC, Ylaya K, Pechhold K, Wilson A, Adem A, Hewitt SM, et al. Multiple endocrine neoplasia type 1 deletion in pancreatic alpha-cells leads to development of insulinomas in mice. Endocrinology. 2010;151:4024–30. doi: 10.1210/en.2009-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen HC, Adem A, Ylaya K, Wilson A, He M, Lorang D, et al. Deciphering von Hippel-Lindau (VHL/Vhl)-associated pancreatic manifestations by inactivating Vhl in specific pancreatic cell populations. PloS one. 2009;4:e4897. doi: 10.1371/journal.pone.0004897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–64. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 40.Rindi G, Efrat S, Ghatei MA, Bloom SR, Solcia E, Polak JM. Glucagonomas of transgenic mice express a wide range of general neuroendocrine markers and bioactive peptides. Virchows Archiv A, Pathological anatomy and histopathology. 1991;419:115–29. doi: 10.1007/BF01600225. [DOI] [PubMed] [Google Scholar]
- 41.Sigmund CD, Jones CA, Mullins JJ, Kim U, Gross KW. Expression of murine renin genes in subcutaneous connective tissue. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:7993–7. doi: 10.1073/pnas.87.20.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sigmund CD, Okuyama K, Ingelfinger J, Jones CA, Mullins JJ, Kane C, et al. Isolation and characterization of renin-expressing cell lines from transgenic mice containing a renin-promoter viral oncogene fusion construct. The Journal of biological chemistry. 1990;265:19916–22. [PubMed] [Google Scholar]
- 43.Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–22. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- 44.Franklin DS, Godfrey VL, O’Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Molecular and cellular biology. 2000;20:6147–58. doi: 10.1128/mcb.20.16.6147-6158.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nature genetics. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 46.Sotillo R, Dubus P, Martin J, de la Cueva E, Ortega S, Malumbres M, et al. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. The EMBO journal. 2001;20:6637–47. doi: 10.1093/emboj/20.23.6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glenn ST, Jones CA, Pan L, Gross KW. In vivo analysis of key elements within the renin regulatory region. Physiological genomics. 2008;35:243–53. doi: 10.1152/physiolgenomics.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sparwasser T, Gong S, Li JY, Eberl G. General method for the modification of different BAC types and the rapid generation of BAC transgenic mice. Genesis. 2004;38:39–50. doi: 10.1002/gene.10249. [DOI] [PubMed] [Google Scholar]
- 49.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes & development. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 50.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nature genetics. 2001;29:418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 51.Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer research. 2003;63:3459–63. [PubMed] [Google Scholar]
- 52.Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–44. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.