

Abstract
Background
Salmonella enterica subsp. enterica serovar Virchow has been recognized as a significant health burden in Asia, Australia and Europe. In addition to its global distribution, S. Virchow is clinically significant due to the frequency at which it causes invasive infections and its association with outbreaks arising from food-borne transmission. Here, we examine the genome of an invasive isolate of S. Virchow SVQ1 (phage type 8) from an outbreak in southeast Queensland, Australia. In addition to identifying new potential genotyping targets that could be used for discriminating between S. Virchow strains in outbreak scenarios, we also aimed to carry out a comprehensive comparative analysis of the S. Virchow genomes.
Results
Genome comparisons between S. Virchow SVQ1 and S. Virchow SL491, a previously published strain, identified a high degree of genomic similarity between the two strains with fewer than 200 single nucleotide differences. Clustered Regularly Interspaced Palindromic Repeats (CRISPR) regions were identified as a highly variable region that could be used to discriminate between S. Virchow isolates. We amplified and sequenced the CRISPR regions of fifteen S. Virchow isolates collected from seven different outbreaks across Australia. We observed three allelic types of the CRISPR region from these isolates based on the presence/absence of the spacers and were able to discriminate S. Virchow phage type 8 isolates originating from different outbreaks. A comparison with 27 published Salmonella genomes found that the S. Virchow SVQ1 genome encodes 11 previously described Salmonella Pathogenicity Islands (SPI), as well as additional genomic islands including a remnant integrative conjugative element that is distinct from SPI-7. In addition, the S. Virchow genome possesses a novel prophage that encodes the Type III secretion system effector protein SopE, a key Salmonella virulence factor. The prophage shares very little similarity to the SopE prophages found in other Salmonella serovars suggesting an independent acquisition of sopE.
Conclusions
The availability of this genome will serve as a genome template and facilitate further studies on understanding the virulence and global distribution of the S. Virchow serovar, as well as the development of genotyping methods for outbreak investigations.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-389) contains supplementary material, which is available to authorized users.
Background
Salmonella enterica subsp. enterica serovar Virchow is commonly associated with gastroenteritis, but it is also known to cause invasive systemic infections [1–4]. Outbreaks of serovar Virchow are a significant public health risk in many European, Asian and Oceanic countries [3, 5–8]. Currently, S. Virchow is one of most prevalent Salmonella serovars in Australia and outbreaks can occur through food-borne transmission via contaminated fruit and vegetables and poor food handling practices [2, 9, 10]. In order to track outbreaks of S. Virchow, molecular subtyping methods are needed to discriminate between strains, however, no such typing scheme currently exits.
Phage typing is a well-established method for discriminating between Salmonella strains based on their susceptibility to lytic infection by specific bacteriophages [11, 12]. However, discrepancies in phage typing results between different laboratories have been reported [13]. There are also several nucleic acid-based typing methods, including pulsed-field gel electrophoresis (PFGE), which involves using restriction enzymes to cut bacterial DNA into fragments and analysing the banding patterns following gel electrophoresis [14, 15]. However, PFGE has limitations in reproducibility and the results can be ambiguous, and is also limited in its ability to discriminate between different strains [16]. On the other hand, multiple-loci variable-number tandem repeat analysis (MLVA), a PCR based method used to detect variation in the number of repeat units in tandem repeat sequences [17, 18], provides improved level of discrimination for many Salmonella serovars compared to PFGE [19]. In addition, Multi Locus Sequence Typing (MLST) also allows greater discrimination between serovars. It involves detecting allelic differences in the sequences of various housekeeping genes [20–22] and can also been extended to include virulence genes [23]. Even greater resolution can be achieved by identifying single nucleotide polymorphisms (SNPs) as genotyping targets from whole genome sequence (WGS) data, with schemes available for serovars like S. Typhimurium [24, 25].
Whilst there are MLVA and SNP typing schemes available for many Salmonella serovars there are currently none available for discriminating between the different S. Virchow phage type (PT) strains [26–30]. By MLST, S. Virchow strains belong to the eBurst Group BG9, however, the majority of BG9 strains in the MLST database are classified as sequence type 16 (ST16) [31]. Therefore, additional genotyping targets with a greater degree of discrimination between strains are required for subtyping S. Virchow.
Clustered regularly interspaced short palindromic repeats (CRISPRs) have recently been used to subtype more than 9 major Salmonella serovars including Typhimurium, Newport and Enteritidis [32–34]. CRISPRs are sequences consisting of 21–47 nucleotides that are repeated in tandem separated by non-repetitive sequences of a similar size [35]. A cluster of genes known as CRISPR-associated genes (cas) is often found near the start of CRISPR regions [36]. CRISPRs function as a defense system against foreign DNA such as plasmids and bacteriophage by producing small RNA (sRNA) that can silence foreign mRNA, similar to a RNA interference system [37]. Generally, CRISPRs vary between Salmonella strains in the number of repeats/spacers [32, 33, 38]. Prophages are also useful for genotyping Salmonella but have not as yet been applied to S. Virchow as a routine epidemiological tool [39, 40].
Like the majority of bacteria, mobile genetic elements such as plasmids, bacteriophages and insertion sequence elements are the main drivers of gene flux in Salmonella [41–44]. This organism has acquired many of its virulence genes from mobile elements and they are often found within islands, referred to as Salmonella Pathogenicity Islands (SPIs) [45]. Other virulence factors such as Type III secreted effectors can be found encoded in prophage regions [42, 46]. Until recently only one other S. Virchow genome had been reported [38]. The genome for S. Virchow SL491, a PT25 strain that was isolated in the United States, was studied as part of a broader comparative study of 28 S. enterica strains [38]. Similarly, during the preparation of this manuscript, a second S. Virchow draft genome was reported as part of a large WGS phylogenetic analysis of 78 Salmonella serovars. These studies showed that S. Virchow strains were most closely related to strains of the Heidelberg serovar and carry distinctive CRISPR regions [38, 47], however, a comprehensive genomic comparison of different S. Virchow strains has yet to be reported.
Here we report our comparative analyses of the genome of an Australian isolate of S. Virchow PT 8 (SVQ1) with the published genome of S. Virchow PT25 (SL491). We report a comparative analysis with 27 other Salmonella genomes that reveals the mobile element content of S. Virchow strains and furthers our understanding of the evolution of this important food-borne pathogen. We have also identified new discriminatory genotyping targets that can be combined with existing Salmonella genotyping schemes to elucidate the relatedness of individual S. Virchow isolates.
Results
Whole genome comparison of S. Virchow SVQ1 and S. Virchow SL491
The draft genome of S. Virchow SVQ1 (PT8) consists of a 4.67 Mbp chromosome and four plasmids that range from 2.5 to 37 kb (Additional file 1: Table S1). Differences between the S. Virchow SVQ1 chromosome and S. Virchow SL491 chromosome include 13 genes that makes up a remnant prophage in SVQ1. The S. Virchow SL491 genome is larger than S. Virchow SVQ1 genome with addition of 280 genes that are distributed amongst three prophage and a genomic island that were likely acquired via lateral gene transfer (LGT) (Figure 1). Read mapping was used to confirm that the observed absence of S. Virchow SL491 prophage and islands regions in S. Virchow SVQ1 was genuine and not as the result of assembly errors (data not shown). The genomic island encodes the aminoglycoside resistance gene rmtC and a partial mercury resistance transposon operon [38]. S. Virchow SVQ1 carries four plasmids that are absent in S. Virchow SL491. The largest SVQ1 plasmid shares 96–98% identity across 78% of the non-virulence plasmid pOU1114 found S. Dublin and encodes a conjugative transfer system [48]. The other three plasmids are non-conjugative and are each unambiguously assembled into a single circular contig (Additional file 1: Table S1). We detected 195 variants within coding regions between the two S. Virchow genomes, including 166 SNPs, 13 single nucleotide frame-shift indels, and 5 three-nucleotide in-frame indels (Additional file 2: Table S2). By comparison, the genome of S. Heidelberg SL497 differs from the genome of S. Virchow SVQ1 by approximately 34,000 SNPs.
Figure 1.
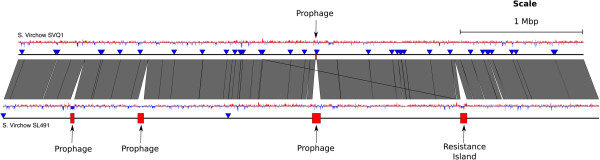
Comparison between the draft genomes of Salmonella Virchow SVQ1 and Salmonella Virchow SL491. Regions of differences are shown as red boxes and labeled accordingly. Vertical blocks between the genomes represents regions of shared similarity according to BLASTn (Nucleotide identity >99%) [49]. The GC content for both genomes is shown as graphs, red indicates above average GC content while blue indicates below average GC content. Contig boundaries are marked with blue triangles. The resistance island carries the rmtC gene, which confers resistance to aminoglycosides [38]. The image was prepared using EasyFig [50].
SNP containing genes provide limited discrimination of S. Virchow isolates
We tested 11 genes that contained SNPs between the genomes of S. Virchow SVQ1 and S. Virchow SL491 that may be potential discriminatory genotyping targets (Additional file 3: Table S3). Amplicons were sequenced from 45 S. Virchow isolates that had been collected from various outbreaks in Australia (Table 1), as well as S. Virchow SVQ1 and S. Virchow SL491. Only one gene out of 11, encoding a probable pyruvate-flavodoxin oxidoreductase (locus tag: Sesv_1374), was determined to be a potential target for discriminating Australian S. Virchow PT8 isolates. In S. Virchow SVQ1, and four other related isolates from same outbreak, this gene contained a Cytosine (C) at position 1428 in the 3.5 kb gene, whereas the remaining isolates (including several PT8 strains from other outbreaks) had a Thymine (T) in this position. The remaining 10 genes were found to have a conserved sequence in all 45 Australian S. Virchow isolates. In all cases the sequencing of SVQ1 and SL491 genotyping candidates was consistent with the original SNP prediction.
Table 1.
List of S. Virchow isolates that were used in this study
| Num of strains | Phage type | Cluster (C) or outbreak (O)1 | Source: faecal (F) or blood (B)2 | Origin3 | Year | Reference4 |
|---|---|---|---|---|---|---|
| 1 | PT8 | C | F | QLD | 2008 | This study |
| 5 | PT8 | O | 4 F, 1B | QLD | 2007 | This study |
| 1 | PT25 | U | F | USA | 2005 | [38] |
| 9 | PT8 | C | F | NT | 2006 | NEPSS, 2006, p11 |
| 9 | PT8 | C | F | WA | 2005 | NEPSS, 2005, p13 |
| 5 | PT8 | C | F | QLD | 2008 | This study |
| 3 | PT8 | C | F | QLD | 2004 | NEPSS, 2005, p9 |
| 7 | PT17 | C | 4 F, 3B | QLD | 2001 | NEPSS 2001 |
| 6 | PT34 | O | F | VIC | 2001 | NEPSS, 2001 |
| SEPT2002, p13 | ||||||
| 1 | PT25 | C | F | QLD | 2005 | NEPPS annual report 2005, 2006 1/06, p12 |
1A cluster (C) is a group of cases that occurred in a specific place and time. An outbreak (O) is an incident of cases where the source of the infection is known. The University of Calgary (U) provided this isolate.
2 F, Faecal isolate; B, Blood isolate.
3QLD, Queensland; NT, Northern Territory; WA, Western Australia; VIC, Victoria.
4National Enteric Pathogens Surveillance Scheme. Annual Reports 2001–2008. Melbourne: Microbiological Diagnostic Unit, University of Melbourne.
CRISPRs as potential targets for discrimination of S. Virchow isolates
Like the majority of Salmonella serovars, S. Virchow SVQ1 has two CRISPRs: CRISPR-1, which is 2.7 kb in length and has 45 spacers, and CRISPR-2, which is 1 kb in length and has 16 spacers (Figure 2a). Comparisons of CRISPRs in S. Virchow SVQ1 and S. Virchow SL491 revealed that CRISPR-1 is substantially larger in SL491 with 55 spacers. However, only the first 21 spacers are conserved between both S. Virchow genomes, indicating that there may be sufficient variability within this region to sub-type S. Virchow strains. CRISPR-2 is identical between the two S. Virchow genomes.
Figure 2.
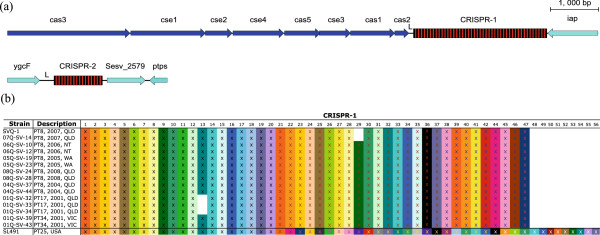
Comparison of S. Virchow CRISPR regions. a. Schematic representation of the two CRISPR regions in S. Virchow SVQ1. Direct repeats are shown as black rectangles and the spacers are shown as red rectangles. CRISPR-associated genes (cas) and other flanking genes are represented by dark-blue and light-blue arrows, respectively. L stands for the leader sequence. The genes that are flanking CRISPR-1 are associated with the locus tags Sesv_2565 to Sesv_2573 and the genes flanking CRISPR-2 are associated with the locus tags Sesv_2578 to Sesv_2580, respectively. This image was prepared using Easyfig [50]. b. Representation of spacer arrangement in CRISPR-1 in 15 Australian S. Virchow isolates. Each unique spacer is represented by a combination of background colour and the colour of the X character. White gaps represent the absence of a particular spacer. Strains are grouped by phage type, the year it was collected and location. The spacer arrangement of CRISPR-1 from the genome of S. Virchow SL491 is also shown. QLD = Queensland, NT = Northern Territory, WA = Western Australia and VIC = Victoria.
The CRISPR-1 region was sequenced in fifteen Australian S. Virchow isolates consisting of various phage types (PT8, PT17 and PT34) to determine the level of variation between strains (Table 2). The fifteen strains selected include at least two strains from each of seven different outbreaks, that have occurred between 2001 and 2008. Three allelic types of CRISPR-1 were observed based on the presence/absence of particular spacer sequences (Figure 2b). CRISPR typing was able to distinguish S. Virchow SVQ1 (PT8) and a second PT8 isolate from the same outbreak (07Q-SV-14) from other Australian PT8 isolates due to the absence of spacer 29. The absence of this spacer distinguishes S. Virchow SVQ1 and 07Q-SV-14 from the other PT8 isolates, demonstrating that CRISPRs can be used to help discriminate between S. Virchow strains within a phage type. Notably, PT17 isolates are characterized by the absence of a different spacer (Figure 2b).
Table 2.
List of Australian S. Virchow isolates used in the CRISPR analysis
| Strain | Phage type | Year | State | Source1 | Accession number |
|---|---|---|---|---|---|
| SVQ1 | PT8 | 2007 | Queensland | This study | [GenBank:AZMP01000000] |
| 07-SV-14 | PT8 | 2007 | Queensland | This study | [GenBank:KF931136] |
| 06-SV-10 | PT8 | 2006 | Northern Territory | NEPSS, 2006, p11 | [GenBank:KF931134] |
| 06Q-SV-12 | PT8 | 2006 | Northern Territory | NEPSS, 2006, p11 | [GenBank:KF931135] |
| 05Q-SV-19 | PT8 | 2005 | Western Australia | NEPSS, 2005, p13 | [GenBank:KF931132] |
| 05Q-SV-23 | PT8 | 2005 | Western Australia | NEPSS, 2005, p13 | [GenBank:KF931133] |
| 08Q-SV-24 | PT8 | 2008 | Queensland | This study | [GenBank:KF931137] |
| 08Q-SV-28 | PT8 | 2008 | Queensland | This study | [GenBank:KF931138] |
| 04Q-SV-37 | PT8 | 2004 | Queensland | NEPSS, 2005, p9 | [GenBank:KF931130] |
| 04Q-SV-44 | PT8 | 2004 | Queensland | NEPSS, 2005, p9 | [GenBank:KF931131] |
| 01Q-SV-32 | PT17 | 2001 | Queensland | This study | [GenBank:KF931125] |
| 01Q-SV-33 | PT17 | 2001 | Queensland | This study | [GenBank:KF931126] |
| 01Q-SV-34 | PT17 | 2001 | Queensland | This study | [GenBank:KF931127] |
| 01Q-SV-39 | PT34 | 2001 | Victoria | NEPSS, 2001 SEPT2002, p13 | [GenBank:KF931128] |
| 01Q-SV-43 | PT34 | 2001 | Victoria | NEPSS, 2001 SEPT2002, p13 | [GenBank:KF931129] |
1National Enteric Pathogens Surveillance Scheme. Annual Reports 2001–2008. Melbourne: Microbiological Diagnostic Unit, University of Melbourne.
Genomic analysis of S. Virchow SVQ1
The genome of S. Virchow SVQ1 was compared with 27 Salmonella genomes to determine genetic differences between Virchow and the other serovars (Table 3). The comparison revealed that the genomic backbone of S. Virchow is similar to the genomes of other Salmonella serovars, including key virulence factors. The S. Virchow genome encodes the two Type III secretion systems that are conserved in all Salmonella serovars and are encoded on Salmonella Pathogenicity Islands (SPI-1 and SPI-2). S. Virchow also carries nine other known SPIs that are conserved within other Salmonella genomes, with the exception of SPI-6 (Figure 3 and Additional file 4: Table S4). The intact SPI-6 island in S. Typhi CT18 carries a Type VI Secretion System (T6SS), two fimbrial gene clusters (safABCD and tcfABCD) and the invasin, PagN [51, 52]. However, the SPI-6 in the S. Virchow genome is missing the T6SS but it still possesses the two fimbrial clusters and pagN (Figure 4).
Table 3.
Genome sequences used in the genomic comparison
| Ring1 | Genome | Strain | GenBank accession | Reference |
|---|---|---|---|---|
| 6 | Salmonella enterica subsp. enterica serovar Virchow | SL491 | ABFH00000000 | [38] |
| 7 | Salmonella enterica subsp. enterica serovar Heidelberg | SL476 | CP001120 | [38] |
| Salmonella enterica subsp. enterica serovar Heidelberg | SL486 | ABEL00000000 | [38] | |
| 8 | Salmonella enterica subsp. enterica serovar Newport | SL254 | CP001113 | [38] |
| Salmonella enterica subsp. enterica serovar Newport | SL317 | ABEW00000000 | [38] | |
| 9 | Salmonella enterica subsp. enterica serovar Typhimurium | LT2 | AE006468 | [53] |
| Salmonella enterica subsp. enterica serovar Typhimurium | UK-1 | CP002614 | [54] | |
| 10 | Salmonella enterica subsp. enterica serovar Saintpaul | SARA23 | ABAM02000001 | [38] |
| Salmonella enterica subsp. enterica serovar Saintpaul | SARA29 | ABAN00000000 | [38] | |
| 11 | Salmonella enterica subsp. enterica serovar Hadar | RI_05P066 | ABFG01000000 | [38] |
| 12 | Salmonella enterica subsp. enterica serovar Choleraesuis | SC-B67 | AE017220 | [55] |
| 13 | Salmonella enterica subsp. enterica serovar Paratyphi C | RKS4594 | CP000857 | [56] |
| 14 | Salmonella enterica subsp. enterica serovar Agona | SL483 | CP001138 | [38] |
| 15 | Salmonella enterica subsp. enterica serovar Kentucky | CDC 191 | ABEI01000000 | [38] |
| Salmonella enterica subsp. enterica serovar Kentucky | SL475 | ABAK02000001 | [38] | |
| 16 | Salmonella enterica subsp. enterica serovar Weltevreden | HI_N05-537 | ABFF00000000 | [38] |
| 17 | Salmonella enterica subsp. enterica serovar Dublin | CT_02021853 | CP001144 | [38] |
| 18 | Salmonella enterica subsp. enterica serovar Enteritidis | P125109 | AM933172 | [57] |
| 19 | Salmonella enterica subsp. enterica serovar Gallinarum | 287/91 | AM933173 | [57] |
| 20 | Salmonella enterica subsp. enterica serovar Paratyphi B | SPB7 | CP000886 | W.U. Genome Sequencing Centre |
| 21 | Salmonella enterica subsp. enterica serovar Schwarzengrund | SL480 | ABEJ01000000 | [38] |
| Salmonella enterica subsp. enterica serovar Schwarzengrund | CVM19633 | CP001127 | [38] | |
| 22 | Salmonella enterica subsp. enterica serovar Javiana | SL478 | ABEH00000000 | [38] |
| 23 | Salmonella enterica subsp. enterica serovar Paratyphi A | ATCC9150 | CP000026 | [53] |
| 24 | Salmonella enterica subsp. enterica serovar Typhi | CT18 | AL513382 | [41] |
| Salmonella enterica subsp. enterica serovar Typhi | Ty2 | AE014613 | [58] | |
| 25 | Salmonella enterica subsp. arizonae serovar 62:z4,z23 | RKS2980 | CP000880 | W.U. Genome Sequencing Centre |
1Genomes are listed as they appear in Figure 3, from innermost to outermost. Rings 1 to 5 correspond to S. Virchow SVQ1 genome position, GC skew, GC content, coverage and contig boundaries, respectively.
Figure 3.
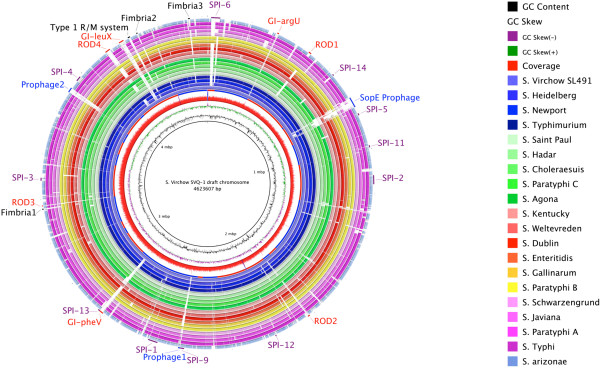
Blast ring image of S. Virchow SVQ1 genome. The innermost rings show S. Virchow SVQ1 genome position (mbp = Megabases), GC content (black) and GC skew (purple/green) and read coverage (red). The contig boundaries for the S. Virchow SVQ1 genome are shown as alternating red and blue bars on the fifth innermost ring. The remaining rings show BLASTn comparison of the 27 other Salmonella genomes listed in Table 3, against S. Virchow SVQ1 (in some cases multiple genomes are grouped into a single ring). BLASTn matches with an identity between 90% and 100% are coloured, while non-matching regions appear as blank spaces in each ring. The outer ring contains annotations, coloured according to function: regions variable in other Salmonella genomes such as fimbrial usher/chaperone operons and a Type I restriction-modification system (black); prophage regions (blue); genomic islands in recognised integration sites (GI-argU, GI-pheV and GI-leuX) and other regions of difference (ROD1-4) (red). Green labels refer to the Salmonella Pathogenicity Islands present in S. Virchow. The image was prepared using BRIG [59].
Figure 4.
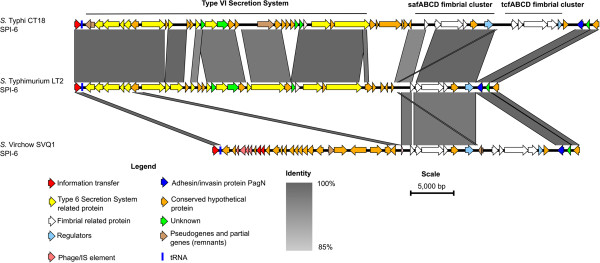
Nucleotide comparison of SPI-6 from S. Typhi CT18, S. Typhimurium LT2 and S. Virchow SVQ1. Grey vertical blocks indicate regions of shared similarity shaded according to BLASTn. The coloured arrows represent genes. The functions of the genes have been inferred from BLAST searches. The intact SPI-6 in S. Typhi CT18 carries a Type VI secretion system (T6SS) and two fimbrial clusters (saf and tcf) and encodes the adhesin/invasin protein PagN. In S. Typhimurium LT2 the tcf fimbrial cluster is absent. In S. Virchow the T6SS is missing but both fimbrial clusters are present. The image was prepared using Easyfig [50].
The whole genome comparison with other Salmonella serovars revealed that S. Virchow SVQ1 contains several regions of difference (RODs) (Additional file 5: Table S5). RODs represent sequences that are present in S. Virchow SVQ1 but absent in most other Salmonella genomes. These RODs include three putative genomic islands, defined as RODs that contained integrase genes or evidence of integrase mediated insertion (i.e. flanking direct repeats) in chromosomal integration hot-spots (GI-argU, GI-pheV, and GI-leuX), and three putative prophage elements (Figure 3 and Additional file 5: Table S5). Putative prophage elements could be distinguished from genomic islands by the presence phage structural or replication genes. Flanking direct repeats could be defined at the boundaries of the three prophage regions and GI-argU (Additional file 5: Table S5). There are also several other RODs including the 9.4 kb O-antigen biosynthetic gene cluster between gln and galF (ROD2), as well as three chaperone-usher fimbrial clusters that are sporadically distributed amongst other Salmonella serovars (Additional file 5: Table S5). S. Virchow also encodes the R-, M- and S- subunits characteristic of a Type I restriction modification system: Sesv_4171, Sev_4170, Sesv_4169 each exhibit 91%, 97% and 52% amino acid identity with the corresponding subunits of the EcoAI enzyme (http://rebase.neb.com/rebase/enz/EcoAI.html), respectively. The DNA-recognition domain (pfam: Methylase_S) of the S-subunit is unique to S. Virchow suggesting that the S. Virchow heteromeric enzyme may resemble EcoAI mechanistically, but may have different sequence specificity.
GI-pheV is a 19.6 kb genomic island that is only found in two other Salmonella genomes (Figure 3) and has likely been acquired by LGT followed by integration into tRNApheV. Notably it carries an orphan cytosine C5-methyltransferase (Sesv_2771) that may play a role in global regulation by site-specific DNA methylation throughout the genome. GI-pheV is inserted directly adjacent to SPI-13, which is a 7.4 kb pathogenicity island encoding a putative lyase, a hydrolase, an oxidase, and an arylsulphatase regulator and is known to be involved in systemic infection of mice and replication inside murine macrophages [60, 61]. All 47 S. Virchow isolates in this study were found to contain GI-pheV on the basis of PCR amplification across the 5′ and 3′ boundaries of the island (Additional file 6: Table S6).
GI-leuX is a 22.2 kb region inserted next to tRNAleuX in place of the SPI-10 which is found in S. Typhi CT18 and S. Enteritidis P125109 (Figure 3). The island encodes an integrase and a degraded genomic island type IV secretion system (GI-T4SS), indicating that it appears to be a remnant integrative conjugative element (ICE) ([62]). Although the majority of conjugal transfer genes are missing, the S. Virchow GI-leuX encodes the archetypal GI-T4SS conjugative coupling protein traD/virD4 gene (locus tag: sesv_4134), albeit with a frame-shift that truncates VirD4 by 53 amino acids. When compared with previously defined representative T4SS sequences [62], S. Virchow VirD4 shares the most similarity (57-60% amino acid identity) with VirD4 homologs from the related and previously characterized ICEs S. Typhi CT18 SPI-7 (locus tag: Sty_4562) [63], H. influenzae ICEHin1056 (locus tag: p1056.35) [64] and P. aeruginosa PAP-I (locus tag: RL047) [65]. Interestingly, the degraded GI-T4SS region encoded in GI-leuX shares ~90% nucleotide identity with GI-T4SS regions within the complete genomes of Klebsiella pneumoniae strains 1084 [GenBank:CP003785] and NTUH-K2044 [GenBank:AP006725], suggesting that GI-leuX belongs to a larger sub-group of uncharacterized ICEs. Like GI-pheV, a GI-leuX was identified in all 47 S. Virchow isolates in this study using PCR (Additional file 6: Table S6); however, further whole-genome sequencing would be required to determine the variability of this region amongst other strains of S. Virchow.
S. Virchow SVQ1 carries a SopE prophage
S. Virchow SVQ1 has three prophage regions encoded on the chromosome, only one of which is intact (Figure 3). Prophage 1 and 2 are incomplete ~8.9 kb and ~21 kb phage remnants, respectively, and prophage 1 is absent from the S. Virchow SL491 genome. Both S. Virchow strains contain an intact prophage which harbors the virulence gene sopE and shares 92–99% nucleotide identity over 67% of the S. Typhimurium Gifsy-1 prophage which carries the GogB Type III secreted effector protein (Figure 5). SopE is a Type III secreted effector protein that induces membrane ruffling and promotes bacterial entry into host cells [66–68]. A previous study has revealed that the sopE gene and 200 bp of flanking sequences (referred to as the sopE cassette) is sporadically distributed on a lambdoid prophage similar to the Gifsy-2 prophage among other Salmonella serovars including S. Gallinarum, S. Dublin and S. Enteritidis and on a non-Gifsy prophage in S. Typhi [69]. It has been proposed that the sopE cassette was transferred between bacteriophage families by homologous recombination [69], a contention that is supported by our observation in S. Virchow. Although the SopE prophage in Virchow is significantly different to the other known SopE prophages in other Salmonella genomes, the 1.2 kb SopE cassette is 97% identical to the cassettes in S. Typhi CT18 and 93% identical to the ones found in S. Gallinarum, S. Dublin and S. Enteritidis. PCR amplification of the sopE gene and across the boundaries of the sopE cassette was used to confirm the presence of this SopE prophage in SVQ1, SL491 and the other 45 S. Virchow isolates (Additional file 6: Table S6). The prevalence of the SopE prophage in the S. Virchow SL491 genome and in all Australian isolates tested suggests that it is a defining feature of S. Virchow.
Figure 5.
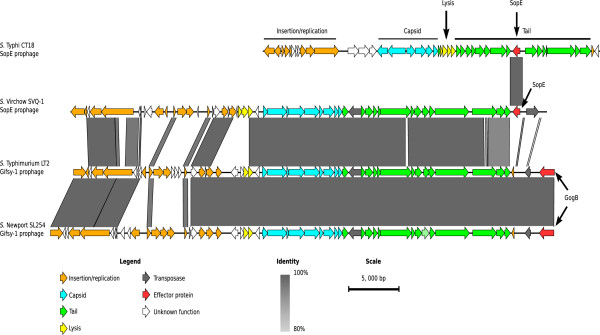
Visual representation of the S. Virchow SVQ1 SopE prophage compared to other prophages. Nucleotide comparison of the SopE prophages from S. Typhi CT18, and S. Virchow SVQ1 and the Gifsy-1 prophages from S. Typhimurium LT2 and S. Newport SL254, respectively. Grey vertical blocks indicate regions of shared similarity shaded according to BLASTn identity. The coloured arrows represent genes. The genes are coloured according to their predicted general functions, which has been inferred from BLAST searches, and are illustrated in the legend. The sopE gene and its conserved flanking sequence, which is called the sopE cassette, is 1.2 kb in length. The percentage identity between the sopE cassette of S. Typhi and S. Virchow is 97%. The image was prepared using Easyfig [50].
Discussion
S. Virchow is of significant public health importance and has a high prevalence in Australia, Asia and Europe [6, 70]. Isolates within this serovar show high levels of genetic relatedness that make discriminating between strains in outbreak investigations difficult [14]. The S. Virchow SVQ1 genome has provided the opportunity to investigate potential targets for sub-typing closely related S. Virchow isolates. Based on our comparisons of the genomes of S. Virchow SVQ1 and S. Virchow SL491 we identified 178 genes that contain SNP or small indel differences between these strains. However, when testing a subset of these candidate targets using a collection of 45 S. Virchow isolates collected from different outbreaks within Australia only one out of the 11 genes tested could discriminate SVQ1 from all other S. Virchow strains in this study. The gene is predicted to encode a pyruvate-flavodoxin oxidoreductase, a conserved housekeeping gene in Salmonella, which we propose could be used as part of a typing scheme to discriminate between S. Virchow PT8 isolates. The lack of other discriminatory SNPs in the set of genes examined is likely due to the selection bias introduced by comparing only two genome sequences. Given the growing availability of high-throughput sequencing technologies we expect that further discriminatory SNPs will be identified by WGS of multiple S. Virchow isolates rather than through PCR-based validation of the remaining candidate genes identified in this study. Furthermore, although routine pathogen surveillance and outbreak investigation will increasingly be carried out by WGS [71], there remains a need for simple molecular genotyping tests.
In this study we found that the CRISPR-1 region could be used to discriminate S. Virchow PT8 isolates. CRISPRs were selected as a genotyping target because they were found to be one of the most rapidly evolving regions in bacterial genomes [72]. CRISPR typing has also been successfully applied to more than nine other Salmonella serovars [32, 33]. Despite sharing nearly all CRISPR-1 spacer sequences in common, three allelic types of CRISPR-1 were observed in the Australian strains including one associated with PT17 strains. However, the different alleles of CRISPR-1 are caused by deletions of single spacers rather than the acquisition of new spacers. In contrast, CRISPR-1 from S. Virchow SL491 contains 34 spacers not found in Australian isolates suggesting evolutionary differences. Even though CRISPRs are considered to be rapidly evolving elements both CRISPR-1 and CRISPR-2 in the Australian S. Virchow strains have not accumulated new spacer sequences over a seven year period, suggesting that the US strain S. Virchow SL491 has been exposed to a greater variety of mobile DNA.
There is sufficient variation within CRISPR-1 that it can be used to discriminate between closely related S. Virchow strains. Despite their repetitive nature, CRISPRs can also be compared using WGS data, as the repeat units in the CRISPRs are short (only about 30 bp long) and are separated by variable, non-repetitive spacer sequences. Most currently available sequencers can produce reads long enough to span these repeat units and overlap with the spacers allowing for correct assembly, as has been recently demonstrated in a study of 102 newly sequenced S. enterica genomes [38, 47]. Due to relatively large size of the CRISPR-1, using Sanger sequencing to sequence the whole region is time-consuming, however, we have observed three allelic types of CRISPR-1 in local S. Virchow strains that differ by the deletion of a single spacer sequence. Therefore, it a PCR based assay to determine the presence/absence of a specific spacer sequence by designing primers that bind to conserved spacers that flanks a deletion site.
Genomic comparisons between S. Virchow SVQ1 and S. Virchow SL491 revealed that lateral gene transfer is the major contributor for variation in the chromosome, as for other enteric bacteria. Excluding plasmids, 0.2% of SVQ1 genome is not shared with SL491. Conversely, 4.1% of the SL491 genome is not present in SVQ1. The bulk of the non-shared DNA in SL491 is associated with prophage regions, which are absent in SVQ1. This is a common theme in Salmonella, as prophages are known to contribute significantly to variation in strains of the same serovar [73]. Comparisons with other published Salmonella genomes revealed several regions of difference in S. Virchow genomes, including genomic islands located within regions in the chromosome that are common DNA integration sites in other serovars. For example, tRNA-leuX is a region of the chromosome that is often associated with foreign DNA in other Salmonella and E. coli strains [74]. In S. Virchow, the GI-leuX appears to encode the remnants of an integrative conjugative element that is distinct from other well-characterized ICE representatives, including the Salmonella SPI-7 family [75]. Although the degradation of the GI-T4SS conjugal transfer region indicates that the S. Virchow GI-leuX is no longer self-transmissible, the island is present in all 47 S. Virchow isolates tested in this study suggesting that there may be a selective advantage to retaining one or more of the encoded cargo genes.
Salmonella employs the SPI-1 Type III secretion system to translocate effector proteins into host cells [76]. These effectors then manipulate host cellular function to enhance the invasiveness and survival of Salmonella. SopE is an effector that is responsible for entry into epithelial cells by inducing cytoskeleton rearrangement and membrane ruffling causing the membrane of the cell to wrap around and engulf the bacterium, a process called macropinocytosis [66, 68, 77]. Knocking out the SopE effector in S. Dublin prevented invasion and attenuated disease [78]. Thus, it is believed that the acquisition of the sopE gene was an important step in the emergence of epidemic Salmonella serovars [79, 80]. Here we report that S. Virchow encodes SopE on a Gifsy-1-like prophage that is dissimilar to the SopE prophages found in other Salmonella serovars. The presence of this virulence factor in different bacteriophages might increase the efficiency of horizontal transfer of sopE between different strains by increasing the host range and helping to evade immunity imposed by other resident prophages and CRISPRs [69].
Conclusions
We have undertaken a comparative analysis of the S. Virchow SVQ1 genome and identified several genomic islands, prophages and other regions of difference that are characteristic of S. Virchow. We have demonstrated that Sesv_1374 and the CRISPR-1 region are genotyping targets that can discriminate between closely related S. Virchow isolates of the same phage type. The genotyping targets described in this study could be used in conjunction with other Salmonella genotyping targets to provide enhanced resolution of S. Virchow strains involved in different outbreaks. Additional genome sequencing of S. Virchow strains will help to evaluate the effectiveness of CRISPR typing for outbreak investigations and identify other potential genotyping targets. Considering S. Virchow’s public health importance as a human pathogen, the availability of the S. Virchow SVQ1 genome is a vital step for understanding the evolution and global distribution of this serovar and the mechanisms in which it causes invasive infections.
Methods
Bacterial strains
The strain sequenced in this study, S. Virchow strain SVQ1 (phage type 8) is a clinical isolate obtained in 2007. The strain was isolated from an outbreak in Queensland, Australia [9]. S. Virchow SL491, for which a genome sequence is available [GenBank:ABFH00000000.2], was included in this study and was phage typed by the Microbiological Diagnostic Unit (MDU), Victoria. S. Virchow SL491 was isolated in 2005 from a patient in the USA, however, prior to onset of illness the patient had visited India [38]. A total of 45 other S. Virchow isolates belonging to four phage types (PT8, PT17, PT25 and PT34) from various locations in Australia were also in this study (see Table 1).
Whole genome sequencing, assembly and annotation
The genome of S. Virchow SVQ1 (PT8) was sequenced using Roche 454 GS-FLX (Australian Genome Research Facility, Brisbane, Australia) producing 340,790 single-end shotgun reads of an average length of 240 bp. The genome was assembled using 454/Roche gsAssembler 2.3.1 (Newbler) into 54 contigs between 293 bp and 432,538 bp in length (N50 contig size, 205,097 bp) with an average 17-fold read coverage depth. Contig scaffolds were built and ordered based on an optical map (Opgen Inc, Gaitherburg MD, 20878) that was generated for the genome [81, 82]. The optical map was also used to check for misassemblies or genome rearrangements and to confirm contig order. Consed [83] was used to check the underlying reads to determine any collapsed repeats that separate adjacent contigs. This approach allowed us to assemble 46 of the 54 contigs into 10 scaffolds that were ordered according to the S. Virchow SL491 genome. The remaining eight unscaffolded contigs corresponded to four plasmids and collapsed repeat contigs that encode rRNA operons, respectively. BLAST comparison of all S. Virchow SVQ1 contigs with S. Virchow SL491 identified scaffold gaps corresponding to each of the 7 rRNA operons in S. Virchow SL491. Examination of paired-end read location from edge of each contig gap suggested that like S. Virchow SL491, S. Virchow SVQ1 encodes 7 rRNA operons. The draft genome was automatically annotated using SUGAR (Simple Unfinished Genome Annotation Resource) as previously described [84]. Automatic annotation was carried out using BLASTp [49] in a hierarchical approach that prioritised a high-quality manually curated annotations by using a diminishing BLASTp identity thresholds against databases comprising proteins from i) Salmonella Typhi str. CT18 genome [GenBank:AL513382] [41], ii) all Salmonella genomes iii) swiss-prot or iv) uniprot. tRNA genes were predicted using TE-SCAN [85]. Subsequent manual annotation of genomic islands, prophage and CRISPR sequences was carried out using Artemis [86] and the results of Pfam [87], TIGRfam [88] and COGs [89] searches. Prophages were also characterized using the PHAST phage annotation server [90]. This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession [GenBank:AZMP00000000] (Bioproject: PRJNA178788). The version described in this paper is version AZMP01000000.
Variant prediction
The draft genome of S. Virchow SVQ1 (PT8) was compared to the previously published draft genome of S. Virchow SL491 (PT25) to identify genes with at least one single nucleotide polymorphism (SNP) that may be suitable genotyping markers. The MUMmer package [91] was used to align the contigs from the genome of S. Virchow SVQ1 to the genome sequence of S. Virchow SL491 and identify indel and SNP variants. This approach was also used to predict SNPs between S. Virchow SVQ1 and S. Heidelberg SL497 [GenBank:CP001120] [92]. A custom Perl script was used to remove any SNPs inside or flanking homopolymer tracts of longer than four nucleotides, as errors in base calling can occur at homopolymeric tracts with 454 sequencing [93]. A final filter step removed SNPs with a read coverage of less than five reads or which were located within 10 nucleotides of contig ends.
PCR amplification and sequencing
Polymerase Chain Reaction (PCR) was used to amplify 11 genes predicted to contain SNPs in 47 S. Virchow isolates including the sequenced strains, S. Virchow SVQ1 and S. Virchow SL491. PCR was used also used to validate the presence of the SopE prophage and selected genomic islands in local S. Virchow isolates by amplifying regions within each island and the boundaries at both ends. The CRISPR-1 region was also amplified from 15 strains and were sequenced both forward and reverse using Big Dye V3.1 Sequencing Kits (Applied Biosystems, Life Technologies) and analyzed on the ABI 3130 Sequencer (Applied Biosystems, Life Technologies, Australia). The primers for amplifying CRISPR-1 were designed to bind to the location 5′ and 3′ outside of the CRISPR loci and to conserved spacers between the two S. Virchow genomes. Primers used in this study are listed in Table 4.
Table 4.
List of primers used to validate genotyping target and genomic features in S. Virchow
| Name | Primer sequence | Length (bp) | Direction | Product size (bp) | Target |
|---|---|---|---|---|---|
| SopE-A/F | GAGTCGGCATAGCACACTCA | 20 | Forward | 474 | SopE (Sesv_0764) |
| SopE-A/R | CAACACACTTTCACCGAGGA | 20 | Reverse | ||
| SopE-B/F | GGCGTGGGAAAGTTTCAGTA | 20 | Forward | 1328 | SopE cassette (3′ region) |
| SopE-B/R | ATGACGTTTTTACGCCAAGC | 20 | Reverse | ||
| SopE-C/F | CGGGGTCTTTACTCGCACTA | 20 | Forward | 923 | SopE cassette (5′ region) |
| SopE-C/R | CACTCAACCACCACAACAGG | 20 | Reverse | ||
| leuX-A/F | TTAAATGTGGCGAACAGCAG | 20 | Forward | 2239 | GI-leuX (internal) |
| leuX-A/R | AGTGCCCGGAAAGAAACTCT | 20 | Reverse | ||
| leuX-B/F | CGGACGCCATATCCATATTC | 20 | Forward | 1120 | GI-leuX (5′ boundary) |
| leuX-B/R | CCTGAATACTGGTCGGGAAA | 20 | Reverse | ||
| leuX-C/F | GTAGATTGGCAACCGAAAGG | 20 | Forward | 876 | GI-leuX (3′ boundary) |
| leuX-C/R | GAGATGAAACGTTCGTGCAA | 20 | Reverse | ||
| pheV-A/F | GCGGCAAGGTAAAATGTGTT | 20 | Forward | 1687 | GI-pheV (internal) |
| pheV-A/R | GGTGATTTACGTGCGGTCTT | 20 | Reverse | ||
| pheV-B/F | TTCTGCTGGTGATGAAGTGC | 20 | Forward | 1138 | GI-pheV (5′ boundary) |
| pheV-B/R | TCCAGATATGGGCTTTCAGG | 20 | Reverse | ||
| pheV-C/F | GATAGTTTCCGCCACCTGAA | 20 | Forward | 1337 | GI-pheV (3′ boundary) |
| pheV-C/R | GAGAGAACTGGAGCCACAGG | 20 | Reverse | ||
| SV-0065-F | GCAGAAAGCCTGTCAGGAAC | 20 | Forward | 856 | Sesv_0065 |
| SV-0065-R | CACCGGGTTAAAAGGGATCT | 20 | Reverse | ||
| SV-1374-F | TTTTACGGTCTGGGAAGCGAC | 21 | Forward | 623 | Sesv_1374 |
| SV-1374-R | TATGCGGATTAACCGCCTGC | 20 | Reverse | ||
| SV-0106-F | GGGCCTGCATTTCTTGTCTA | 20 | Forward | 935 | Sesv_0106 |
| SV-0106-R | GCCCTTTCTGGATAAGACGA | 20 | Reverse | ||
| SV-0279-F | CGCAGGTACGCGTGTTATTA | 20 | Forward | 814 | Sesv_0279 |
| SV-0279-R | CCGTCGGTGATATTTTCCAC | 20 | Reverse | ||
| SV-0317-F | GCGCTTAGTCGGCTATTGAC | 20 | Forward | 805 | Sesv_0317 |
| SV-0317-R | TACAACCGAATTCACGGACA | 20 | Reverse | ||
| SV-1243-F | GTTTTGCTGGTTTGGCATTTG | 21 | Forward | 742 | Sesv_1243 |
| SV-1243-R | GTCGAACGAACCCAGTCCATG | 21 | Reverse | ||
| SV-1046-F | GTATGGCGGCAATCATCGTTG | 21 | Forward | 804 | Sesv_1046 |
| SV-1046-R | CCTCCTTGAGGACAGCCAACG | 21 | Reverse | ||
| SV-1509-F | CCAACCGCCTGTACACTTCT | 20 | Forward | 720 | Sesv_1509 |
| SV-1509-R | TCGCAGACAACGACTTCATC | 20 | Reverse | ||
| SV-0512-F | GAAGGTGTACCCGCCAGATA | 20 | Forward | 714 | Sesv_0512 |
| SV-0512-R | GGTGGTAACGCTGATGGACT | 20 | Reverse | ||
| SV-1129-F | CGTTGTTAAATGCGTGGTTG | 20 | Forward | 987 | Sesv_1129 |
| SV-1129-R | GGCTGGTAACGACTGGAAAA | 20 | Reverse | ||
| SV-0619-F | TTTCACCGATGAACCCGTGAC | 21 | Forward | 760 | Sesv_0619 |
| SV-0619-R | CGACGGATATGATCGCTCCAG | 21 | Reverse | ||
| C1-F1 | GATGTAGTGCGGATAATGCT | 20 | Forward | 1405 | CRISPR-1 |
| C1-R1 | CTCATCTCCCCAGATTTTTG | 20 | Reverse | ||
| C1-F2 | CGTAACGTTTAAGCGTGGAAAG | 22 | Forward | 399 | CRISPR-1 |
| C1-R2 | CGCTTACGATACAATGATGGTC | 22 | Reverse | ||
| C1-F3 | CAGTCACAATCTTTTGCGGC | 20 | Forward | 1497 | CRISPR-1 |
| C1-R3 | GTTTCTTTTCTTCCTGTTG | 19 | Reverse | ||
| C1-F4 | TCCCACTTATCAAATTTAGCC | 21 | Forward | 578 | CRISPR-1 |
| C1-R4 | GCCATCGTAGCGGATTTCAGA | 21 | Reverse |
Bioinformatics analysis
Pairwise whole genome comparisons of S. Virchow SVQ1 with 27 Salmonella genomes (Table 3) were performed using BLASTn and visualized using the Artemis Comparison Tool [94]. Circular visualization figures were made using BRIG (BLAST Ring Image Generator) [59] and linear visualization figures were made using Easyfig [50]. CRISPR amplicon sequences were assembled using CLC Genomic Workbench (http://www.clcbio.com/). Similarity searches of the non-redundant nucleotide database and whole-genome shotgun contigs were carried out using the NCBI BLAST portal. The absence in S. Virchow SVQ1 of genomic regions present in S. Virchow SL491 was confirmed by mapping the 454 reads against the S. Virchow SL491 genome as a reference. Prior to mapping, the quality of the 454 reads was checked with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads that were shorter than 200 bp were removed and the remaining reads were trimmed by 10 nucleotide from the 5′ end and 30 nucleotide from the 3 end using PrinSeq-Lite [56]. Read mapping was performed using BWA-SW (Smith Waterman) [57] with default parameters.
Electronic supplementary material
Additional file 1: Table S1: List of plasmids in the genome of S. Virchow SVQ1. (XLSX 37 KB)
Additional file 2: Table S2: List of protein coding sequences that contain at least one SNP or indel between SVQ1 and SL491. (XLSX 52 KB)
Additional file 3: Table S3: List of Single nucloetide polymorphisms test. (XLSX 41 KB)
Additional file 4: Table S4: Salmonella Pathogenicity Island in S. Virchow SVQ1. (XLSX 39 KB)
Additional file 5: Table S5: Regions of differences identified in S. Virchow SVQ1. (XLSX 50 KB)
Additional file 6: Table S6: PCR results of S. Virchow unique regions. (XLSX 52 KB)
Acknowledgements
We acknowledge the support given by the Australian public health reference laboratories (Microbiological Diagnostic Unit, University of Melbourne VIC, IMVS Pathology SA and Path West Laboratory Medicine WA) for provision of requested Salmonella Virchow outbreak isolates and Dr K. E. Sanderson, SGSC, University of Calgary for the S. Virchow SL491 isolates. Queensland Health Scientific Services for funding the whole genome sequencing of S. Virchow SVQ1 and molecular microbiology laboratory work, as well as the provision of isolates. This work was supported by project grants 511224 and 566883 from the Australian National Health and Medical Research Council. We acknowledge the support provided by S. Virchow MLST data are publicly available at http://mlst.ucc.ie, which is currently supported by a grant from the Science Foundation of Ireland (05/FE1/B882). We thank Dr Jayde Gawthorne for providing a critical review of the manuscript.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SAB and JRS conceived the study; NLB and SAB carried out the comparative genomics analyses; NLB and JRS carried out the molecular genetics analyses; JMS developed and used the auto-annotation and SNP-calling software; SAB, JRS and NKP participated in the design and coordination of the study; NLB, SAB, JRS, NKP and NBZ participated in the analysis of the data and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Nathan L Bachmann, Email: nathan.bachmann@uqconnect.edu.au.
Nicola K Petty, Email: nicola.petty@uts.edu.au.
Nouri L Ben Zakour, Email: n.benzakour@uq.edu.au.
Jan M Szubert, Email: marek.szubert@gmail.com.
John Savill, Email: johnr_savill@health.qld.gov.au.
Scott A Beatson, Email: s.beatson@uq.edu.au.
References
- 1.Ashdown LR, Ryan PJ. Invasive disease due to Salmonella virchow: a North Queensland problem. Med J Aust. 1990;153:330–335. doi: 10.5694/j.1326-5377.1990.tb136941.x. [DOI] [PubMed] [Google Scholar]
- 2.Bennett CM, Dalton C, Beers-Deeble M, Milazzo A, Kraa E, Davos D, Puech M, Tan A, Heuzenroeder MW. Fresh garlic: a possible vehicle for Salmonella Virchow. Epidemiol Infect. 2003;131(3):1041–1048. doi: 10.1017/S0950268803001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matheson N, Kingsley RA, Sturgess K, Aliyu SH, Wain J, Dougan G, Cooke FJ. Ten years experience of Salmonella infections in Cambridge, UK. J Infect. 2010;60(1):21–25. doi: 10.1016/j.jinf.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 4.Salisbury AM, Bronowski C, Wigley P. Salmonella Virchow isolates from human and avian origins in England - molecular characterization and infection of epithelial cells and poultry. J Appl Microbiol. 2011;111(6):1505–1514. doi: 10.1111/j.1365-2672.2011.05152.x. [DOI] [PubMed] [Google Scholar]
- 5.Folster JP, Rickert R, Barzilay EJ, Whichard JM. Identification of the aminoglycoside resistance determinants armA and rmtC among non-Typhi Salmonella isolates from humans in the United States. Antimicrob Agents Chemother. 2009;53(10):4563–4564. doi: 10.1128/AAC.00656-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ispahani P, Slack RC. Enteric fever and other extraintestinal salmonellosis in University Hospital, Nottingham, UK, between 1980 and 1997. Eur J Clin Microbiol Infect Dis. 2000;19(9):679–687. doi: 10.1007/s100960000341. [DOI] [PubMed] [Google Scholar]
- 7.Hendriksen RS, Vieira AR, Karlsmose S, Lo Fo Wong DM, Jensen AB, Wegener HC, Aarestrup FM. Global monitoring of Salmonella serovar distribution from the World Health Organization Global Foodborne Infections Network Country Data Bank: results of quality assured laboratories from 2001 to 2007. Foodborne Pathog Dis. 2011;8(8):887–900. doi: 10.1089/fpd.2010.0787. [DOI] [PubMed] [Google Scholar]
- 8.Galanis E, Lo Fo Wong DM, Patrick ME, Binsztein N, Cieslik A, Chalermchikit T, Aidara-Kane A, Ellis A, Angulo FJ, Wegener HC. Web-based surveillance and global Salmonella distribution, 2000–2002. Emerg Infect Dis. 2006;12(3):381–388. doi: 10.3201/eid1205.050854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.OzFoodNet OzFoodNet Quarterly report, 1 April to 30 June 2007. Communciable Dis Intell. 2007;31(3):314–318. doi: 10.33321/cdi.2007.31.35. [DOI] [PubMed] [Google Scholar]
- 10.Powling J. National Enteric Pathogens Surveillance Scheme, vol. Human Annual Report 2010. Victoria, Australia: Microbiological Diagnostic Unit, The University of Melbourne; 2010. [Google Scholar]
- 11.Ward LR, de Sa JD, Rowe B. A phage-typing scheme for Salmonella enteritidis. Epidemiol Infect. 1987;99(2):291–294. doi: 10.1017/S0950268800067765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chambers RM, Mcadam P, Desa JDH, Ward LR, Rowe B. A phage-typing scheme for Salmonella-Virchow. FEMS Microbiol Lett. 1987;40(2–3):155–157. doi: 10.1111/j.1574-6968.1987.tb02016.x. [DOI] [Google Scholar]
- 13.Baggesen DL, Sorensen G, Nielsen EM, Wegener HC. Phage typing of Salmonella Typhimurium - is it still a useful tool for surveillance and outbreak investigation? Euro Surveillance. 2010;15(4):19471. [PubMed] [Google Scholar]
- 14.Kerouanton A, Marault M, Lailler R, Weill FX, Feurer C, Espie E, Brisabois A. Pulsed-field gel electrophoresis subtyping database for foodborne Salmonella enterica serotype discrimination. Foodborne Pathog Dis. 2007;4(3):293–303. doi: 10.1089/fpd.2007.0090. [DOI] [PubMed] [Google Scholar]
- 15.Gerner-Smidt P, Hise K, Kincaid J, Hunter S, Rolando S, Hyytia-Trees E, Ribot EM, Swaminathan B. PulseNet USA: a five-year update. Foodborne Pathog Dis. 2006;3(1):9–19. doi: 10.1089/fpd.2006.3.9. [DOI] [PubMed] [Google Scholar]
- 16.Cooke FJ, Wain J, Fookes M, Ivens A, Thomson N, Brown DJ, Threlfall EJ, Gunn G, Foster G, Dougan G. Prophage sequences defining hot spots of genome variation in Salmonella enterica serovar typhimurium can be used to discriminate between field isolates. J Clin Microbiol. 2007;45(8):2590–2598. doi: 10.1128/JCM.00729-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torpdahl M, Sorensen G, Lindstedt BA, Nielsen EM. Tandem repeat analysis for surveillance of human Salmonella Typhimurium infections. Emerg Infect Dis. 2007;13(3):388–395. doi: 10.3201/eid1303.060460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sintchenko V, Wang Q, Howard P, Ha CW, Kardamanidis K, Musto J, Gilbert GL. Improving resolution of public health surveillance for human Salmonella enterica serovar Typhimurium infection: 3 years of prospective multiple-locus variable-number tandem-repeat analysis (MLVA) BMC Infect Dis. 2012;12:78. doi: 10.1186/1471-2334-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergamini F, Iori A, Massi P, Pongolini S. Multilocus variable-number of tandem-repeats analysis of Salmonella enterica serotype Gallinarum and comparison with pulsed-field gel electrophoresis genotyping. Vet Microbiol. 2011;149(3–4):430–436. doi: 10.1016/j.vetmic.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Fakhr MK, Nolan LK, Logue CM. Multilocus sequence typing lacks the discriminatory ability of pulsed-field gel electrophoresis for typing Salmonella enterica serovar Typhimurium. J Clin Microbiol. 2005;43(5):2215–2219. doi: 10.1128/JCM.43.5.2215-2219.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sukhnanand S, Alcaine S, Warnick LD, Su WL, Hof J, Craver MP, McDonough P, Boor KJ, Wiedmann M. DNA sequence-based subtyping and evolutionary analysis of selected Salmonella enterica serotypes. J Clin Microbiol. 2005;43(8):3688–3698. doi: 10.1128/JCM.43.8.3688-3698.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kotetishvili M, Stine OC, Kreger A, Morris JG, Jr, Sulakvelidze A. Multilocus sequence typing for characterization of clinical and environmental salmonella strains. J Clin Microbiol. 2002;40(5):1626–1635. doi: 10.1128/JCM.40.5.1626-1635.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tankouo-Sandjong B, Sessitsch A, Liebana E, Kornschober C, Allerberger F, Hachler H, Bodrossy L. MLST-v, Multilocus sequence typing based on virulence genes, for molecule typing of Salmonella enterica subsp enterica serovars. J Microbiol Methods. 2007;69:23–36. doi: 10.1016/j.mimet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 24.Kingsley RA, Msefula CL, Thomson NR, Kariuki S, Holt KE, Gordon MA, Harris D, Clarke L, Whitehead S, Sangal V, Marsh K, Achtman M, Molyneux ME, Cormican M, Parkhill J, Maclenna CA, Heyderman RS, Dougan G. Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 2009;19(12):2279–2287. doi: 10.1101/gr.091017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pang S, Octavia S, Feng L, Liu B, Reeves PR, Lan R, Wang L. Genomic diversity and adaptation of Salmonella enterica serovar Typhimurium from analysis of six genomes of different phage types. BMC Genomics. 2013;14(1):718. doi: 10.1186/1471-2164-14-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindstedt BA, Torpdahl M, Nielsen EM, Vardund T, Aas L, Kapperud G. Harmonization of the multiple-locus variable-number tandem repeat analysis method between Denmark and Norway for typing Salmonella Typhimurium isolates and closer examination of the VNTR loci. J Appl Microbiol. 2007;102(3):728–735. doi: 10.1111/j.1365-2672.2006.03134.x. [DOI] [PubMed] [Google Scholar]
- 27.Lindstedt BA, Vardund T, Aas L, Kapperud G. Multiple-locus variable-number tandem-repeats analysis of Salmonella enterica subsp. enterica serovar Typhimurium using PCR multiplexing and multicolor capillary electrophoresis. J Microbiol Methods. 2004;59(2):163–172. doi: 10.1016/j.mimet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Pang S, Octavia S, Reeves PR, Wang Q, Gilbert GL, Sintchenko V, Lan R. Genetic relationships of phage types and single nucleotide polymorphism typing of salmonella enterica serovar typhimurium. J Clin Microbiol. 2012;50(3):727–734. doi: 10.1128/JCM.01284-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guard J, Morales CA, Fedorka-Cray P, Gast RK. Single nucleotide polymorphisms that differentiate two subpopulations of Salmonella enteritidis within phage type. BMC Res Notes. 2011;4:369. doi: 10.1186/1756-0500-4-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holt KE, Dutta S, Manna B, Bhattacharya SK, Bhaduri B, Pickard DJ, Ochiai RL, Ali M, Clemens JD, Dougan G. High-resolution genotyping of the endemic Salmonella Typhi population during a Vi (typhoid) vaccination trial in Kolkata. PLoS Negl Trop Dis. 2012;6(1):e1490. doi: 10.1371/journal.pntd.0001490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Achtman M, Wain J, Weill FX, Nair S, Zhou Z, Sangal V, Krauland MG, Hale JL, Harbottle H, Uesbeck A, Dougan G, Harrison LH, Brisse S. Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 2012;8(6):e1002776. doi: 10.1371/journal.ppat.1002776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu F, Barrangou R, Gerner-Smidt P, Ribot EM, Knabel SJ, Dudley EG. Novel virulence gene and CRISPR multilocus sequence typing scheme for subtyping the major serovars of Salmonella enterica subspecies enterica. Appl Environ Microbiol. 2011;77(6):1946–1956. doi: 10.1128/AEM.02625-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu F, Kariyawasam S, Jayarao BM, Barrangou R, Gerner-Smidt P, Ribot EM, Knabel SJ, Dudley EG. Subtyping Salmonella serovar Enteritidis isolates from different sources using sequence typing based on virulence genes and CRISPRs. Appl Environ Microbiol. 2011;77(13):4520–4526. doi: 10.1128/AEM.00468-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fabre L, Zhang J, Guigon G, Le Hello S, Guibert V, Accou-Demartin M, de Romans S, Lim C, Roux C, Passet V, Diancourt L, Guibourdenche M, Issenhuth-Jeanjean S, Achtman M, Brisse S, Sola C, Weill FX. CRISPR typing and subtyping for improved laboratory surveillance of salmonella infections. PLoS One. 2012;7(5):e36995. doi: 10.1371/journal.pone.0036995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vale PF, Little TJ. CRISPR-mediated phage resistance and the ghost of coevolution past. Proc Biol Sci. 2010;277(1691):2097–2103. doi: 10.1098/rspb.2010.0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinforma. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fricke WF, Mammel MK, McDermott PF, Tartera C, White DG, Leclerc JE, Ravel J, Cebula TA. Comparative genomics of 28 salmonella enterica isolates: evidence for CRISPR-mediated adaptive sublineage evolution. J Bacteriol. 2011;193(14):3556–3568. doi: 10.1128/JB.00297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang NX, Huang B, Hiley L, Bates J, Savill J. A rapid multiplex DNA suspension array method for Salmonella typhimurium subtyping using prophage-related markers. J Microbiol Methods. 2012;88(1):19–27. doi: 10.1016/j.mimet.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 40.Young CC, Ross IL, Heuzenroeder MW. A new methodology for differentiation and typing of closely related Salmonella enterica serovar Heidelberg isolates. Curr Microbiol. 2012;65(5):481–487. doi: 10.1007/s00284-012-0179-3. [DOI] [PubMed] [Google Scholar]
- 41.Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, Churcher C, Mungall KL, Bentley SD, Holden MT, Sebaihia M, Baker S, Basham D, Brooks K, Chillingworth T, Connerton P, Cronin A, Davis P, Davies RM, Dowd L, White N, Farrar J, Feltwell T, Hamlin N, Haque A, Hien TT, Holroyd S, Jagels K, Krogh A, Larsen TS, et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature. 2001;413(6858):848–852. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- 42.Thomson N, Baker S, Pickard D, Fookes M, Anjum M, Hamlin N, Wain J, House D, Bhutta Z, Chan K, Falkow S, Parkhill J, Woodward M, Ivens A, Dougan G. The role of prophage-like elements in the diversity of Salmonella enterica serovars. J Mol Biol. 2004;339(2):279–300. doi: 10.1016/j.jmb.2004.03.058. [DOI] [PubMed] [Google Scholar]
- 43.Doublet B, Boyd D, Mulvey MR, Cloeckaert A. The Salmonella genomic island 1 is an integrative mobilizable element. Mol Microbiol. 2005;55(6):1911–1924. doi: 10.1111/j.1365-2958.2005.04520.x. [DOI] [PubMed] [Google Scholar]
- 44.Rychlik I, Gregorova D, Hradecka H. Distribution and function of plasmids in Salmonella enterica. Vet Microbiol. 2006;112(1):1–10. doi: 10.1016/j.vetmic.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 45.Mills DM, Bajaj V, Lee CA. A 40 kb chromosomal fragment encoding Salmonella typhimurium invasion genes is absent from the corresponding region of the Escherichia coli K-12 chromosome. Mol Microbiol. 1995;15(4):749–759. doi: 10.1111/j.1365-2958.1995.tb02382.x. [DOI] [PubMed] [Google Scholar]
- 46.Ehrbar K, Hardt WD. Bacteriophage-encoded type III effectors in Salmonella enterica subspecies 1 serovar Typhimurium. Infect Genet Evol. 2005;5(1):1–9. doi: 10.1016/j.meegid.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 47.Timme RE, Pettengill JB, Allard MW, Strain E, Barrangou R, Wehnes C, Van Kessel JS, Karns JS, Musser SM, Brown EW. Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp. enterica inferred from genome-wide reference-free SNP characters. Genome Biol Evol. 2013;5(11):2109–2123. doi: 10.1093/gbe/evt159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chu C, Feng Y, Chien AC, Hu S, Chu CH, Chiu CH. Evolution of genes on the Salmonella Virulence plasmid phylogeny revealed from sequencing of the virulence plasmids of S. enterica serotype Dublin and comparative analysis. Genomics. 2008;92(5):339–343. doi: 10.1016/j.ygeno.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 49.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 50.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Folkesson A, Advani A, Sukupolvi S, Pfeifer JD, Normark S, Lofdahl S. Multiple insertions of fimbrial operons correlate with the evolution of Salmonella serovars responsible for human disease. Mol Microbiol. 1999;33(3):612–622. doi: 10.1046/j.1365-2958.1999.01508.x. [DOI] [PubMed] [Google Scholar]
- 52.Lambert MA, Smith SG. The PagN protein mediates invasion via interaction with proteoglycan. FEMS Microbiol Lett. 2009;297(2):209–216. doi: 10.1111/j.1574-6968.2009.01666.x. [DOI] [PubMed] [Google Scholar]
- 53.McClelland M, Sanderson KE, Clifton SW, Latreille P, Porwollik S, Sabo A, Meyer R, Bieri T, Ozersky P, McLellan M, Harkins CR, Wang C, Nguyen C, Berghoff A, Elliott G, Kohlberg S, Strong C, Du F, Carter J, Kremizki C, Layman D, Leonard S, Sun H, Fulton L, Nash W, Miner T, Minx P, Delehaunty K, Fronick C, Magrini V, et al. Comparison of genome degradation in Paratyphi A and Typhi, human-restricted serovars of Salmonella enterica that cause typhoid. Nat Genet. 2004;36(12):1268–1274. doi: 10.1038/ng1470. [DOI] [PubMed] [Google Scholar]
- 54.Luo Y, Kong Q, Yang J, Golden G, Wanda SY, Jensen RV, Ernst PB, Curtiss R., 3rd Complete Genome Sequence of the Universal Killer Salmonella enterica Serovar Typhimurium UK-1 (ATCC 68169) J Bacteriol. 2011;193(15):4035–4036. doi: 10.1128/JB.05224-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chiu CH, Tang P, Chu C, Hu S, Bao Q, Yu J, Chou YY, Wang HS, Lee YS. The genome sequence of Salmonella enterica serovar Choleraesuis, a highly invasive and resistant zoonotic pathogen. Nucleic Acids Res. 2005;33(A-5):1690–1698. doi: 10.1093/nar/gki297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu WQ, Feng Y, Wang Y, Zou QH, Chen F, Guo JT, Peng YH, Jin Y, Li YG, Hu SN, Johnston RN, Liu GR, Liu SL. Salmonella paratyphi C: genetic divergence from Salmonella choleraesuis and pathogenic convergence with Salmonella typhi. PLoS One. 2009;4(2):e4510. doi: 10.1371/journal.pone.0004510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomson NR, Clayton DJ, Windhorst D, Vernikos G, Davidson S, Churcher C, Quail MA, Stevens M, Jones MA, Watson M, Barron A, Layton A, Pickard D, Kingsley RA, Bignell A, Clark L, Harris B, Ormond D, Abdellah Z, Brooks K, Cherevach I, Chillingworth T, Woodward J, Norberczak H, Lord A, Arrowsmith C, Jagels K, Moule S, Mungall K, Sanders M, et al. Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res. 2008;18(10):1624–1637. doi: 10.1101/gr.077404.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deng W, Liou SR, Plunkett G, 3rd, Mayhew GF, Rose DJ, Burland V, Kodoyianni V, Schwartz DC, Blattner FR. Comparative genomics of Salmonella enterica serovar Typhi strains Ty2 and CT18. J Bacteriol. 2003;185(7):2330–2337. doi: 10.1128/JB.185.7.2330-2337.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12(1):402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shah DH, Lee MJ, Park JH, Lee JH, Eo SK, Kwon JT, Chae JS. Identification of Salmonella gallinarum virulence genes in a chicken infection model using PCR-based signature-tagged mutagenesis. Microbiology. 2005;151(Pt 12):3957–3968. doi: 10.1099/mic.0.28126-0. [DOI] [PubMed] [Google Scholar]
- 61.Shi L, Adkins JN, Coleman JR, Schepmoes AA, Dohnkova A, Mottaz HM, Norbeck AD, Purvine SO, Manes NP, Smallwood HS, Wang H, Forbes J, Gros P, Uzzau S, Rodland KD, Heffron F, Smith RD, Squier TC. Proteomic analysis of Salmonella enterica serovar typhimurium isolated from RAW 264.7 macrophages: identification of a novel protein that contributes to the replication of serovar typhimurium inside macrophages. J Biol Chem. 2006;281(39):29131–29140. doi: 10.1074/jbc.M604640200. [DOI] [PubMed] [Google Scholar]
- 62.Wee BA, Woolfit M, Beatson SA, Petty NK. A distinct and divergent lineage of genomic island-associated type IV secretion systems in Legionella. PLoS One. 2013;8(12):e82221. doi: 10.1371/journal.pone.0082221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baker S, Pickard D, Whitehead S, Farrar J, Dougan G. Mobilization of the incQ plasmid R300B with a chromosomal conjugation system in Salmonella enterica serovar typhi. J Bacteriol. 2008;190(11):4084–4087. doi: 10.1128/JB.00065-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mohd-Zain Z, Turner SL, Cerdeno-Tarraga AM, Lilley AK, Inzana TJ, Duncan AJ, Harding RM, Hood DW, Peto TE, Crook DW. Transferable antibiotic resistance elements in Haemophilus influenzae share a common evolutionary origin with a diverse family of syntenic genomic islands. J Bacteriol. 2004;186(23):8114–8122. doi: 10.1128/JB.186.23.8114-8122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klockgether J, Wurdemann D, Reva O, Wiehlmann L, Tummler B. Diversity of the abundant pKLC102/PAGI-2 family of genomic islands in Pseudomonas aeruginosa. J Bacteriol. 2007;189(6):2443–2459. doi: 10.1128/JB.01688-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Galan JE, Zhou D. Striking a balance: modulation of the actin cytoskeleton by Salmonella. Proc Natl Acad Sci U S A. 2000;97(16):8754–8761. doi: 10.1073/pnas.97.16.8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rahman H, Hardt WD, Murugkar HV, Bhattacharyya DK. Occurrence of sopE gene and its phenotypic expression among different serovars of Salmonella enterica isolated from man and animals. Indian J Exp Biol. 2005;43(7):631–634. [PubMed] [Google Scholar]
- 68.Humphreys D, Davidson A, Hume PJ, Koronakis V. Salmonella virulence effector SopE and host GEF ARNO cooperate to recruit and activate WAVE to trigger bacterial invasion. Cell Host Microbe. 2012;11(2):129–139. doi: 10.1016/j.chom.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mirold S, Rabsch W, Tschape H, Hardt WD. Transfer of the Salmonella type III effector sopE between unrelated phage families. J Mol Biol. 2001;312(1):7–16. doi: 10.1006/jmbi.2001.4950. [DOI] [PubMed] [Google Scholar]
- 70.Sullivan AM, Ward LR, Rowe B, Woolcock JB, Cox JM. Phage types of Australian isolates of Salmonella enterica subsp. enterica serovar Virchow. Lett Appl Microbiol. 1998;27(4):216–218. doi: 10.1046/j.1472-765X.1998.00421.x. [DOI] [PubMed] [Google Scholar]
- 71.Walker MJ, Beatson SA. Epidemiology. Outsmarting outbreaks. Science. 2012;338(6111):1161–1162. doi: 10.1126/science.1232327. [DOI] [PubMed] [Google Scholar]
- 72.Sorek R, Kunin V, Hugenholtz P. CRISPR - a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6(3):181–186. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- 73.Vernikos GS, Thomson NR, Parkhill J. Genetic flux over time in the Salmonella lineage. Genome Biol. 2007;8(6):R100. doi: 10.1186/gb-2007-8-6-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bishop AL, Baker S, Jenks S, Fookes M, Gaora PO, Pickard D, Anjum M, Farrar J, Hien TT, Ivens A, Dougan G. Analysis of the hypervariable region of the Salmonella enterica genome associated with tRNA(leuX) J Bacteriol. 2005;187(7):2469–2482. doi: 10.1128/JB.187.7.2469-2482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Seth-Smith HM, Fookes MC, Okoro CK, Baker S, Harris SR, Scott P, Pickard D, Quail MA, Churcher C, Sanders M, Harmse J, Dougan G, Parkhill J, Thomson NR. Structure, diversity, and mobility of the Salmonella pathogenicity island 7 family of integrative and conjugative elements within Enterobacteriaceae. J Bacteriol. 2012;194(6):1494–1504. doi: 10.1128/JB.06403-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kubori T, Matsushima Y, Nakamura D, Uralil J, Lara-Tejero M, Sukhan A, Galan JE, Aizawa S. Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science. 1998;280(5363):602–605. doi: 10.1126/science.280.5363.602. [DOI] [PubMed] [Google Scholar]
- 77.Schlumberger MC, Hardt W. Salmonella type 3 secretion effectors: pulling the host cell’s strings. Curr Opin Microbiol. 2006;9:46–54. doi: 10.1016/j.mib.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 78.Wood MW, Rosqvist R, Mullan PB, Edwards MH, Galyov EE. SopE, a secreted protein of Salmonella dublin, is translocated into the target eukaryotic cell via a sip-dependent mechanism and promotes bacterial entry. Mol Microbiol. 1996;22(2):327–338. doi: 10.1046/j.1365-2958.1996.00116.x. [DOI] [PubMed] [Google Scholar]
- 79.Prager R, Mirold S, Tietze E, Strutz U, Knuppel B, Rabsch W, Hardt WD, Tschape H. Prevalence and polymorphism of genes encoding translocated effector proteins among clinical isolates of Salmonella enterica. Int J Med Microbiol. 2000;290(7):605–617. doi: 10.1016/S1438-4221(00)80009-0. [DOI] [PubMed] [Google Scholar]
- 80.Hopkins KL, Threlfall EJ. Frequency and polymorphism of sopE in isolates of Salmonella enterica belonging to the ten most prevalent serotypes in England and Wales. J Med Microbiol. 2004;53(Pt 6):539–543. doi: 10.1099/jmm.0.05510-0. [DOI] [PubMed] [Google Scholar]
- 81.Saunders MP, Wu G, Abuoun M, Pan Z, Anjum M, Woodward MJ. Optical genetic mapping defines regions of chromosomal variation in serovars of S. enterica subsp. enterica of concern for human and animal health. Epidemiol Infect. 2010;139(7):1065–1074. doi: 10.1017/S0950268810002086. [DOI] [PubMed] [Google Scholar]
- 82.Petersen RF, Litrup E, Larsson JT, Torpdahl M, Sorensen G, Muller L, Nielsen EM. Molecular characterization of Salmonella Typhimurium highly successful outbreak strains. Foodborne Pathog Dis. 2011;8(6):655–661. doi: 10.1089/fpd.2010.0683. [DOI] [PubMed] [Google Scholar]
- 83.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8(3):195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 84.Wilkinson P, Paszkiewicz K, Moorhouse A, Szubert JM, Beatson S, Gerrard J, Waterfield NR, Ffrench-Constant RH. New plasmids and putative virulence factors from the draft genome of an Australian clinical isolate of Photorhabdus asymbiotica. FEMS Microbiol Lett. 2010;309(2):136–143. doi: 10.1111/j.1574-6968.2010.02030.x. [DOI] [PubMed] [Google Scholar]
- 85.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 87.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;38(Database issue):D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Selengut JD, Haft DH, Davidsen T, Ganapathy A, Gwinn-Giglio M, Nelson WC, Richter AR, White O. TIGRFAMs and Genome Properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res. 2007;35:D260–D264. doi: 10.1093/nar/gkl1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28(1):33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39(Web Server issue):W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30(11):2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13(1):36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT. Direct comparisons of illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS One. 2012;7(2):e30087. doi: 10.1371/journal.pone.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis comparison tool. Bioinformatics. 2005;21(16):3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: List of plasmids in the genome of S. Virchow SVQ1. (XLSX 37 KB)
Additional file 2: Table S2: List of protein coding sequences that contain at least one SNP or indel between SVQ1 and SL491. (XLSX 52 KB)
Additional file 3: Table S3: List of Single nucloetide polymorphisms test. (XLSX 41 KB)
Additional file 4: Table S4: Salmonella Pathogenicity Island in S. Virchow SVQ1. (XLSX 39 KB)
Additional file 5: Table S5: Regions of differences identified in S. Virchow SVQ1. (XLSX 50 KB)
Additional file 6: Table S6: PCR results of S. Virchow unique regions. (XLSX 52 KB)