

Abstract
Both androgen and phosphatidylinositol 3-kinase (PI3K) signaling are critical for cell proliferation of androgen receptor (AR)–positive prostate cancer cells, but the underlying mechanisms are still not fully understood. Here we report that serum- and glucocorticoid-inducible kinase 3 (SGK3), a Ser/Thr kinase functioning downstream of PI3K, is an AR transcriptional target and promotes prostate cancer cell proliferation. SGK3 expression is up-regulated by androgen DHT via AR. We identified an AR-binding region at the sgk3 locus, which confers androgen responsiveness of sgk3 promoters. Interestingly, we found that androgen/AR-dependent SGK3 expression requires estrogen receptor (ER) (including both isoforms, ERα and ERβ). Depletion of ER blocked DHT-induced SGK3 expression. Functionally, knockdown of SGK3 expression significantly decreased LNCaP prostate cancer cell proliferation by inhibiting G1 to S phase cell cycle progression. We further provided evidence that SGK3 promotes p70 S6 kinase (p70S6K) activation and increases cyclin D1 levels. In summary, our study identifies SGK3 as an AR target and provides a novel androgen-induced cell proliferation mechanism mediated by the AR-SGK3-p70S6K-cyclin D1 pathway in prostate cancer cells.
Prostate cancer is the most frequently diagnosed malignancy and the second leading cause of cancer death among men in the United States (1). Androgen hormone plays a significant role in both the initiation and progression of prostate cancer (2–4). Androgen exerts its biological effects via androgen receptor (AR), which induces a gene expression program promoting prostate cancer cell proliferation and survival (5–7). Although it is well established that androgen promotes AR-positive prostate cancer cell proliferation, the molecular mechanisms of androgen-mediated cell proliferation are still not totally understood.
Serum- and glucocorticoid-inducible kinase 3 (SGK3) is one of the SGKs that belong to the “AGC” kinase family (protein kinase A, protein kinase G, and protein kinase C). SGKs have 3 isoforms in mammals (SGK1, SGK2, and SGK3), which share great homology with protein kinase B/Akt in the kinase domain but are coded by 3 distinct genes (8). Like Akt, SGKs function downstream of phosphatidylinositol 3-kinase (PI3K) and are the direct substrates of phosphoinositide-dependent kinase-1 (8). SGKs have been implicated in the regulation of ion channels, glucose homeostasis, and cell proliferation, survival, and migration (9–11). Of note, SGK3 has been suggested to play a pivotal role in Akt-independent signaling in human cancer (12, 13). However, very little is known about regulation of SGK3. Recently, we have demonstrated that SGK3 is transcriptionally regulated by estrogen receptor (ER) and promotes estrogen-mediated cell survival of breast cancer cells (14). The observation that SGK3 expression is increased upon androgen stimulation (15, 16) prompted us to hypothesize that SGK3 is also an AR direct target.
The PI3K pathway is constitutively activated due to phosphatase and tensin homolog loss in prostate cancer (17, 18). The PI3K pathway is critical for proliferation and survival of prostate cancer cells (19), but the underlying mechanisms are still not fully understood. It has been reported that reciprocal feedback regulation of PI3K and AR signaling is critical for prostate cancer cell survival (20). Because SGK3 is a downstream kinase of PI3K and its expression is increased upon androgen treatment (15, 16), we hypothesized that SGK3 may mediate androgen-induced cell proliferation of AR-positive prostate cancer. The data presented in this study demonstrate that SGK3 is transcriptionally regulated by AR and promotes G1 to S phase cell cycle progression of prostate cancer cells through activation of p70 S6 kinase (p70S6K) and up-regulation of cyclin D1. Our study provides a new link between PI3K and AR signaling as well as a new androgen-induced cell proliferation mechanism mediated by SGK3 in prostate cancer cells.
Materials and Methods
Cell culture
Human prostate cancer cell lines LNCaP, 22Rv1, and PC3 and human benign prostatic hyperplasia (BPH) epithelial cell line BPH-1 were propagated in RPMI 1640 medium (HyClone Laboratories, Inc) supplemented with 2 mM l-glutamine, 10% fetal bovine serum (FBS) (Omega Scientific), and 100 U/mL penicillin-streptomycin. Human breast cancer cell lines MCF-7 and T47D were cultured in Eagle's MEM (HyClone Laboratories) medium supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 1% nonessential amino acids, and 100 U/mL penicillin-streptomycin. Cells of the human AR-positive breast cancer cell line MDA-MB-453 were propagated in Leibovitz's L-15 (ATCC) medium supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 1% nonessential amino acids, and 100 U/mL penicillin-streptomycin. All cell lines were incubated at 37°C with a 5% CO2 humidified atmosphere. To evaluate the effect of steroid hormone on SGK3 expression or cell proliferation, cells were switched to phenol red–free RPMI 1640 (for LNCaP, 22RV1, PC3, and BPH-1) or MEM (for MCF-7 and T47D) or L-15 (for MDA-MB-453) medium with 10% charcoal/dextrin (CD)-treated FBS (Omega Scientific) 48 hours before hormone treatments.
Plasmid constructs
The preparation of the different sgk3 promoter luciferase reporter constructs used in the study, including pGL3-sgk3−401/+250 (contains the basal promoter for transcript variant 1&2), pGL3-sgk3−401/+250plus+1401/+1650 (basal promoter linked to the ER-binding region 1), pGL3-sgk3−600/+1650 (contains the promoter for transcript variant 3 and the ER-binding region 1), and pGL3-sgk3−600/+1400 (contains promoter for variant 3 without the ER-binding region 1), was described previously (14). To generate the pGL3-sgk3+1417/+1650 luciferase reporter, the sgk3+1417/+1650 fragment was amplified by PCR using genomic DNA from HeLa cells as the template and primers 5′-GCCGGTACCTTAACTGCCACTGCATGGAG-3′ and 5′-GCCAAGCTTGCTGACCTACCAATTCAATG-3′. The PCR product was then digested and cloned into the pGL3 basic vector (Promega) through the KpnI/HindIII sites. To delete the putative androgen response element (ARE) (nt [nucleotide] +1401/+1416) from the construct pGL3-sgk3−600/+1650, site-directed mutagenesis was performed according to the protocol described in the QuikChange Site-Directed Mutagenesis Kit (Stratagene) using the primers 5′-GGCTAACTCTGGAGACACTTAACTGACCACTGCATGG-3′ and 5′-CCATGCAGTGGCAGTTAAGTGTCTCCAGAGTTAGCC-3′. The same mutagenesis method was used to delete the estrogen response element (ERE) (nt +1523/+1541) and/or the half ARE site (nt +1558/+1563) from the constructs pGL3-sgk3−600/+1650 and pGL3-sgk3−401/+250plus+1401/+1650, respectively, using the following primer sets: for deletion of ERE, 5′-GACATAGTTTAGATATGGAAATGTACTATAAA-3′ and 5′-TTTTATAGTACATTTCCATATCTAAACTATGTC-3′; and for deletion of the half ARE site, 5′-GAAATGTACTATAAAATTTTTTGTTTGCACAC-3′ and 5′-GATGTGTGCAAACAAAAAATTTTATAGTAC-3′. All of the sgk3 mutation fragments were subcloned into pGL3 basic vector. To generate the pRetroX-Tight-Pur-SGK3 plasmid for the inducible expression of SGK3 using the retroviral expression system, the complete coding region for the SGK3 gene was amplified by PCR using the primer set 5′-ATTGCGGCCGCCACCATGCAAAGAGATCACACCATGGAC-3′ and 5′-CCTTCAGAAGACTTATTTTTGTGAACGCGTGCC-3′. The PCR product was digested and subcloned into the pRetroX-Tight-Pur vector (Clontech) via the Not1 and Mlu1 sites. All of the inserts were verified by DNA sequencing.
RNA preparation and real-time quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from cells using TRIzol (Invitrogen). The RNA (5 μg) was then reverse transcribed into cDNA by the SuperScript III first-strand synthesis system (Invitrogen) following the manufacturer's instructions. qRT-PCR was performed as described previously (21). The primers used in real-time PCR were as follows: for SGK3, 5′- CCAGGAGTGAGTCTTACAG-3′ and 5′-CCAGCCACATTAGGATTA-3′; for ERβ, 5′-GTTTGGGTGATTGCCAAGAG-3′ and 5′-CACTGGGACCACATTTTTTGC-3′, as reported previously (22); and for β-actin, 5′-CACCAACTGGGACGACAT-3′ and 5′-GCACAGCCTGGATAGCAAC-3′.
Western blotting
Western blotting was performed as described previously (23). The following antibodies were used: SGK3 antibody (MCA5480Z) from AbD Serotec; ERα antibodies (HC-20), AR antibody (AR 441), sterol regulatory element-binding protein 1 (SREBP-1) antibody (2A4), and β-actin antibody (I-19R) from Santa Cruz Biotechnology; ERβ antibody (N2C2) from GeneTex; cyclin D1 (DCS6) antibody (no. 2926), cyclin E1 (HE12) antibody (no. 4129), cyclin-dependent kinase (CDK) 2 (78B2) antibody (no. 2546), CDK4 (DCS156) antibody (no. 2906), CDK6 (DCS83) antibody (no. 3136), poly(ADP-ribose) polymerase (PARP) antibody (no. 9542), cleaved caspase 3 (5A1E) antibody (no. 9664), phospho-GSK3β (Ser9) antibody (no. 9336), GSK3β (27C10) antibody (no. 9315), p27/Kip1 antibody (no. 2552), phospho-p70S6K (Thr389) (108D2) antibody (no. 9234), and p70S6K antibody (no. 9202) from Cell Signaling Technology; and phosphor-p27/Kip1 (Thr157) antibody (AF1555) from R&D Systems Inc.
Chromatin immunoprecipitation (ChIP) assay
LNCaP cells were cultured in phenol red–free medium containing 10% CD-treated FBS for 72 hours and then were treated with dimethyl sulfoxide (DMSO) or 10 nM 5α-DHT for 4 hours. The ChIP assay was performed as described previously (24), except that the antibody used for immunoprecipitation in this study is anti-AR mouse monoclonal antibody (AR 441; Santa Cruz Biotechnology). Normal mouse IgG (Santa Cruz Biotechnology) was used as a control in ChIP analysis. The sequences of the primers used in the subsequent qPCR are the following: for SGK3, 5′-GACTTGTGTAACATGGTCTCTTTCA-3′ and 5′-CAAGTTCAATCTGACCCTCATATCT-3′; for TATA box binding protein, 5′-GTGGCGGTCCACATAAAAAC-3′ and 5′-GTCCTCTCATGCCCTGTGTT-3′; and for prostate-specific antigen, 5′-AAGACAGCAACACCTTTTT-3′ and 5′-TGTTCTTTGAGGACACTGGA-3′.
Transient transfection and luciferase reporter assay
LNCaP and MDA-MB-453 cells were seeded in 24-well plates at 1.5 × 105 cells/well and cultured in phenol red–free RPMI medium 1640 or MEM containing 5% CD-treated FBS overnight. The cells in each well were transfected with 1.5 μg of Lipofectamine 2000 (Invitrogen) and 0.25 μg of reporter plasmids in 250 μL of Opti-MEM. Four hours after transfection, 250 μL of phenol red–free RPMI medium 1640 or MEM with 5% CD-treated FBS containing either DMSO or 10 nM DHT was added to each corresponding well. After 24 hours of incubation, the cells were washed once with PBS. Then 150 μL of passive lysis buffer (Promega) was added to each well, and the plate was shaken at room temperature until cells were completely lysed. Luciferase activities of the cell lysates were assayed using luciferase assay substrate (Promega), and the protein concentrations of cell lysates were measured using the Bio-Rad protein assay according to the manufacturer's protocol. The relative luciferase activity was calculated by dividing luciferase activity by the protein concentration. Each transfection was performed in triplicate. The data are expressed as means ± SD of 3 independent experiments.
Small interfering RNA (siRNA) transfection
Reverse siRNA transfection was performed using siPORT NeoFX transfection agent (Ambion) according to the manufacturer's instructions. For the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, siRNA transfection was performed in 24-well plates. For cell cycle analysis and for immunoblotting, 60-mm dishes were used. In brief, cells were trypsinized and diluted in the medium at 1 × 105 cells/mL. For each well of the 24-well plate, 1 μL of siPORT NeoFX transfection agent and 1.5 μL of SGK3 siRNA (10 μM) (Santa Cruz Biotechnology), or negative control siRNA (Ambion) were diluted in 25 μL of OPTI-MEM I medium, respectively. For each 60-mm dish, 10 μL of siPORT NeoFX transfection agent and 15 μL of siRNA negative control or siRNA against SGK3, ERα, ERβ, or p70S6K were diluted in 250 μL of OPTI-MEM I medium, respectively. After being mixed and incubated for 10 minutes, the mixtures of siRNA and transfection agent were dispersed into 24-well plates or 60-mm dishes. Cell suspensions (1 × 105 cells/mL) were overlaid onto the transfection complexes at 0.5 mL of cell suspension/well or 5 mL of cell suspension/dish. The plates or dishes were gently rocked back and forth to evenly distribute the complexes and were incubated at 37°C until ready to assay.
Generation of LNCaP/TO/SGK3 cell line for the inducible expression of SGK3 using a retroviral expression system
The Retro-X-Tet-On Advanced inducible expression system (Clontech Laboratories, Inc) was used to establish the double-stable RetroX-Tet On LnCaP cells that express SGK3 under the control of tetracycline according to the manufacturer's instructions. In brief, 293T cells cultured in 10-cm dishes were transiently cotransfected with pCMV-GP (gag-pol–expressing vector), a retroviral expression vector (pRetroX-Tet-On Advanced vector for the expression of the tetracycline-controlled transactivator rtTA-Advanced or pRetroX-Tight-Pur-SGK3 vector for the expression of SGK3 under the inducible response promoter, PTight), and an envelope (env)-expressing vector (pVSV-G) using Lipofectamine 2000 (Invitrogen). At 8 to 10 hours after transfection, the culture medium containing DNA and Lipofectamine 2000 was replaced with 10 mL of fresh complete medium, and the cells were continuously cultured for an additional 48 to 72 hours before the crude viral supernatant was collected. The virus supernatant was briefly centrifuged and aliquoted and stored at −70°C until use.
LNCaP cells were seeded in a 6-well plate at a density to produce about 50% confluence 12 to 18 hours after seeding. To generate LNCaP/TetOn cells that constitutively express the tetracycline-controlled transactivator rtTA-Advanced, the LNCaP cells in the 6-well plate were infected with the RetroX-Tet-On Advanced virus stock. In brief, the medium was removed, 1 mL of virus stock was added to each well, and Polybrene was added to a final concentration of 4 μg/mL. Twenty-four hours after infection, cells were cultured in the fresh complete medium containing 600 μg/mL G418 for about 2 weeks. The selective medium was replaced every 2 days. After G418 selection for 2 weeks, the G418-resistant clones were pooled and were further infected with the RetroX-Tight-Pur-SGK3 virus stock. Twenty-four hours after infection, the cells were cultured in the medium containing 2 μg/mL puromycin for 4 to 5 days. The individual LNCaP/TO/SGK3 cell clone was isolated using limiting dilution in 96-well plates. The puromycin-resistant clones were then tested for inducible SGK3 expression in the absence and presence of 500 ng/mL doxycyline (DOX) to screen the clones that generate the highest overall induction and lowest background expression of SGK3.
Cell proliferation assay
Cell proliferation was measured by an MTT assay. At the indicated time, the medium in each well was removed from the cells and replaced with 0.5 mL of fresh phenol red–free medium containing 0.5 mg/mL MTT, and then the cells were incubated at 37°C for 1 hour. The medium was discarded, and 0.5 mL of DMSO was added to each well to dissolve the formazan dye trapped in the living cells. Then 100 μL of the supernatant was transferred into a 96-well plate and read in a SpectraMax M5 plate reader (Molecular Devices) at A570.
Cell cycle analysis
Seventy-two hours after transfection with the siRNA negative control or SGK3 siRNA, LNCaP cells cultured in 60-mm dishes were harvested, fixed in 70% ethanol overnight, and treated with RNase (10 mg/mL). Propidium iodide (250 μg/mL) was then added to the samples, and the samples were mixed and incubated in the dark for 30 minutes. Flow cytometry analysis was performed using the EPICS Elite ESP high-performance cell sorter (Coulter Electronics). The raw data collected were analyzed by ModFit LT (version 2.0; Verity Software) to eliminate aggregated cells for determination of cell cycle distribution.
Statistical analysis
Data were from 3 independent experiments were analyzed using the Student t test and are expressed as means ± SD. A P value of < .05 was considered statistically significant.
Results
Androgen stimulates sgk3 gene transcription via AR
The microarray data from DePrimo et al (16) showed that the SGK3 mRNA level was increased after LNCaP cells were exposed to 1 nM synthetic androgen R1881. To confirm that SGK3 is transcriptionally regulated by androgen, cells of the AR-positive human prostate cancer cell line LNCaP were hormone-stripped for 48 hours and then were treated with 1 nM DHT, a nonaromatizable form of androgen. The SGK3 mRNA level was measured by RT-qPCR (note that the primers used in qPCR are nontranscript variant specific and are able to amplify all 3 of the SGK3 transcript variants). As shown in Figure 1A, DHT treatment increased SGK3 mRNA levels in a time-dependent manner, and the enhancement of the SGK3 mRNA levels in response to DHT was evident as early as 1 hour posttreatment. Moreover, the increase in SGK3 mRNA levels by DHT could be blocked by the general transcription inhibitor actinomycin D, but not by the protein synthesis inhibitor cycloheximide (Figure 1B), suggesting that the sgk3 gene is a direct transcriptional target of androgen/AR.
Figure 1.

DHT stimulates sgk3 gene transcription via AR. Cells were hormone stripped for 2 days before treatment. A, LNCaP cells were treated with 1 nM DHT for the indicated time. RT-qPCR was performed to quantify mRNA levels of SGK3 and β-actin. Relative expression level was calculated by dividing SGK3 mRNA level by β-actin level. qPCRs were performed in triplicate, and data were expressed as means ± SD. *, P < .05 vs 0 hours of DHT treatment by a Student t test. B, LNCaP cells were pretreated with 1 μg/mL actinomycin D (ActD) or 100 μg/mL cycloheximide (CHX) for 30 minutes and then were treated with either DMSO or 1 nM DHT for 4 hours. Cells were harvested for RT-qPCR. *, P < .05 vs DMSO control. C, LNCaP cells were treated with increasing doses of DHT as indicated for 48 hours. Cell extracts were subjected to Western blotting with anti-SGK3 antibody, and the blot was reprobed with anti-β-actin antibody. D, LNCaP cells were treated with or without 1 nM DHT alone or plus 5 μM bicalutamide (BIC) for 48 hours and then harvested for Western blotting analysis. E and F, PC-3 cells (E) and BPH-1 cells (F) were treated with the increasing doses of DHT as indicated for 48 hours. Cell extracts were harvested for Western blotting. G, LNCaP cells were transfected with siRNA negative control or SGK3 siRNA and then were treated with or without 1 nM DHT for 48 hours. Cells were harvested for Western blotting analysis. H, 22Rv1 cells were treated with increasing doses of DHT as indicated for 48 hours and then harvested for Western blotting analysis. NS, nonspecific band(s).
Western blotting analysis was also performed to test the up-regulation of SGK3 protein levels by DHT via AR. As shown in Figure 1, C and D, DHT increased SGK3 protein levels in a dose-dependent manner in LNCaP cells, and up-regulation of SGK3 expression by DHT was blocked by the AR antagonist bicalutamide. In contrast, DHT did not induce SGK3 expression in the cells of the AR-negative prostate cancer cell line PC3 or the AR-negative BPH epithelial cell line BPH-1 (Figure 1, E and F), suggesting that androgen-induced SGK3 expression requires AR. To further confirm that up-regulation of SGK3 by DHT is mediated by AR, LNCaP cells were transfected with either an siRNA negative control or AR siRNA and then treated with or without DHT. As shown in Figure 1G, depletion of AR expression blocked DHT-induced SGK3 expression, further confirming that androgen-induced SGK3 expression is mediated by AR.
To show that the up-regulation of SGK3 expression by DHT was not a single cell–type phenomenon, we treated cells of another AR-positive prostate cancer cell line, 22Rv1, as well as those of 2 AR-positive breast cancer cell lines, MCF-7 and T47D, with DHT. As shown in Figure 1H and Supplemental Figure 1, A and B, SGK3 expression was dose dependently increased by DHT in all 3 cell lines tested.
Identification of AR-binding region at the sgk3 locus
To determine the molecular basis for the induction of SGK3 expression by androgen/AR, we went on to identify the AR-binding region at the sgk3 locus, which is responsible for androgen/AR transcriptional regulation of sgk3 promoters. As illustrated in Figure 2A, the human sgk3 gene has 3 transcript variants transcribed by 2 different promoters, one for transcript variant 3 and the other for transcript variants 1 and 2. We have previously mapped these 2 promoters (14); nt −401/+250 contain a promoter for transcript variants 1 and 2, and nt −600/+300 contain a promoter for transcript variant 3. Moreover, we found that the ER-binding region 1 (nt +1401/+1650) previously identified by us in a ChIP assay with direct Solexa ChIP-sequencing approach, which locates in the intron 1 of transcript variant 3 and 62 kb away from the transcription start site of transcript variants 1 and 2, is responsible for the estrogen responsiveness of both promoters (14). Of note, there is a putative ARE, CAGAACTTCCCCTTCT (nt +1401 to +1416), within this ER-binding region (Figure 2A), which is predicted by the transcription factor binding site software TRANSFAC (BIOBASE Biological Databases). To test whether this ER-binding region is also responsible for the androgen responsiveness of sgk3 promoters, LNCaP cells were transiently transfected with the constructs containing sgk3 promoters linked with or without this ER-binding region (nt +1401/+1650) as illustrated in the left panel of Figure 2B. As shown in the right panel of Figure 2B, DHT treatment resulted in 32- and 104-fold increases in the promoter activity of the reporter constructs containing the sgk3 promoters linked with the ER-binding region 1 (pGL3basic-sgk3−401/+250plus+1401/+1650-Luc and pGL3basic-sgk3−600/+1650-Luc), respectively. Moreover, the DHT-induced activity of both promoters could be suppressed by bicalutamide (Figure 2B), demonstrating that the activity of both sgk3 promoters can be up-regulated by DHT and further supporting the fact that sgk3 is an androgen-responsive gene. In contrast, DHT failed to induce the promoter activity of either reporter construct containing sgk3 promoters without the ER-binding region 1 (pGL3basic-sgk3−401/+250-Luc and pGL3basic-sgk3−600/+1400-Luc), supporting the fact that this ER-binding region (nt +1401/+1650) is responsible for the androgen responsiveness of both sgk3 promoters.
Figure 2.

Identification of the AR-binding region at the sgk3 locus. A, Schematic representation of SGK3 transcript variants (lower panel) and the previously identified ER-binding region 1 on sgk3 gene. The ER-binding region 1 contains a putative ARE, an ERE, and a half ARE site (upper panel). B, Left panel, schematic representation of constructs containing sgk3 promoter reporters used in the right panel. The nucleotides are numbered from the transcriptional start site that is assigned as +1. Right panel, LNCaP cells were transfected with the luciferase reporters as indicated and then cultured in the presence or absence of 1 nM DHT and 100 nM bicalutamide (BIC) alone or in combination. Twenty-four hours after transfection, luciferase activity of cell extracts was measured and normalized to protein concentration. Data were expressed as means ± SD from 3 independent experiments. **, P < .01, by the Student t test. C, ChIP-qPCR analysis of the recruitment of AR to the ER-binding region 1 (nt +1401/+1650) in chromatin. LNCaP cells were hormone-stripped for 72 hours and then treated with DMSO or 10 nM DHT for 4 hours. ChIP experiments were carried out using either normal IgG (control) or anti-AR antibody. The precipitated DNAs were used as templates for the qPCR. The primers for each qPCR were described in Materials and Methods. TATA box binding protein (TBP) is a known non-AR target serving as negative control, whereas prostate-specific antigen (PSA) is a known AR target serving as positive control of ChIP-qPCR experiment. **, P < .01, by the Student t test.
To further verify that this ER-binding region is also the AR-binding region, we performed a ChIP analysis. LNCaP cells were hormone-stripped for 72 hours and then treated with 10 nM DHT or DMSO (solvent control) for 4 hours. As shown in Figure 2C, the binding of AR to the ER-binding region was significantly increased after DHT treatment compared with that for the DMSO control, confirming that this region is the bona fide AR-binding region at the sgk3 locus.
Involvement of ER in androgen/AR-dependent SGK3 expression
As mentioned earlier, there is a putative ARE (nt +1401/+1416) within this ER/AR-binding region (Figure 2A). However, deletion of this putative ARE from the nt −600/+1650 (pGL3basic-sgk3−600/+1650delARE-Luc) or from nt −401/+250plus+1401/+1650 (pGL3basic-sgk3−401/+250plus+1417/+1650-Luc) did not affect the DHT responsiveness of either sgk3 promoter (Figure 3A), suggesting that this putative ARE does not mediate the androgen responsiveness of either sgk3 promoter. To our surprise, deletion of the ERE from nt −600/+1650 (pGL3basic-sgk3−600/+1650delERE-Luc) or from nt −401/+250plus+1401/+1650 (pGL3basic-sgk3−401/+250plus+1401/+1650delERE-Luc) resulted in a dramatic reduction of the DHT responsiveness of both promoters by 82% (the promoter for variant 3) and 70% (promoter for variants 1 and 2), respectively (Figure 3B), implying that the ERE is important for androgen induction of SGK3 expression. Of note, several AR ChIP-sequencing and ChIP-chip studies using prostate cancer cell lines have showed that most genome-wide AR DNA binding sites consist of only a half ARE site (25–28). Given that finding, we reexamined this ER/AR-binding region (+1401/+1650) and found a half ARE site (5′-AGAACA-3′) at position +1558/+1563, which is 16 nt downstream of ERE (Figure 2A). To test whether this half ARE site is functional, we deleted this site from the region and examined its effect on androgen responsiveness of both sgk3 promoters. As shown in Figure 3C, deletion of this half ARE site significantly reduced but did not eliminate the androgen responsiveness of either sgk3 promoter, suggesting that this half ARE site is functional but might not be the only site responsible for androgen induction of sgk3 promoter activity. We went on to delete both the ERE and the half ARE site from the region. A luciferase transactivation assay showed that simultaneous deletions of the 2 sites almost completely eliminated the androgen responsiveness of both sgk3 promoters (Figure 3D), suggesting that these 2 sites coordinately regulate androgen induction of sgk3 promoter activity.
Figure 3.

Identification of the elements within the AR-binding region responsible for androgen responsiveness of sgk3 promoters. A–D, LNCaP cells were transfected with the luciferase reporters as indicated and then cultured in the presence or absence of 1 nM DHT. Twenty-four hours after transfection, luciferase activity of cell extracts was measured and normalized to protein concentration. Data are expressed as means ± SD from 3 independent experiments. n.s, not statistically significant; **, P < .01, by the Student t test.
Given that this ERE is responsible for estrogen/ER regulation of sgk3 promoters (14), we hypothesized that ER (including ERα and ERβ) might involve androgen/AR-dependent SGK3 expression. In support of this hypothesis, knockdown of ERβ expression as determined by Western blotting (Figure 4A) and RT-qPCR (Supplemental Figure 2A) resulted in a dramatic decrease in DHT-induced promoter activity of both sgk3 promoters in LNCaP cells (Figure 4B), which express ERβ, but no significant ERα (29). Accordingly, DHT-induced SGK3 expression was greatly inhibited after ERβ was depleted (Figure 4C). Moreover, transfection of siRNA-resistant ERβ cDNA largely rescued ERβ siRNA-mediated down-regulation of SGK3 expression (Figure 4D).
Figure 4.

Androgen/AR-dependent SGK3 transcription involves ER. A, LNCaP cells were transfected with the siRNA negative control (siNC) or ERβ siRNA (siERβ) for 72 hours. Cell extracts were subjected to Western blotting. B, After being hormone-stripped for 2 days, LNCaP cells were transfected with the siRNA negative control or ERβ siRNA for 48 hours and then were transfected with the luciferase reporters as indicated and cultured in the presence or absence of 1 nM DHT for 24 hours. Luciferase activity of cell extracts was measured and normalized to protein concentration. Data are expressed as means ± SD from 3 independent experiments. **, P < .01, by the Student t test. C, LNCaP cells were transfected with siRNA negative control or ERβ siRNA. Twenty-four hours after transfection, the cells were treated with DMSO or 10 nM DHT for 48 hours. The cells were harvested and subjected to Western blotting with the relevant antibodies. D, LNCaP cells were reversely transfected with siRNA negative control or ERβ siRNA and cultured in the presence of 10 nM DHT for 24 hours and then were transfected with empty vector pCI or ERβ expression vector for 48 hours. Cell extracts were subjected to Western blotting analysis. E, MCF-7 cells were hormone-stripped for 2 days and then were added with or without 10 nM DHT alone or plus 100 nM ICI182,780 (ICI). RT-qPCR was performed to evaluate the SGK3 mRNA level. **, P < .01, by the Student t test. F, MCF-7 cells were transfected with the siRNA negative control or ERα siRNA. Seventy-two hours after transfection, the cells were treated with DMSO or 10 nM DHT for 24 hours. The cells were harvested and subjected to Western blotting with the relevant antibodies. G, After being hormone-stripped for 2 days, MDA-MB-453 cells were transfected with empty vector pSG5 or ERα expression vector for 48 hours and then were transfected with the luciferase reporters as indicated and cultured in the presence or absence of 1 nM DHT for 24 hours. Luciferase activity of cell extracts was measured and normalized to protein concentration. Data are expressed as mean ± SD from 3 independent experiments. n.s., not statistically significant; **, P < .01, by the Student t test. H, MDA-MB-453 cells were hormone-stripped for 2 days and transfected with empty vector or ERα expression vector and then were cultured in the presence or absence of 10 nM DHT for 48 hours. Cell extracts were subjected to Western blotting with the relevant antibodies. NS, nonspecific band(s).
To further support the finding that androgen/AR-dependent SGK3 expression requires ER, we took advantage of two other cell lines, MCF-7 and MDA-MB-453; the former cell line expresses both AR and ER (including ERα and ERβ, but ERα is dominant), and the latter cell line expresses AR but not ER. We pretreated MCF-7 cells with ICI182,780, which is a known antiestrogen and ER destabilizer. The RT-qPCR result showed that ICI182,780 blocked DHT-induced SGK3 expression in MCF-7 cells (Figure 4E). Moreover, depletion of ERα abrogated DHT-induced SGK3 expression (Figure 4F), demonstrating that inhibition of ERα disrupts androgen-induced SGK3 expression in MCF-7 cells. The ER involvement in the androgen-dependent SGK3 expression was further supported by the results that DHT treatment did not induce sgk3 promoter activity (Figure 4G) and SGK3 expression (Supplemental Figure 2B) in MDA-MB-453 cells. However, after introduction of ERα or ERβ into this cell line, DHT was able to induce sgk3 promoter activity (Figure 4G) and SGK3 expression (Figure 4H and Supplemental Figure 2C). Taken together, these results suggest that androgen/AR-dependent SGK3 expression requires ERα or ERβ, which depends on cell context.
SGK3 promotes cell proliferation of AR-positive prostate cancer cells
To explore the physiological significance of androgen/AR-dependent SGK3 expression, we first knocked down SGK3 expression and examined its effect on the prostate cancer cell proliferation. To this end, LNCaP cells were transfected with either pooled (sc-44852; Santa Cruz Biotechnology) or 3 individual SGK3 siRNA duplexes (sc-44852A, sc-44852B, and sc-44852C; Santa Cruz Biotechnology). As shown in Figure 5A and Supplemental Figure 3A, transfection with either pooled SGK3 siRNA or 3 individual SGK3 siRNA duplexes resulted in a dramatic decrease in SGK3 expression and a significant suppression of cell proliferation. Similar results were obtained when cells of another AR-positive prostate cancer cell line, 22Rv1, were transfected with 3 individual SGK3 siRNA duplexes (Figure 5B). In contrast, knockdown of SGK3 had no significant effect on cells of the AR-negative prostate cancer cell line PC3 (Supplemental Figure 3B). These data indicate that SGK3 promotes cell proliferation of AR-positive prostate cancer cells.
Figure 5.

SGK3 promotes cell proliferation of AR-positive prostate cancer cells. A and B, LNCaP cells (A) or 22Rv1 cells (B) were transfected with the siRNA negative control (siNC) or 3 individual SGK3 siRNA duplexes (siSGK), respectively. Western blotting (insert) was performed to examine SGK3 level after the cells were transfected with siRNA for 72 hours. An MTT assay was performed to measure cell proliferation at the indicated time after siRNA transfection. Error bars represent SDs of 3 independent experiments. C, LNCaP/TetON/SGK3 cells cultured in phenol red–free RPMI 1640 medium with 2% CD-treated FBS were added with or without 100 ng/mL DOX. An MTT assay was performed to measure cell proliferation at the indicated time. D, LNCaP cells were transfected with the siRNA negative control or SGK3 siRNA and cultured in the phenol red–free RPMI 1640 medium with 5% CD-treated FBS in the presence or absence of DHT (1 nM). SGK3 protein levels were examined by Western blotting at 72 hours after siRNA transfection. The cell growth rate was measured by an MTT assay at the indicated time. **, P < .01, by the Student t test.
To further confirm the results, we went on to test whether forced overexpression of SGK3 promotes cell proliferation of AR-positive prostate cancer cells. To this end, we used LNCaP cells to generate a SGK3-inducible expression cell line designated LNCaP/TetOn/SGK3, in which SGK3 expression is inducible by DOX. The DOX-inducible expression of SGK3 in LNCaP/TetOn/SGK3 cells was confirmed by Western blotting (Figure 5C, inset). When the cells were cultured in the medium containing 2% CD-treated FBS, induction of SGK3 expression by DOX caused a significant increase in proliferation of LNCaP/TetOn/SGK3 cells (Figure 5C), supporting the observation that SGK3 could promote LNCaP cell proliferation.
As we demonstrated above, SGK3 is an androgen-inducible kinase. Figure 5D shows that knockdown of SGK3 expression significantly inhibited the DHT-induced proliferation of LNCaP cells, demonstrating that SGK3 promotes androgen-dependent cell proliferation.
SGK3 promotes G1 to S phase cell cycle progression by up-regulation of cyclin D1
Light microscopy indicated that there was a significant decrease in cell proliferation but no obvious visible increase in cell death after LNCaP cells were transfected with SGK3 siRNA (Figure 6A). Western blotting results showed that depletion of SGK3 did not increase PARP cleavage and cleaved caspase 3 levels (Figure 6B), confirming that SGK3 depletion does not result in apoptosis in LNCaP cells. Therefore, we speculated that SGK3 regulates cell proliferation by affecting the cell cycle rather than cell viability. To test this hypothesis, flow cytometry analysis was performed after LNCaP cells were transfected with SGK3 siRNA for 72 hours. As shown in Figure 6, C and D, the G1 population was significantly increased, whereas the S phase population was dramatically decreased in the SGK3 siRNA-transfected cells compared with that in the siRNA negative control transfected cells, suggesting that knockdown of SGK3 expression inhibits G1 to S phase cell cycle progression.
Figure 6.

SGK3 promotes the G1 to S phase cell cycle progression of LNCaP cells through up-regulation of cyclin D1. A and B, LNCaP cells were transfected with the siRNA negative control (siNC) or pooled SGK3 siRNA duplexes (siSGK3) for 4 days. The transfected cells were imaged under a microscope (A) or harvested for Western blotting (B) to determine PARP cleavage and cleaved caspase 3 levels. Scale bar corresponds to 50 μm; ×100 magnification. For Western blotting analysis of apoptosis markers, we also included a positive control, which are LNCaP cells treated with 20 μM proteasome inhibitor MG132 for 24 hours. C, LNCaP cells were transfected with siRNA negative control (LNCaP/siNC) or pooled SGK3 siRNA duplexes (LNCaP/siSGK3). Seventy-two hours after transfection, cells were harvested, fixed in 70% ethanol, and stained with propidium iodide and analyzed by flow cytometry. The percentage of DNA content in each phase was analyzed with ModFit LT 2.0 software. The representative result of 3 experiments was shown. D, Quantitative result of the percentage of DNA content in each cell cycle phase from 3 experiments. The data are expressed as means ± SD. *, P < .05; **, P < .01, by the Student t test. E and F, LNCaP cells were transfected with the siRNA negative control, pooled (E) or 3 individual (F) SGK3 siRNA duplexes, respectively. At 72 hours after transfection, the cells were harvested for Western blotting analysis with the relevant antibodies.
To understand the molecular basis of this finding, we examined the effect of SGK3 depletion on the levels of G1-S phase regulators such as cyclin D1, cyclin E1, CDK2, CDK4, and CDK6. As shown in Figure 6, E and F, knockdown of SGK3 expression in LNCaP cells significantly decreased cyclin D1 levels but had no significant effect on levels of cyclin E1, CDK2, CDK4, and CDK6, suggesting that knockdown of SGK3 selectively down-regulates the levels of cyclin D1, which is a major cyclin controlling the G1/S checkpoint. It has been reported that its related isoform, SGK1, regulates phosphorylation of p27/Kip that is involved in G1-S phase progression regulation (30). We asked whether SGK3 also regulates p27/Kip phosphorylation. As shown in Figure 6F, knockdown of SGK3 had no effect on p27/Kip phosphorylation, suggesting that alteration of p27 phosphorylation is not a mechanism for SGK3-mediated proliferation of LNCaP cells.
Knockdown of SGK3 expression decreases p70S6K phosphorylation
We went on to investigate the mechanisms for regulation of cyclin D1 by SGK3. Given that GSK3β is a substrate of SGK3 (31, 32) and controls cyclin D1 stability (33), we first examined the effect of SGK3 depletion on GSK3β phosphorylation. As shown in Figure 6D, knockdown of SGK3 expression did not decrease the phospho-GSK3β level in LNCaP cells, indicating that the alteration in GSK3β activity is not a major mechanism for up-regulation of cyclin D1 by SGK3.
Several studies have shown that p70S6K regulates cell growth and the cyclin D1 level (34–36). Of note, Xu et al (35) reported that androgen induces prostate cancer cell proliferation through the Akt-independent activation of mammalian target of rapamycin complex 1 (mTORC1)/p70S6K signaling and subsequent increase in cyclin D1 levels. Because SGK3 functions in parallel to Akt in the PI3K pathway and its expression is induced by androgen, we asked whether SGK3 regulates mTORC1/p70S6K signaling. Given that phosphorylation of p70S6K at Thr389 is exerted by mTORC1 and is essential for p70S6K activity (37), we knocked down SGK3 expression and examined its effect on phospho-p70S6K (Thr389) levels. As shown in Figure 7, A and B, knockdown of SGK3 expression in LNCaP cells by either pooled or 3 individual SGK3 siRNA duplexes dramatically decreased phospho-p70S6K (T389) levels, whereas it slightly decreased total p70S6K levels and had no inhibitory effect on Akt phosphorylation, suggesting that SGK3 promotes mTORC1-dependent p70S6K activation and this effect is independent of Akt. To confirm that mTORC1-dependent p70S6K activation is critical for LNCaP prostate cancer proliferation and modulates cyclin D1 levels, we treated LNCaP cells with the mTORC1 inhibitor rapamycin that inactivates p70S6K. As shown in Figure 7, C and D, rapamycin treatment dephosphorylated p70S6K, reduced cyclin D1 expression, and inhibited LNCaP cell proliferation. Up-regulation of cyclin D1 levels by p70S6K was further demonstrated by the siRNA experiment in which knockdown of p70S6K expression significantly decreased cyclin D1 levels in LNCaP cells (Figure 7E).
Figure 7.

Knockdown of SGK3 expression results in a decrease in p70S6K phosphorylation. A and B, LNCaP cells were transfected with the siRNA negative control (siNC), pooled (A) or individual (B) SGK3 siRNA duplexes (siSGK3), respectively. At 72 hours after transfection, the cells were harvested for Western blotting analysis with the relevant antibodies. C and D, LNCaP cells were treated with DMSO or the indicated concentrations of rapamycin (RAPA). An MTT assay (D) was performed to measure cell proliferation at the indicated time posttreatment. C, Western blotting was performed at 24 hours after treatment. **, P < .01, by the Student t test. E, LNCaP cells were transfected with siRNA negative control or p70S6K siRNA (sip70S6K) for 72 hours and harvested for Western blotting with the relevant antibodies.
Discussion
In contrast to SGK1, which is predominantly regulated at the transcriptional level by a huge number of extracellular signals, little is known about the regulation of SGK3 expression. SGK3 mRNA was not induced by serum or glucocorticoids in rat2 fibroblasts (8). It has even been considered that SGK3 expression is constitutive and is not regulated at the transcriptional level (8, 38). We have previously reported that SGK3 is an estrogen-inducible kinase and promotes estrogen-mediated cell survival of ER-positive breast cancer cells (14). Sherk et al (15) mentioned that SGK3 expression was induced by androgen in AR-positive prostate cancer cells, whereas one study showed that SGK3 remains unchanged after androgen stimulation in LNCaP cells (39). By examining the previously reported transcriptome analyses of androgen-responsive genes, we found a microarray analysis from DePrimo et al (16) showing that SGK3 mRNA levels were increased after LNCaP cells were exposed to 1 nM synthetic androgen R1881 (16). In the present study, we confirmed that SGK3 expression is induced by androgen DHT and demonstrated that SGK3 is an AR direct target. Moreover, we found that SGK3 promotes prostate cancer cell proliferation by regulation of G1 to S phase cell cycle progression. This is the first study on the molecular mechanisms by which androgen regulates sgk3 transcription and on the physiological significance of this regulation.
Promoter analysis showed that our previously identified ER-binding region 1 (14) is also the AR-binding region and is responsible for androgen responsiveness of both sgk3 promoters. Of note, the AR-binding region identified in this study is located on chromosome 8 from nt 67788896 to 67789146 and is within the AR-binding region (chromosome 8 from nt 67788621 to 67789401) that Massie et al (40) found at the sgk3 locus using ChIP-seq genome-wide approach (Supplemental Table S7 in Ref. 40). We further identified a functional half ARE site in this region. Interestingly, we found that the ERE, which is 16 nt upstream of the half ARE site, is also important for androgen responsiveness of both sgk3 promoters, because deletion of this ERE dramatically reduced androgen-induced activity of both sgk3 promoters. Moreover, our data showed that androgen/AR-dependent SGK3 expression involves ER. It has been shown that AR could interact with ER (including ERα and ERβ) (41, 42); therefore, it is likely that androgen/AR regulates sgk3 transcription by association with ER that binds to the ERE of the sgk3 gene. Our data suggest that either ERα or ERβ could assist AR to up-regulate SGK3 expression and that it depends on cell context; ie, depletion of ERβ blocks DHT-induced sgk3 promoter activity and SGK3 expression in LNCaP cells that express ERβ but no significant ERα, whereas inhibition of ERα by ICI182,780 or ERα siRNA significantly impairs DHT-induced SGK3 expression in MCF-7 cells that mainly express ERα. Moreover, we showed that DHT treatment does not induce sgk3 promoter activity and SGK3 expression in MDA-MB-453 cells that express AR but no ER, and after introduction of ERα or ERβ into this cell line, DHT is able to induce sgk3 promoter activity and SGK3 expression.
ERα and ERβ are two types of ER. They are coded by two separate genes and have distinct tissue-specific expression. Both ERα and ERβ are expressed in the adult human prostate. Knockout studies of receptors in mice revealed that ERα promotes whereas ERβ suppresses prostate cancer progression (43, 44). LNCaP cells express ERβ and naturally lack ERα (29). Although forced overexpression of ERβ could induce apoptosis in LNCaP cells (45), the studies from different laboratories have shown that endogenous ERβ indeed is required for androgen-stimulated LNCaP cell proliferation (41, 46, 47). We also found that knockdown of ERβ expression significantly decreases LNCaP cell proliferation (data not shown). The underlying mechanisms by which ERβ promotes LNCaP cell proliferation are not well understood. Given that ERβ is required for androgen/AR-dependent SGK3 expression in LNCaP cells, we hypothesize that ERβ promotes androgen-dependent LNCaP cell proliferation, at least in part, through up-regulation of SGK3 expression.
SGK3 plays an important role in human cancer. Liu et al (32) reported that SGK3 is amplified and acts as an oncogene in hepatocellular carcinoma. Several studies, including our own, have shown that SGK3 is up-regulated in breast cancer, especially in ER-positive breast cancer (14, 48–50) and promotes estrogen-mediated cell survival (14, 51). We also showed that forced overexpression of SGK3 in estrogen-dependent T47D breast cancer cells could result in antiestrogen ICI182,780 resistance (52). The current study has demonstrated that SGK3 is transcriptionally regulated by AR and promotes androgen-mediated prostate cancer cell proliferation, suggesting that SGK3 may play a role in AR-positive prostate cancer development.
Previously, Xu et al (35) reported that androgen induces prostate cancer cell proliferation through Akt-independent activation of mTORC1/p70S6K signaling and subsequent increases in cyclin D protein levels. Moreover, they found that mTORC1 activation by androgen is dependent on AR-stimulated mRNA synthesis. The current study has demonstrated that SGK3, a PI3K downstream kinase, is an AR transcriptional target. Moreover, we showed that knockdown of SGK3 expression in LNCaP cells reduced mTORC1 dependent-p70S6K phosphorylation and cyclin D1 levels. These data suggest that SGK3 is likely to be the kinase mediating androgen-induced Akt-independent mTORC1 activation and the subsequent increase in the cyclin D level (Figure 8). Of note, the isoform SGK1 has also been reported to promote androgen-induced prostate cancer cell proliferation (15), which might occur through regulation of p27/Kip phosphorylation (30). In the current study, we did not observe a significant alteration in p27 phosphorylation after knockdown of SGK3, indicating that the alteration of p27 phosphorylation is not a mechanism for SGK3-mediated proliferation of LNCaP cells. Given that SGK3 functions in parallel with Akt and shares some substrates such as GSK3β, FoxO3a, and Bad with Akt (31, 53), it would be interesting to test whether SGK3 uses the same mechanisms as Akt to activate mTORC1/p70S6K signaling in LNCaP prostate cancer cells.
Figure 8.
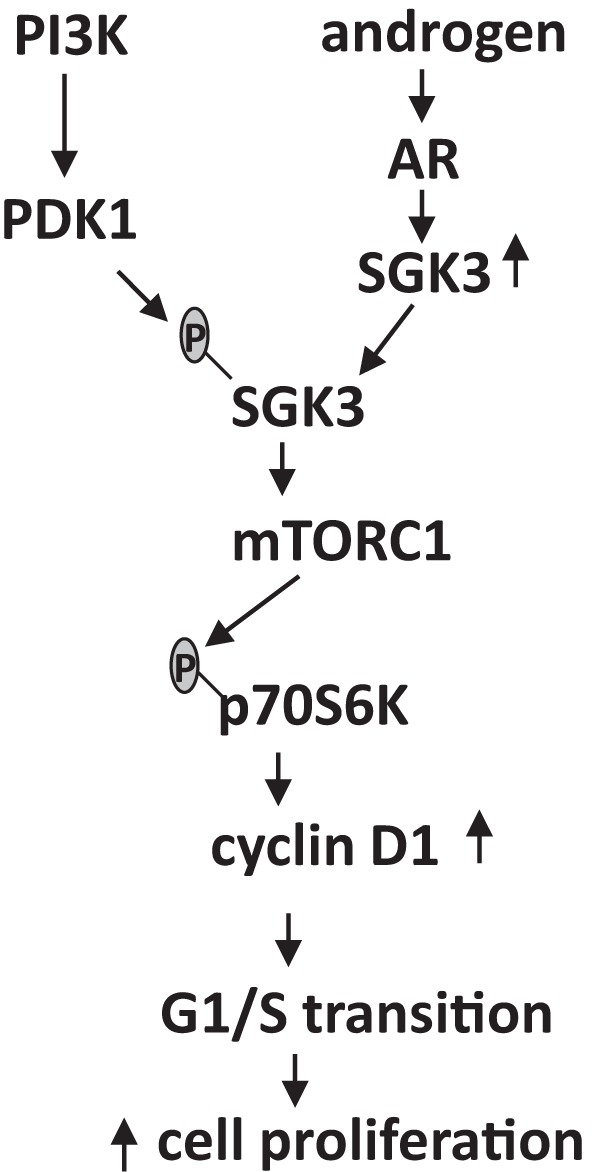
A diagram of the proposed androgen-dependent cell proliferation mediated by SGK3 in AR-positive prostate cancer. SGK3 is induced by androgen/AR through a transcriptional mechanism and is activated by phosphoinositide-dependent kinase-1 (PDK1) downstream of PI3K. Activated SGK3 promotes prostate cancer cell proliferation through up-regulation of mTORC1/p70S6K signaling and a subsequent increase in cyclin D1 levels.
In summary, our study demonstrates that SGK3 is transcriptionally regulated by AR and promotes AR-positive prostate cancer cell proliferation. Because it is induced by both estrogen and androgen and is activated by PI3K signaling, SGK3 is a unique kinase connecting both sex hormones and the PI3K pathway.
Additional material
Supplementary data supplied by authors.
Acknowledgments
We thank Lucy Brown from Analytical Cytometry Core supported by the National Cancer Institute of the National Institutes of Health under award number P30CA33572 for flow cytometry analysis.
This work was supported by a Carr Baird grant (to Y.W.), Hope Idol 2012 (to S.C.), the ThinkCure grant (to S.C.) and National Institutes of Health Grant CA44735 (to S.C.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AR
- androgen receptor
- ARE
- androgen response element
- BPH
- benign prostatic hyperplasia
- CD
- charcoal/dextrin
- CDK
- cyclin-dependent kinase
- ChIP
- chromatin immunoprecipitation
- DMSO
- dimethyl sulfoxide
- DOX
- doxycycline
- ER
- estrogen receptor
- ERE
- estrogen response element
- FBS
- fetal bovine serum
- mTORC1
- mammalian target of rapamycin complex 1
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- nt
- nucleotide
- p70S6K
- p70 S6 kinase
- PARP
- poly(ADP-ribose) polymerase
- PI3K
- phosphatidylinositol 3-kinase
- qRT-PCR
- quantitative RT-PCR
- SGK3
- serum- and glucocorticoid-inducible kinase 3
- siRNA
- small interfering RNA.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. [DOI] [PubMed] [Google Scholar]
- 2. Green SM, Mostaghel EA, Nelson PS. Androgen action and metabolism in prostate cancer. Mol Cell Endocrinol. 2012;360:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Labrie F. Blockade of testicular and adrenal androgens in prostate cancer treatment. Nat Rev Urol. 2011;8:73–85. [DOI] [PubMed] [Google Scholar]
- 4. Sharifi N. Minireview: Androgen metabolism in castration-resistant prostate cancer. Mol Endocrinol. 2013;27:708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. [DOI] [PubMed] [Google Scholar]
- 6. Dehm SM, Tindall DJ. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol. 2007;21:2855–2863. [DOI] [PubMed] [Google Scholar]
- 7. Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC. Molecular cell biology of androgen receptor signalling. Int J Biochem Cell Biol. 2010;42:813–827. [DOI] [PubMed] [Google Scholar]
- 8. Kobayashi T, Deak M, Morrice N, Cohen P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J. 1999;344(Pt 1):189–197. [PMC free article] [PubMed] [Google Scholar]
- 9. Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. Second AKT: the rise of SGK in cancer signalling. Growth Factors. 2010;28:394–408. [DOI] [PubMed] [Google Scholar]
- 10. He P, Lee SJ, Lin S, et al. Serum- and glucocorticoid-induced kinase 3 in recycling endosomes mediates acute activation of Na+/H+ exchanger NHE3 by glucocorticoids. Mol Biol Cell. 2011;22:3812–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yao LJ, McCormick JA, Wang J, et al. Novel role for SGK3 in glucose homeostasis revealed in SGK3/Akt2 double-null mice. Mol Endocrinol. 2011;25:2106–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vasudevan KM, Barbie DA, Davies MA, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. AKT-independent PI3-K signaling in cancer—emerging role for SGK3. Cancer Manag Res. 2013;5:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y, Zhou D, Phung S, Masri S, Smith D, Chen S. SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol Endocrinol. 2011;25:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sherk AB, Frigo DE, Schnackenberg CG, et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008;68:7475–7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DePrimo SE, Diehn M, Nelson JB, et al. Transcriptional programs activated by exposure of human prostate cancer cells to androgen. Genome Biol. 2002;3:RESEARCH0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cairns P, Okami K, Halachmi S, et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57:4997–5000. [PubMed] [Google Scholar]
- 18. Whang YE, Wu X, Suzuki H, et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci USA. 1998;95:5246–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15:4799–4805. [DOI] [PubMed] [Google Scholar]
- 20. Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou D, Shen R, Ye JJ, et al. Nuclear receptor coactivator PNRC2 regulates energy expenditure and adiposity. J Biol Chem. 2008;283:541–553. [DOI] [PubMed] [Google Scholar]
- 22. Green AR, Young P, Krivinskas S, et al. The expression of ERα, ERβ and PR in lobular carcinoma in situ of the breast determined using laser microdissection and real-time PCR. Histopathology. 2009;54:419–427. [DOI] [PubMed] [Google Scholar]
- 23. Zhou D, Quach KM, Yang C, Lee SY, Pohajdak B, Chen S. PNRC: a proline-rich nuclear receptor coregulatory protein that modulates transcriptional activation of multiple nuclear receptors including orphan receptors SF1 (steroidogenic factor 1) and ERRα1 (estrogen related receptor α-1). Mol Endocrinol. 2000;14:986–998. [DOI] [PubMed] [Google Scholar]
- 24. Zhou D, Masri S, Ye JJ, Chen S. Transcriptional regulation of the mouse PNRC2 promoter by the nuclear factor Y (NFY) and E2F1. Gene. 2005;361:89–100. [DOI] [PubMed] [Google Scholar]
- 25. Perets R, Kaplan T, Stein I, et al. Genome-wide analysis of androgen receptor targets reveals COUP-TF1 as a novel player in human prostate cancer. PLoS One. 2012;7:e46467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Denayer S, Helsen C, Thorrez L, Haelens A, Claessens F. The rules of DNA recognition by the androgen receptor. Mol Endocrinol. 2010;24:898–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tewari AK, Yardimci GG, Shibata Y, et al. Chromatin accessibility reveals insights into androgen receptor activation and transcriptional specificity. Genome Biol. 2012;13:R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Q, Li W, Liu XS, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27:380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lau KM, LaSpina M, Long J, Ho SM. Expression of estrogen receptor (ER)-α and ER-β in normal and malignant prostatic epithelial cells: regulation by methylation and involvement in growth regulation. Cancer Res. 2000;60:3175–3182. [PubMed] [Google Scholar]
- 30. Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell. 2008;30:701–711. [DOI] [PubMed] [Google Scholar]
- 31. Dai F, Yu L, He H, et al. Human serum and glucocorticoid-inducible kinase-like kinase (SGKL) phosphorylates glycogen syntheses kinase 3 beta (GSK-3β) at serine-9 through direct interaction. Biochem Biophys Res Commun. 2002;293:1191–1196. [DOI] [PubMed] [Google Scholar]
- 32. Liu M, Chen L, Chan TH, et al. Serum and glucocorticoid kinase 3 at 8q13.1 promotes cell proliferation and survival in hepatocellular carcinoma. Hepatology. 2012;55:1754–1765. [DOI] [PubMed] [Google Scholar]
- 33. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grewe M, Gansauge F, Schmid RM, Adler G, Seufferlein T. Regulation of cell growth and cyclin D1 expression by the constitutively active FRAP-p70s6K pathway in human pancreatic cancer cells. Cancer Res. 1999;59:3581–3587. [PubMed] [Google Scholar]
- 35. Xu Y, Chen SY, Ross KN, Balk SP. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 2006;66:7783–7792. [DOI] [PubMed] [Google Scholar]
- 36. Akar U, Ozpolat B, Mehta K, et al. Targeting p70S6K prevented lung metastasis in a breast cancer xenograft model. Mol Cancer Ther. 2010;9:1180–1187. [DOI] [PubMed] [Google Scholar]
- 37. Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA. 1998;95:1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tessier M, Woodgett JR. Serum and glucocorticoid-regulated protein kinases: variations on a theme. J Cell Biochem. 2006;98:1391–1407. [DOI] [PubMed] [Google Scholar]
- 39. Shanmugam I, Cheng G, Terranova PF, Thrasher JB, Thomas CP, Li B. Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival. Cell Death Differ. 2007;14:2085–2094. [DOI] [PubMed] [Google Scholar]
- 40. Massie CE, Lynch A, Ramos-Montoya A, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30:2719–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arnold JT, Liu X, Allen JD, Le H, McFann KK, Blackman MR. Androgen receptor or estrogen receptor-β blockade alters DHEA-, DHT-, and E2-induced proliferation and PSA production in human prostate cancer cells. Prostate. 2007;67:1152–1162. [DOI] [PubMed] [Google Scholar]
- 42. Panet-Raymond V, Gottlieb B, Beitel LK, Pinsky L, Trifiro MA. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol Cell Endocrinol. 2000;167:139–150. [DOI] [PubMed] [Google Scholar]
- 43. Slusarz A, Jackson GA, Day JK, et al. Aggressive prostate cancer is prevented in ERαKO mice and stimulated in ERβKO TRAMP mice. Endocrinology. 2012;153:4160–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ricke WA, McPherson SJ, Bianco JJ, Cunha GR, Wang Y, Risbridger GP. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor α signaling. FASEB J. 2008;22:1512–1520. [DOI] [PubMed] [Google Scholar]
- 45. Dey P, Strom A, Gustafsson JA. Estrogen receptor β upregulates FOXO3a and causes induction of apoptosis through PUMA in prostate cancer [published online ahead of print September 30, 2013]. Oncogene. doi: 10.1038/onc.2013.384. [DOI] [PubMed] [Google Scholar]
- 46. Maggiolini M, Recchia AG, Carpino A, et al. Oestrogen receptor beta is required for androgen-stimulated proliferation of LNCaP prostate cancer cells. J Mol Endocrinol. 2004;32:777–791. [DOI] [PubMed] [Google Scholar]
- 47. Migliaccio A, Castoria G, Di Domenico M, et al. Steroid-induced androgen receptor-oestradiol receptor β-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19:5406–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Capra M, Nuciforo PG, Confalonieri S, et al. Frequent alterations in the expression of serine/threonine kinases in human cancers. Cancer Res. 2006;66:8147–8154. [DOI] [PubMed] [Google Scholar]
- 49. Speers C, Tsimelzon A, Sexton K, et al. Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clin Cancer Res. 2009;15:6327–6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu J, Wan M, He Q, et al. SGK3 is associated with estrogen receptor expression in breast cancer. Breast Cancer Res Treat. 2012;134:531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu J, Liao L, Qin J, Xu J, Liu D, Songyang Z. Identification of Flightless-I as a substrate of the cytokine-independent survival kinase CISK. J Biol Chem. 2009;284:14377–14385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang Y, Xu W, Zhou D, Neckers L, Chen S. Coordinated regulation of serum- and glucocorticoid-inducible kinase 3 by a C-terminal hydrophobic motif and Hsp90-Cdc37 chaperone complex. J Biol Chem. 2014;289:4815–4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu D, Yang X, Songyang Z. Identification of CISK, a new member of the SGK kinase family that promotes IL-3-dependent survival. Curr Biol. 2000;10:1233–1236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.