

Abstract
Protein-based methods of targeted short-interfering RNA (siRNA) delivery have the potential to solve some of the problems faced by nanoparticle-based methods, such as poor pharmacokinetics and biodistribution, low tumor penetration, and polydispersity. However, protein-based targeted delivery has been limited to fusion proteins with polycationic peptides as siRNA carriers, whose high charge density in some cases results in undesirable biophysical and in vivo properties. Here, we present a fully proteinaceous, multiagent approach for targeted siRNA delivery to epidermal growth factor receptor (EGFR), using a nonpolycationic carrier for siRNA. Each agent contributes a fundamentally different mechanism of action that work together for potent targeted RNA interference. The first agent is an EGFR-targeted fusion protein that uses a double-stranded RNA-binding domain as a nonpolycationic siRNA carrier. This double-stranded RNA-binding domain fusion protein can deliver siRNA to the endosomes of an EGFR-expressing cell line. A second agent delivers the cholesterol-dependent cytolysin, perfringolysin O, in a targeted manner, which enhances the endosomal escape of siRNA and induces gene silencing. A third agent that clusters EGFR increases gene-silencing potency and decreases cytolysin toxicity. Altogether, this system is potent, with only 16 nmol/l siRNA required for gene silencing and a therapeutic window that spans two orders of magnitude of targeted cytolysin concentrations.
Keywords: cholesterol-dependent cytolysin, dsRBD, protein-based siRNA delivery
Introduction
The most established methods for nonviral targeted delivery of short-interfering RNA (siRNA) employ the use of nanoparticles,1 with small molecule or macromolecular ligands tethered to the nanoparticle surface for targeting and internalization. Although nanoparticle-based delivery vehicles can carry large siRNA payloads, they suffer from several problems that limit their efficacy and that protein-based delivery can potentially solve. For example, nanoparticles are rapidly phagocytosed by the reticuloendothelial system and accumulate in the liver and spleen, leading to poor pharmacokinetics and biodistribution.2 They also exhibit poor extravasation and penetration into solid tumors due to their large size.3 On the other hand, protein-based systems can be engineered to fall within the window of 60 and 500 kDa, which would be large enough to avoid rapid renal clearance, yet small enough for efficient extravasation and avoidance of phagocytic clearance. Also, nanoparticle formulations can be difficult to prepare in a reproducible and monodisperse manner. In contrast, proteins are relatively straightforward to synthesize using recombinant DNA technology and can generally be purified in a straightforward, reproducible manner to monodispersity.
While limited in number, there do exist protein-based delivery vehicles for siRNA that combine a targeting agent, such as an antibody fragment, with an siRNA complexation agent, usually a short polycationic peptide.4,5,6,7 However, these methods have limitations that have prevented them from reaching the potential of protein-based delivery methods. For example, in some cases, polycationic peptides can suffer from poor pharmacokinetics and biodistribution, due to high global organ uptake as a result of their high positive charge.8,9 They also tend to be inefficient and require large amounts of siRNA for silencing.4,6 These agents require complex preparation or purification schemes, such as protein refolding7 or chemical conjugation.4 Also, in our experience, these polycationic peptides are prone to aggregation and difficult to work with.
In this work, we present the use of a multiagent protein-based siRNA delivery system for targeted siRNA delivery (Figure 1). The first agent, termed E6N2, employs the double-stranded RNA-binding domain (dsRBD) of human protein kinase R10 as an alternative siRNA carrier with low charge density and an engineered 10th type 3 fibronectin (Fn3) that binds epidermal growth factor receptor (EGFR) for targeting.11 Although dsRBD has also been used previously for siRNA delivery, these prior works relied on polycationic cell penetrating peptides for cytoplasmic entry in an untargeted manner.12,13,14
Figure 1.
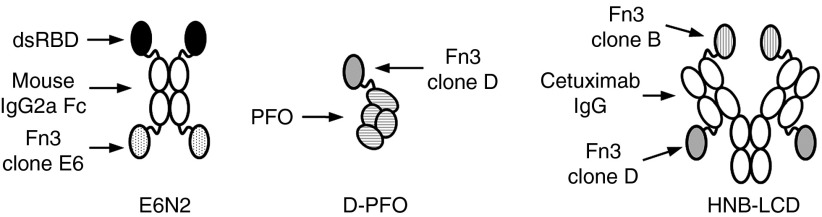
Schematic of protein constructs used in this work. E6N2 is a homodimeric fusion protein containing the dsRBD from human protein kinase R, the mouse IgG2a Fc fragment, and the EGFR-binding Fn3 clone, E6. D-PFO is a fusion protein with EGFR-binding Fn3 clone D and the cholesterol-dependent cytolysin, PFO. HNB-LCD is a multispecific construct described by Spangler et al.16 and contains EGFR-binding Fn3 clones B and D on the N terminus of the heavy chain and the C terminus of the light chain of cetuximab, respectively. All proteins are drawn with the N-terminus on top and the C-terminus on the bottom. dsRBD, double-stranded RNA-binding domain; PFO, perfringolysin O.
E6N2 is able to deliver siRNA to the endosomes of EGFR-expressing cells. In order to enhance the endosomal escape of siRNA, a second agent is used, consisting of an alternate EGFR-binding Fn3 clone fused to the cholesterol-dependent cytolysin, perfringolysin O15 (PFO) (Figure 1). PFO is delivered in a targeted fashion and disrupts endosomal compartments to allow the escape of internalized siRNA to access the cytoplasm. Successful gene silencing is achieved with the two-agent approach, but the addition of a third agent that was developed by Spangler et al.16 to induce EGFR clustering (Figure 1) can significantly widen the therapeutic window, through the simultaneous enhancement in gene-silencing potency and protection from cytotoxicity of EGFR-binding Fn3–PFO fusion proteins.
Results
Preparation of E6N2
E6N2 was expressed as a homodimeric protein containing three components: E6, an engineered Fn3 variant that binds EGFR11 for EGFR-specific targeting and internalization; the mouse IgG2a Fc fragment; and the dsRBD of human protein kinase R for siRNA complexation.13 E6N2 was expressed in transient transfections of HEK293F cells, with a single affinity chromatography purification step. Purification by Protein A or cobalt-based resins typically yielded 1–3 mg protein per liter of cell culture, and the resulting protein was monomeric as assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and size exclusion chromatography analysis. Compared with the refolding or chemical conjugation steps of polycationic peptide fusion constructs,4,7 dsRBD fusions were relatively straightforward to purify. The EGFR-binding Fn3 moiety also retained high affinity binding to EGFR in the E6N2 construct, with apparent dissociation constant, KD,app ~ 2.1 nmol/l (Supplementary Table S1; Supplementary Materials and Methods).
Analysis of siRNA/dsRBD interactions
In order to visualize the complexation between E6N2 and siRNA, an agarose gel shift assay was performed. A shift in the siRNA band was visible in the presence of E6N2, with partial complexation at a dsRBD:siRNA ratio of 1, based on the partial disappearance of the free siRNA band (Figure 2a). Complete complexation was observed at dsRBD:siRNA ratios of 2 and above, indicating potent complexation between the siRNA and dsRBD.
Figure 2.
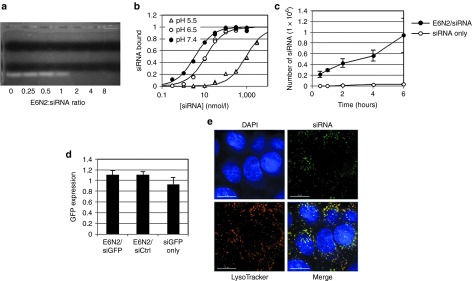
Characterization of E6N2 for short-interfering RNA (siRNA) internalization. The interaction between E6N2 and siRNA was evaluated (a) qualitatively by an agarose gel shift assay and (b) quantitatively using titrations of siRNA-Alexa 488 on E6N2-coated Protein A beads in complete media with 10% fetal bovine serum at 37 °C at varying pH conditions. The KD values were measured to be 3.5 ± 0.2 nmol/l at pH 7.4, 11.4 ± 0.9 nmol/l at pH 6.5, and 925 ± 88 nmol/l at pH 5.5, with 68% confidence intervals reported. (c) siRNA uptake by A431 cells is measured for E6N2/siRNA-Alexa 488 complexed at a 1:1 molar ratio, or free siRNA-Alexa 488 in the absence of E6N2, at a final siRNA concentration of 100 nmol/l (data shown as mean ± SD, n = 6). (d) When gfp siRNA (siGFP) or control siRNA (siCtrl) is delivered in 1:1 complexes with E6N2, or in the absence of E6N2, to A431-d2EGFP cells, no GFP knockdown is observed (data shown as mean ± SD, n = 6). (e) Fluorescence microscopy images were taken of A431 cells treated for 6 hours with E6N2 complexed with Alexa 488 labeled siRNA. The cells are additionally stained with the nuclear marker 4′,6-diamidino-2-phenylindole (DAPI) and the late endosomal and lysosomal marker LysoTracker Red. There is a high level of colocalization of siRNA and LysoTracker Red, with a Pearson correlation coefficient of 0.81. Scale bar represents 15 µm.
For a more quantitative measurement of the dsRBD/siRNA-binding affinity, titrations of fluorescently labeled siRNA were performed on Protein A magnetic beads preloaded with E6N2. The titrations were performed in media containing 10% serum at 37 °C to simulate physiological conditions. Previous reports described a weak siRNA-binding affinity of dsRBD, with KD ~ 200 nmol/l.10 In E6N2, bivalent dsRBD significantly enhanced the avidity of siRNA binding over monovalent dsRBD, with a KD,app of siRNA binding of 3.5 ± 0.2 nmol/l (Figure 2b).
In order to address the possibility that large molecular weight aggregates or multimers could form during complexation of E6N2 and siRNA, dynamic light scattering analysis was performed. E6N2 alone or complexed with siRNA showed monomodal distributions with a radius of 6.0 or 5.9 nm, respectively (Supplementary Figure S1). The lack of increase in molecular radius when complexed with siRNA is consistent with the extended and flexible structure of the unbound state of dsRBD, compared with the bound state, which wraps around and makes contacts on opposite sides of the double helix of double-stranded RNA.17,18 The lack of a high molecular weight peak in the dynamic light scattering analysis indicates that multimeric aggregates did not form when E6N2 was complexed with siRNA.
pH sensitivity of binding of siRNA and dsRBD
In order for the siRNA cargo to be loaded into the RNA-induced silencing complex/Ago2 complex in the cytoplasm for gene silencing, it must be able to dissociate from dsRBD. Therefore, the KD,apps of binding were measured at pH 7.4, 6.5, and 5.5 to determine if siRNA could dissociate at endosomal or lysosomal pH once it is internalized. The KD,app of siRNA/E6N2 binding was pH dependent, with a difference of over two orders of magnitude between pH 7.4 and 5.5 (Figure 2b). This indicated that siRNA would be able to dissociate from dsRBD within the acidic conditions of the endosome or lysosome. Secondary staining using (Fab′)2 fragments against mouse Fc confirmed that equivalent amounts of E6N2 remained bound to Protein A beads after exposure to each pH condition tested (Supplementary Figure S2).
siRNA uptake by E6N2
The amount of fluorescently labeled siRNA taken up by E6N2/siRNA complexes was measured in A431 cells using flow cytometry. In order to differentiate between surface and internalized fluorescence signal, the cells were trypsinized to degrade surface-bound E6N2 and eliminate surface fluorescence. Using a calibration of fluorescence signal to number of siRNA molecules, it was determined that ~106 molecules of siRNA was internalized into A431 cells after 6 hours of treatment with E6N2/siRNA complexes (Figure 2c). There was negligible internalization of siRNA by fluid-phase pinocytosis in the absence of E6N2 (Figure 2c) or by Fc-dsRBD fusions targeted to irrelevant antigens, CD25, or carcinoembryonic antigen (Supplementary Figure S3a). Additionally, there is negligible internalization of siRNA by E6N2 in the EGFR-negative cell line CTLL-2 (Supplementary Figure S3b). Altogether, this indicates that siRNA uptake is mediated by E6N2 and is specific to EGFR.
When Amaxa electroporation was used as a positive control for delivery directly to the cytoplasm, fewer than 104 molecules of siRNA were required for observable knockdown of green fluorescent protein (GFP) protein expression in A431 cells stably transfected with d2EGFP, a destabilized form of GFP with a 2-hour half-life (Supplementary Figure S4). However, no GFP knockdown was observed in these cells with over 106 molecules of gfp siRNA delivered by E6N2 (Figure 2d). Fluorescence microscopy revealed that almost all of the detectable internalized siRNAs were trapped within endosomal and lysosomal compartments (Figure 2e). This implied that endosomal escape was the critical barrier for effective RNAi by siRNA delivered by dsRBD fusion proteins.
PFO fusion proteins for endosomal escape
Previously, Pirie et al.19 showed the enhancement of endosomal escape of immunotoxins using targeted fusion proteins with two cholesterol-dependent cytolysins, listeriolysin O and PFO. In the current work, we investigated the applicability of this approach to enhance the endosomal escape of endocytosed siRNA delivered by E6N2. PFO was chosen for this work based on its improved stability at neutral pH over listeriolysin O.20 Fusion proteins were constructed containing PFO and various EGFR-binding Fn3 clones.16 In total, four PFO fusion constructs with unique EGFR-binding Fn3 clones were evaluated. For knockdown assays, we used the A431 cell line stably transfected with d2EGFP under the cytomegalovirus promoter, A431-d2EGFP. When added to A431-d2EGFP cells along with 100 nmol/l E6N2/siRNA complexes, all EGFR-binding Fn3–PFO fusion proteins mediated GFP knockdown in a dose-dependent manner, most likely due to enhancement of endosomal escape of endocytosed siRNA (Figure 3a and Supplementary Figure S5). Of these, the fusion with clone “D,”16 D-PFO, was found to be most effective and was used for further characterization.
Figure 3.

Characterization of D-PFO fusion protein for endosomal escape. (a) GFP knockdown assays were performed in A431-d2EGFP cells, along with cell viability measurements to obtain a therapeutic window. E6N2 was used to deliver 100 nmol/l of either gfp siRNA (siGFP) or control siRNA (siCtrl) to A431-d2EGFP cells with varying amounts of D-PFO. The therapeutic window of D-PFO is shown with cell viability overlaid with GFP expression (data shown as mean ± SD, n = 9). Viability is normalized to a value of 1 for untreated cells and 0 for wells without cells. GFP expression is normalized to a value of 1 for untreated A431-d2EGFP cells and 0 for A431 cells. (b) The membrane disruptive activity of PFO is measured in a mouse red blood cell hemolysis assay. Hemolysis is normalized to 1 for 10% Triton-X 100–treated cells and 0 for untreated cells (data shown as mean ± SD, n = 3). siRNA, short-interfering RNA.
D-PFO exhibited high EGFR-binding affinity, with KD ~ 6.6 nmol/l (Supplementary Table S1), and PFO lytic activity, as shown in hemolysis assays (Figure 3b). Reductions in GFP expression were not due to a global downregulation of all cell proteins, or an artifact of PFO cytotoxicity, because GFP expression was unchanged when negative control siRNA was delivered (Figure 3a). GFP knockdown was also dependent on delivery by E6N2, as free gfp siRNA could not induce GFP knockdown in the presence of D-PFO (Supplementary Figure S6). The PFO fusion protein with C7, an Fn3 that binds an irrelevant antigen, carcinoembryonic antigen,21 was not capable of mediating GFP knockdown (Supplementary Figure S5). Altogether, these data revealed that gene silencing was dependent on both the delivery of siRNA by E6N2 and EGFR-binding of Fn3–PFO fusion proteins and was not the result of an alternate mechanism of cytoplasmic delivery, such as siRNA diffusion through pores in the cell membrane formed by PFO or nonspecific uptake of PFO fusion proteins into endosomes.
Enhancement of siRNA uptake by multispecific constructs
Successful gene silencing was achieved through EGFR-specific siRNA internalization mediated by E6N2 and endosomal escape mediated by D-PFO. However, the therapeutic window of this method was relatively narrow. In order to expand this therapeutic window, a third agent that induces EGFR clustering was used. Previously, Spangler et al.16 showed that antibody–Fn3 fusion proteins that bind to multiple distinct epitopes of EGFR can induce EGFR clustering and downregulation without activating EGFR signaling pathways. An increased internalization rate due to the simultaneous internalization of clustered EGFR was observed,16 and thus, it was hypothesized that the use of these constructs could enhance gene-silencing potency due to a concentrating effect of EGFR molecules per endosome. Out of three candidate multispecific constructs, HNB-LCD was the most effective in enhancing E6N2-mediated siRNA uptake, with greater than twofold enhancement of siRNA uptake (Figures 1 and 4a and Supplementary Figure S7).
Figure 4.
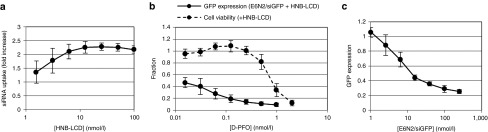
Characterization of HNB-LCD for the expansion of the short-interfering RNA (siRNA) delivery therapeutic window. (a) Varying amounts of HNB-LCD were added to A431 cells treated with 100 nmol/l E6N2/siRNA-Alexa 488 complexes for 6 hours. siRNA uptake was normalized to uptake by E6N2/siRNA-Alexa 488 in the absence of HNB-LCD (data shown as mean ± SD, n = 6). (b) The therapeutic window for D-PFO was determined for A431-d2EGFP cells in the presence of 7.5 nmol/l HNB-LCD and 100 nmol/l E6N2/siRNA complexes (data shown as mean ± SD, n = 9). (c) The potency of E6N2/siGFP is measured in a GFP knockdown assay in the presence of 100 pmol/l D-PFO and 7.5 nmol/l HNB-LCD (data shown as mean ± SD, n = 9). Cell viability and GFP expression are normalized as in Figure 3.
Enhancement of GFP knockdown by multispecific constructs
Next, HNB-LCD was tested to see if the enhancement in siRNA uptake could result in enhanced knockdown of GFP expression. The enhancement of GFP knockdown depended on the Fn3 clone used, with D-PFO showing the greatest amount of enhancement, approximately fourfold (Figure 4b and Supplementary Figure S5). GFP expression was not altered when delivering control siRNA by E6N2 in the presence of HNB-LCD (Supplementary Figure S8). In the presence of 7.5 nmol/l HNB-LCD and 100 nmol/l E6N2/siGFP complexes, D-PFO had a half maximal effective concentration of less than 15 pmol/l for GFP knockdown and could mediate 90% knockdown with less than 20% loss in cell viability (Figure 4b).
Cytotoxicity profiles of Fn3–PFO in the presence of HNB-LCD
It was originally hypothesized that the addition of a multiepitopic EGFR binder would only enhance GFP knockdown through clustering, without any effect on PFO-related cytotoxicity. However, when the cytotoxicity profiles were measured for the various Fn3–PFO fusion proteins, it was found that the presence of HNB-LCD had a protective effect on A431 cells. This effect was consistent across all EGFR-binding Fn3–PFO constructs and provided an effective three- to fourfold reduction in Fn3–PFO cytotoxicity (Figure 4b). When the cytotoxicity of carcinoembryonic antigen–binding C7-PFO was measured in A431 cells, there was no difference in the presence or absence of HNB-LCD. This indicated that the reduction in Fn3–PFO cytotoxicity by HNB-LCD required EGFR binding by the Fn3–PFO construct.
Potency of E6N2 for gene silencing
With 100 pmol/l D-PFO, a subcytotoxic concentration, and 7.5 nmol/l HNB-LCD, the potency of E6N2-mediated gene silencing was measured at a constant 1:1 molar ratio of E6N2/siGFP. GFP knockdown was observed at all concentrations of E6N2 greater than the measured Kd,app of E6N2 binding to EGFR. Greater than 50% GFP knockdown was observed at E6N2 concentrations of 16 nmol/l and greater (Figure 4c).
Discussion
The dsRBD fusion protein was expressed and purified in a straightforward manner, without any chemical conjugation or refolding required. In our experience, it was not prone to aggregation even when complexed with siRNA, presumably due to its relative low charge density, unlike the highly charged polycationic peptides used previously for siRNA complexation.4,5,6,7 The dsRBD moiety was reported to bind specifically to double-stranded RNA and provide protection against siRNA degradation by RNases.13 The dsRBD moiety interacts with the RNA backbone in a double-stranded RNA-dependent and sequence-independent manner, thus allowing siRNA directed against any target to be loaded.10 In combination with PFO fusions, dsRBD fusion proteins delivered enough siRNA to the cytoplasm for potent gene silencing. As low as 16 nmol/l siRNA could induce >50% gene silencing, whereas typically, 1 µmol/l or more is used with polyarginine as an siRNA carrier4 and 100 nmol/l–5 µmol/l is used with protamine as an siRNA carrier.5,6,7 These properties make dsRBD fusion proteins an attractive option to other peptide-based methods for complexing and delivering siRNA.
Endosomal escape has long been recognized as a barrier to effective delivery of nucleic acids.22,23 Protein-based strategies to overcome this barrier have been limited to the use of cell penetrating peptides or fusogenic peptides,23 which are generally polycationic and suffer from similar disadvantages of polycationic peptide siRNA carriers described previously. In this work, we report the application of targeted PFO fusion proteins for the endosomal escape of siRNA delivered by dsRBD fusions in an EGFR-targeted manner. Their potencies were remarkable, considering that GFP knockdown was achieved at Fn3–PFO concentrations less than the KD of EGFR binding, even though Fn3–PFO binding to EGFR was required. On the other hand, despite its potency, E6N2 was required at concentrations greater than the KD,app of EGFR binding for effective siRNA delivery. Therefore, although there is added complexity arising from the use of two agents, optimal concentrations of each component can be used relative to the effective concentrations of E6N2 and Fn3–PFO and the cytotoxic limits of Fn3–PFO. This would not be possible with single agent containing both functions in the form of a fusion protein.
The use of E6N2 for targeted delivery of siRNA to endosomes and Fn3–PFO for enhancing endosomal escape of siRNA was sufficient for gene silencing. However, when comparing the efficacy of GFP knockdown to the cytotoxicity of PFO fusion proteins, a relatively narrow therapeutic window was revealed for all EGFR-binding Fn3 clones tested. In order for this method to be useful in a therapeutic setting, it will be important to expand this therapeutic window.
The use of multispecific antibody–Fn3 fusion constructs was originally motivated by the prospect of enhancing gene-silencing potency by inducing EGFR clustering and increasing the number of EGFR and E6N2/siRNA complexes internalized per endosome. Indeed, HNB-LCD was capable of enhancing siRNA uptake and gene silencing though the degree of enhancement is sensitive to the particular clone used in the Fn3–PFO fusion construct. The use of multispecific constructs was not initially expected to have any effect on PFO-mediated cytotoxicity. The observation that HNB-LCD could reduce the cytotoxicity of EGFR-binding Fn3–PFO constructs was indeed surprising, especially since EGFR downregulation induced by clustering has been shown to reduce viability.16 Initially, the mechanism for this protective effect was hypothesized to be a decreased exposure to EGF from fetal bovine serum (FBS) in the media, caused by EGFR downregulation by HNB-LCD. EGF exposure has been shown to inhibit growth in A431 cells,24 but this explanation is inconsistent with the requirement on EGFR binding of the Fn3–PFO construct for the protective effect of HNB-LCD binding. Instead, the current hypothesized mechanism involves the depletion of extracellular Fn3–PFO by EGFR clustering and internalization. It is believed that the cytotoxicity of Fn3–PFO arises from cell membrane disruption, as opposed to endosomal disruption. This is based on the quantitative similarity in A431 cytotoxicities of all Fn3–PFO constructs, regardless of Fn3 specificity, as well as hemolysis of EGFR-negative red blood cells. It is likely that EGFR clustering and downregulation with HNB-LCD causes cointernalization of EGFR-bound Fn3–PFO. Since the number of EGFR molecules is the same order of magnitude as the number of Fn3–PFO molecules at the concentrations used, extracellular Fn3–PFO available for plasma membrane disruption is likely depleted, resulting in decreased cytotoxicity. Further elucidation of this mechanism of action will be required to better understand the therapeutic applications for this delivery method.
The general strategy of using EGFR-targeted Fn3–PFO to facilitate endosomal escape of separately delivered macromolecules has been successfully shown across other cell lines with different levels of EGFR expression.19 Here, we have expanded the application of Fn3–PFO to enhance cytoplasmic siRNA delivery on the A431 cell line, a model system that has been used extensively to study EGFR biology and EGFR-based therapeutics.25,26,27 Based on the specificity of delivery to EGFR, we anticipate that siRNA uptake and potentially knockdown in other cell lines would correlate with the level of EGFR expression, and this is a topic worth exploring in future work.
In summary, we have shown that dsRBD can be used as a nonpolycationic siRNA carrier for targeted siRNA delivery to endosomes. Targeted delivery of PFO enhanced endosomal escape of siRNA, allowing knockdown in the A431 cell line. The addition of a third agent that clusters EGFR significantly expanded the therapeutic window, with approximately two orders of magnitude difference in the half-maximal lethal dose and the half-maximal effective dose of D-PFO for GFP knockdown. This arose from both the enhancement of GFP knockdown and the decrease in D-PFO toxicity. The feasibility of a two-agent method using targeted PFO to enhance the endosomal escape of macromolecular payloads has been established previously in vivo in a tumor xenograft context.19 As a therapeutic modality for siRNA delivery, a third agent would significantly increase the complexity of this approach, and therefore, follow-up work includes efforts to combine EGFR clustering functionality with one or both of the siRNA delivery agent and endosomal disruption agent. Immunogenicity is another potential concern, especially for the bacteria-derived PFO, but this may be addressed with the use of perforin, a human protein with structural and functional similarities to the cholesterol-dependent cytolysin pore forming proteins.28 The stability of the E6N2/siRNA interaction will also be important for effective delivery in vivo. Although the apparent binding affinity of E6N2 and siRNA is relatively high in the presence of serum at 37 °C (KD,app = 3.5 nmol/l), affinity maturation of this interaction may be required if it is found to be insufficient in vivo. As for the expected pharmacokinetics, the lack of observed aggregation in E6N2/siRNA complexes will hopefully allow the constructs avoid reticuloendothelial clearance and exhibit favorable biodistribution properties. The Fc domain will also allow for extended serum half-life due to recycling via the neonatal Fc receptor. As this system is further optimized and characterized in vivo, it will be interesting to see how the pharmacokinetic, biodistribution, and tumor penetration properties compare with those of nanoparticle and polycationic peptide based delivery systems.
Materials and methods
Protein expression and purification. The gene containing the dsRBD moiety from human protein kinase R was a kind gift from Dr. James Cole (University of Connecticut). Genetic fusions containing E6-mouse IgG2a Fc-dsRBD with an N-terminal His tag were constructed and inserted into the gWiz vector (Genlantis, San Diego, CA) using the method described by Geiser et al.29 The C121V and C135V mutations, which were shown not to be important for RNA binding,30 were incorporated into dsRBD using the Quikchange mutagenesis kit according to the manufacturer's instructions (Agilent, Santa Clara, CA). E6N2 was expressed in transiently transfected HEK293F cells (Invitrogen, Carlsbad, CA) for 8 days. E6N2 was purified from the supernatant using a Talon column according to the manufacturer's instructions (Clontech, Mountain View, CA).
Fn3–PFO genetic fusions with a C-terminal His tag and a C215A mutation were constructed using a modified Quikchange reaction as described by Geiser et al.29 and inserted into the pmal-c2x vector with a TEV cleavage site immediately downstream of the Factor Xa site. In total, four fusions with EGFR-binding Fn3s (E6-PFO, C-PFO, D-PFO, and E-PFO) and one fusion with a carcinoembryonic antigen–binding Fn3 (C7-PFO) were constructed.11,21 Fn3–PFO fusion proteins were transformed into Rosetta 2 (DE3) Escherichia coli (Novagen, San Diego, CA). Cells were grown to an optical density at 600 nm of 0.5 -1.0 and induced with 0.5 mmol/l IPTG for 6 hours at 30 °C. Resuspended cell pellets were sonicated and the lysates were subjected to purification on an amylose column according to the manufacturer's instructions (New England Biolabs, Ipswich, MA). After overnight digestion with TEV protease, Fn3–PFO proteins were purified by ion exchange chromatography.
The multispecific construct HNB-LCD was prepared as described previously.16
Tissue culture. The human epidermoid carcinoma cell line, A431, was cultured in a humidified atmosphere in 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin. A431 cells stably expressing d2EGFP under the cytomegalovirus promoter were generated by transfection of pd2EGFP-N1 (Clontech) using the Amaxa Nucleofector 2b (Lonza, Basel, Switzerland) according to the manufacturer's instructions. Forty-eight hours after transfection, 0.75 mg/ml G418 (Invitrogen) was added to the culture medium. G418-resistant cells were propagated and the GFP-expressing fraction was sorted twice by fluorescence-activated cell sorting using a Mo-Flo sorter (Cytomation, Carpinteria, CA). The resulting cells, termed A431-d2EGFP, were >99% GFP positive and were cultured using a maintenance G418 concentration of 0.1 mg/ml.
Agarose gel shift assay. siRNA (50 pmol) was mixed with varying amounts of E6N2 for 30 minutes at room temperature. The resulting complexes were run on a 2% agarose gel and visualized using SYBR-Gold (Invitrogen).
Measurement of dsRBD and siRNA-binding affinity. In order to quantify the siRNA-binding affinity of the dsRBD portion of E6N2, E6N2 was loaded onto Protein A Dynal Beads (Invitrogen). The Protein A beads were washed in phosphate-buffered saline (PBS) + 0.1% bovine serum albumin (PBSA) and resuspended in Dulbecco's modified Eagle's medium + 10% FBS adjusted to the specified pH with Alexa 488 labeled AllStars Negative Control siRNA (Qiagen, Valencia, CA) at the specified concentration at 37 °C for 1 hour. The beads were washed twice in ice-cold PBSA and analyzed by flow cytometry on an Accuri C6 flow cytometer (Accuri, Ann Arbor, MI). The KD,app of binding was numerically calculated from the data as described previously.31
siRNA cell uptake assay. A431 cells were plated in 96-well flat-bottom plates at 5 × 104 cells per well and serum starved overnight. Alexa 488 labeled Negative AllStars siRNA was complexed with E6N2 at a 1:1 ratio for 30 minutes at room temperature. Complexes or free siRNA in the absence of E6N2 were added to the cells at a 100 nmol/l final concentration, in complete media (10% FBS), in the presence or absence of varying amounts of HNB-LCD. At each time point, the cells were washed twice with PBS and trypsinized for 20 minutes. Cells were washed twice with complete media and resuspended in PBS + 2% FBS for analysis by flow cytometry.
In order to correlate the fluorescence signal with the number of siRNA molecules, a calibration curve was determined using the Quantum Simply Cellular anti-Mouse beads according to the manufacturer's instructions (Bangs Laboratories, Fishers, IN), using E6N2 labeled with Alexa 488 at a 6 dye:1 protein ratio.
Fluorescence microscopy. Cells were plated on MatTek chambers with a 0.13-mm glass coverslip bottom (MatTek, Ashland, MA) and serum starved overnight. Alexa 488 labeled siRNA was complexed with E6N2 at a 1:1 molar ratio for 30 minutes at room temperature and added to the cells at a 100 nmol/l final concentration in complete media. After 6 hours, the cells were washed with complete media and then stained with LysoTracker Red (Invitrogen) and 4′,6-diamidino-2-phenylindole (Roche Applied Science, Indianapolis, IN). Cells were imaged using a Delta Vision fluorescence microscope (Applied Precision, Issaquah, WA).
GFP knockdown assay. A431-d2EGFP cells were plated in 96-well flat-bottom plates and serum starved overnight. E6N2 was mixed with either Negative Control AllStars siRNA or GFP Duplex I siRNA (Thermo Scientific, Lafayette, CO) at a 1:1 molar ratio for 30 minutes at room temperature. Complexes or free siRNA in the absence of E6N2 were added to the cells in complete media with varying amounts of Fn3–PFO, in the presence or absence of 7.5 nmol/l HNB-LCD. After 6 hours, cells were washed and incubated for 24 hours in complete media. The cells were trypsinized, washed twice with PBS + 2% FBS, and analyzed by flow cytometry.
Cytotoxicity of PFO fusion proteins. Cell viability measurements were performed using the Wst-1 reagent with a 1-hour incubation according to the manufacturer's instructions (Roche Applied Science). They were performed either prior to trypsinization for flow cytometry analysis of GFP expression in GFP knockdown assays or in separate cytotoxicity assays. In all cases, cytotoxicity measurements were performed on cells exposed to E6N2, siRNA, and HNB-LCD where stated. When performed prior to GFP analysis, the cells were washed twice with PBS after Wst-1 exposure. The presence of d2EGFP did not affect Wst-1 reagent performance. The Wst-1 reagent also did not affect GFP expression measurements in knockdown assays.
Hemolysis assay. Fresh mouse red blood cells (Fitzgerald Industries, Acton, MA) were washed three times in PBSA. Fifty microliters of a 10% suspension of red blood cells were used per sample. The cells were then incubated for 1 hour at 37 °C with varying amounts of PFO or with 10% Triton-X 100 as a positive control for lysis. The cells were centrifuged for 4 minutes at 14,000g, and the supernatants were measured for hemoglobin release by absorbance at 541 nm.
SUPPLEMENTARY MATERIAL Figure S1. Dynamic light scattering analysis of E6N2/siRNA complexes. Figure S2. Measurement of E6N2 loading on Protein A beads. Figure S3. Specificity of siRNA uptake by Fc-dsRBD fusion proteins. Figure S4. Determination of the cytoplasmic delivery threshold required for GFP knockdown. Figure S5. Therapeutic window of alternate Fn3–PFO fusion proteins. Figure S6. Lack of GFP knockdown from untargeted gfp siRNA. Figure S7. Evaluation of siRNA uptake enhancement by multispecific constructs. Figure S8. Lack of GFP knockdown by control siRNA delivered by E6N2 in the presence of HNB-LCD. Table S1. EGFR binding affinity for dsRBD and PFO fusion constructs. Supplementary Materials and Methods.
Acknowledgments
This work was funded by National Institutes of Health Grants AI065824 and CA101830. The authors thank James Cole (University of Connecticut) for providing the gene encoding dsRBD. The Koch Institute Flow Cytometry and Microscopy core facilities provided technical assistance with fluorescence-activated cell sorting and fluorescence microscopy, respectively. The MIT Biophysical Instrumentation Facility gratefully provided equipment for dynamic light scattering analysis. The authors also thank Chris Pirie for useful discussions.
The authors declare no conflict of interest.
Supplementary Material
Dynamic light scattering analysis of E6N2/siRNA complexes.
Measurement of E6N2 loading on Protein A beads.
Specificity of siRNA uptake by Fc-dsRBD fusion proteins.
Determination of the cytoplasmic delivery threshold required for GFP knockdown.
Therapeutic window of alternate Fn3–PFO fusion proteins.
Lack of GFP knockdown from untargeted gfp siRNA.
Evaluation of siRNA uptake enhancement by multispecific constructs.
Lack of GFP knockdown by control siRNA delivered by E6N2 in the presence of HNB-LCD.
EGFR binding affinity for dsRBD and PFO fusion constructs.
References
- Jarvis L. Delivering the promise. Chem Eng News. 2009;87:18–27. [Google Scholar]
- Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MM, Wittrup KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther. 2009;8:2861–2871. doi: 10.1158/1535-7163.MCT-09-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Ban HS, Kim SS, Wu H, Pearson T, Greiner DL, et al. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell. 2008;134:577–586. doi: 10.1016/j.cell.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- Peer D, Zhu P, Carman CV, Lieberman J, Shimaoka M. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proc Natl Acad Sci USA. 2007;104:4095–4100. doi: 10.1073/pnas.0608491104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J, Martin-Killias P, Plückthun A, Zangemeister-Wittke U. EpCAM-targeted delivery of nanocomplexed siRNA to tumor cells with designed ankyrin repeat proteins. Mol Cancer Ther. 2009;8:2674–2683. doi: 10.1158/1535-7163.MCT-09-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesner U, Halin C, Lozzi L, Günthert M, Neri P, Wunderli-Allenspach H, et al. Quantitation of the tumor-targeting properties of antibody fragments conjugated to cell-permeating HIV-1 TAT peptides. Bioconjug Chem. 2002;13:729–736. doi: 10.1021/bc025517+. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Pardridge WM. Pharmacokinetics and delivery of tat and tat-protein conjugates to tissues in vivo. Bioconjug Chem. 2001;12:995–999. doi: 10.1021/bc0155061. [DOI] [PubMed] [Google Scholar]
- Bevilacqua PC, Cech TR. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry. 1996;35:9983–9994. doi: 10.1021/bi9607259. [DOI] [PubMed] [Google Scholar]
- Hackel BJ, Ackerman ME, Howland SW, Wittrup KD. Stability and CDR composition biases enrich binder functionality landscapes. J Mol Biol. 2010;401:84–96. doi: 10.1016/j.jmb.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N, et al. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat Biotechnol. 2009;27:567–571. doi: 10.1038/nbt.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee SH, Choe J, Park TG. Intracellular small interfering RNA delivery using genetically engineered double-stranded RNA binding protein domain. J Gene Med. 2009;11:804–812. doi: 10.1002/jgm.1365. [DOI] [PubMed] [Google Scholar]
- Geoghegan JC, Gilmore BL, Davidson BL. Gene silencing mediated by siRNA-binding fusion proteins is attenuated by double-stranded RNA-binding domain structure. Mol Ther Nucleic Acids. 2012;1:e53. doi: 10.1038/mtna.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado CJ, Kondos S, Bull TE, Kuiper MJ, Law RH, Buckle AM, et al. The MACPF/CDC family of pore-forming toxins. Cell Microbiol. 2008;10:1765–1774. doi: 10.1111/j.1462-5822.2008.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler JB, Manzari MT, Rosalia EK, Chen TF, Wittrup KD. Triepitopic antibodies inhibit cetuximab-resistant BRAF- and KRAS mutant tumors via EGFR signal repression. J Mol Biol. 2012;28:532–544. doi: 10.1016/j.jmb.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Yuan YA. Structural insights into RISC assembly facilitated by dsRNA-binding domains of human RNA helicase A (DHX9) Nucleic Acids Res. 2013;41:3457–3470. doi: 10.1093/nar/gkt042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17:5458–5465. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirie CM, Liu DV, Wittrup KD. Targeted cytolysins synergistically potentiate cytoplasmic delivery of gelonin immunotoxin. Mol Cancer Ther. 2013;12:1774–1782. doi: 10.1158/1535-7163.MCT-12-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuerch DW, Wilson-Kubalek EM, Tweten RK. Molecular basis of listeriolysin O pH dependence. Proc Natl Acad Sci USA. 2005;102:12537–12542. doi: 10.1073/pnas.0500558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirie CM, Hackel BJ, Rosenblum MG, Wittrup KD. Convergent potency of internalized gelonin immunotoxins across varied cell lines, antigens, and targeting moieties. J Biol Chem. 2010;286:4165–4172. doi: 10.1074/jbc.M110.186973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W, Lam JKW.2012Endosomal escape pathways for non-viral nucleic acid delivery systems. Ceresa B.ed.). Molecular Regulation of Endocytosis Intech; Rijeka; 429–456. [Google Scholar]
- Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. J Cell Sci. 2010;123:1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- Barnes DW. Epidermal growth factor inhibits growth of A431 human epidermoid carcinoma in serum-free cell culture. J Cell Biol. 1982;93:1–4. doi: 10.1083/jcb.93.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Zoncu R, Johns TG, Pypaert M, Lee FT, Mellman I, et al. Internalization, intracellular trafficking, and biodistribution of monoclonal antibody 806: a novel anti-epidermal growth factor receptor antibody. Neoplasia. 2007;9:1099–1110. doi: 10.1593/neo.07721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Rowinsky EK. The ErbB receptor family: a therapeutic target for cancer. Trends Mol Med. 2002;8:S19–S26. doi: 10.1016/s1471-4914(02)02306-7. [DOI] [PubMed] [Google Scholar]
- Wakeling AE. Epidermal growth factor receptor tyrosine kinase inhibitors. Curr Opin Pharmacol. 2002;2:382–387. doi: 10.1016/s1471-4892(02)00183-2. [DOI] [PubMed] [Google Scholar]
- Dunstone MA, Tweten RK. Packing a punch: the mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Curr Opin Struct Biol. 2012;22:342–349. doi: 10.1016/j.sbi.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser M, Cèbe R, Drewello D, Schmitz R. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques. 2001;1:88–92. doi: 10.2144/01311st05. [DOI] [PubMed] [Google Scholar]
- Spanggord RJ, Beal PA. Site-specific modification and RNA crosslinking of the RNA-binding domain of PKR. Nucleic Acids Res. 2000;28:1899–1905. doi: 10.1093/nar/28.9.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DV, Maier LM, Hafler DA, Wittrup KD. Engineered interleukin-2 antagonists for the inhibition of regulatory T cells. J Immunother. 2009;32:887–894. doi: 10.1097/CJI.0b013e3181b528da. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dynamic light scattering analysis of E6N2/siRNA complexes.
Measurement of E6N2 loading on Protein A beads.
Specificity of siRNA uptake by Fc-dsRBD fusion proteins.
Determination of the cytoplasmic delivery threshold required for GFP knockdown.
Therapeutic window of alternate Fn3–PFO fusion proteins.
Lack of GFP knockdown from untargeted gfp siRNA.
Evaluation of siRNA uptake enhancement by multispecific constructs.
Lack of GFP knockdown by control siRNA delivered by E6N2 in the presence of HNB-LCD.
EGFR binding affinity for dsRBD and PFO fusion constructs.