

Abstract
Our laboratory previously has identified soluble guanylyl cyclase α1 (sGCα1) as a direct target of androgen receptor and essential for prostate cancer cell growth via a pathway independent of nitric oxide (NO) signaling. We identified the COP9 signalosome subunit 4 (CSN4) as a novel interacting partner for sGCα1. Importantly, the CSN4-sGCα1 interaction inhibits sGCα1 proteasomal degradation. Consistent with this, disruption of CSN4 led to a significant decrease in prostate cancer cell proliferation, which was significantly but not completely rescued by sGCα1 overexpression, opening the possibility of an additional target of CSN4. Interestingly, immunoprecipitation experiments showed that p53 is found in the CSN4-sGCα1 cytoplasmic protein complex. However, in contrast to sGCα1, p53 protein stability was compromised by CSN4, leading to prostate cancer cell survival and proliferation. Interestingly, we observed that CSN4 was overexpressed in prostate tumors, and its protein level correlates directly with sGCα1 and inversely with p53 proteins, mimicking what was observed in prostate cancer cells. Our data further showed that CSN4 silencing decreased CSN5 protein levels and suggest that the CSN4 effects on sGCα1 and p53 proteins are mediated by CSN5. Lastly, our study showed that caseine kinase-2 (CK2) was involved in regulating p53 and sGCα1 protein stability as determined by both disruption of CK2 expression and inhibition of its kinase activity. Collectively, our study has identified a novel endogenous CSN4-CSN5-CK2 complex with sGCα1and p53 that oppositely controls the stability of these 2 proteins and provides prostate cancer cells an important mechanism for survival and proliferation.
Androgens and androgen receptor (AR) play essential roles in development of prostate cancer. Indeed, one of the most important mechanisms for the development of castration-resistant prostate cancer is overexpression and restoration of AR transcriptional activity (1–3). AR action is mediated by androgen-regulated genes, of which many have been identified in recent years. Our laboratory has focused its recent efforts on one of these newly identified genes, soluble guanylyl cyclase α-1 (sGCα1), (gene name GUCY1A3). This gene is a direct target of AR and mediates the progrowth and prosurvival functions of AR-positive prostate cancer cells (4–6). The classical function of sGCα1 is to heterodimerize with sGCβ1, forming the sGC enzyme, the principal receptor for nitric oxide (NO) and mediator of NO signaling (6–9). Interestingly, both the progrowth and prosurvival functions of sGCα1 in prostate cancer are independent of NO signaling (6, 10). Recently, we also reported that sGCα1 physically associates with and sequesters p53 in the cytoplasm and prevents prostate cancer cells from undergoing p53-dependent apoptosis (10). As one of the most important inducers of apoptosis in mammalian biology (11), p53 is the most commonly mutated gene in human cancers and is under complex regulation (12–15). Although p53 mutations are rare in early-stage prostate cancer, they are far more common in advanced disease (14, 15). Many interacting partners have been identified for p53, from proteins that regulate p53 gene expression to proteins that control p53 stability and to proteins that regulate p53 activity as a transcription factor (16, 17). Among these many interacting proteins, sGCα1 represents a new partner for p53 that blocks its activity by mediating its cytoplasmic sequestration (10). We have also previously shown that p53 can disrupt AR transcriptional activity in prostate cancer cells (18).
To disrupt these progrowth and prosurvival functions of sGCα1, we synthesized an interacting peptide, which exhibited potent cytotoxicity against both androgen-dependent and castration-resistant prostate cancer cells and, more importantly, strong anticancer activity in mouse xenograft studies (19). Furthermore, sGCα1 is overexpressed in castration-resistant prostate tumors, whereas sGCβ1 showed very weak expression (6). In view of all these published data, we hypothesized that sGCα1 may form a protein complex, independent of its complex with sGCβ1, that serves progrowth and prosurvival functions in prostate cancer.
To identify such a protein complex in prostate cancer, mass spectrometric (MS) analysis was used and identified the COP9 signalosome subunit 4 (CSN4) (CSN4; gene name COPS4) as a novel binding partner for sGCα1. The CSN protein complex consists of 8 core subunits (CSN1–CSN8) and is evolutionarily conserved in all eukaryotes from yeast to humans (20, 21). Although first identified as a negative regulator of photo-morphogenesis in plants, the mammalian CSN complex plays very important roles in regulating cell proliferation and survival (22–25). The CSN complex shows structural similarity with the 19S proteasome-regulatory complex (26). The well-characterized role of CSN is to differentially regulate ubiquitin-proteasome-dependent degradation of many tumor suppressors and oncoproteins (27–29), including p53 and p27 (30, 31).
When CSN was purified from mammalian cells, 2 kinases were identified to copurify: serine threonine kinases like protein kinase-D (PKD) and casein kinase-2 (CK2). CSN-associated kinase activity was later shown to be involved in phosphorylating c-Jun and p53, with opposite effects on their subsequent ubiquitin-dependent degradation (32–34). Interestingly Curcumin can inhibit enzyme activity of both kinases and prevent the degradation of p53 (32, 35). In addition, different CSN subunits, such as CSN7, CSN3, and CSN2, are reported to bind and be phosphorylated by CK2 and PKD kinases (32, 36). Recently it has been reported that in neuroblastoma cells, CSN4 regulates the stability of 2 synaptic proteins, Snapin and Stonin 2. Interestingly, Snapin is phosphorylated by PKD, and its expression is decreased in response to PKD kinase inhibition (37).
In this study, we report, for the first time, that CSN4 is a novel binding partner for sGCα1. CSN4 interacts not only with sGCα1, but also p53 in prostate cancer cells, having a positive effect on sGCα1 protein and negative on p53. Our data strongly suggest that CSN5 mediates the CSN4 affects on p53. Furthermore, CK2 kinase activity is involved in regulating the stability of sGCα1 and p53 proteins. Lastly, immunoprecipitation (IP) experiments show the existence in prostate cancer cells of a novel endogenous, cytoplasmic complex, consisting of CSN4, CSN5, sGCα1, p53, and CK2.
Materials and Methods
Cell culture, small interfering RNA (siRNA) transfection, and inhibitors
LNCaP, C81, CWR-22Rv1, and PC-3 cells were grown as previously described (from American Type Culture Collection [ATCC], passage 15–30 for all cell lines) and control siRNA, sGCα1 siRNA (6, 10), CSN4 siRNA ACAGCAUCUUGCA UCUAUAUU), CSN4 siRNA (3′-untranslated region) (GUGAAAUAUCUGUGGCUAAUU), CSN5 siRNA (CUUGAGCUGUUGUGGA AUA), and CK2 siRNA (UCAAGAUGACUACC AGCUGUU) (Dharmacon) were transfected at a final concentration of 50 nM into cells using Lipofectamine siMAX (Invitrogen) or Lipofectamine 2000 (Invitrogen). LNCaP and CWR-22Rv1 cells were treated with 40 μM Curcumin or Emodin (from Sigma) for 24 hours and 48 hours, after which cell extracts were prepared using 2% sodium dodecyl sulfate (SDS) and then subjected to Western blotting.
Reporter assay and plasmid transfection
LNCaP cells were grown to 70%–80% confluence in RPMI-1640 with 10% fetal bovine serum. After 24 hours, cells were transiently transfected with 0.1 μg p53-Luc reporter plasmid, and control siRNA, or CSN4 (3′-untranslated region specific) siRNA, CSN5 siRNA, and/or CSN4/cytomegalovirus plasmid. The pCH110 plasmid (0.5 μg) encoding β-galactosidase was used to control for transfection efficiency (38). The p53-Luc plasmid was kindly provided by Dr William Taylor and contains three p53-responsive elements. Lipofectamine 2000 (Invitrogen) was used for transfection and luciferase activity was measured using Luciferase assay system from Promega. LNCaP and CWR-22Rv1 cells were transiently transfected with CSN4 siRNA, control siRNA, p53 siRNA, CSN5 siRNA, 2 μg sGCα1/pCI-Neo, and 2 μg CSN4/cytomegalovirus and, 48 hours after transfection, cell extracts were prepared using 2% SDS and subjected to Western blotting.
Proliferation and apoptosis assays
For proliferation, cells were transfected with siRNA and then 72 hours after transfection, 20 000 cells were seeded in 24-well plates. The MTT assay (Sigma) was used as described elsewhere (19) to determine cell number. For apoptosis, 5000 cells were seeded in 96-well plates and treated with vehicle (dimethylsulfoxide), CSN4 siRNA (50 μM), Etoposide (20 μM) (Sigma) at different time points. Caspase-3/7 activity was measured using the Apo-ONE Homogeneous Caspase-3/7 assay kit (Promega Corp). Poly (ADP-ribose) polymerase (PARP) cleavage was also used to measure apoptosis in LNCaP and CWR-22Rv1 cells, after being transiently transfected with CSN4 siRNA or control siRNA. Cells were harvested 72 hours after transfection and subjected to Western blotting.
Western blotting and cell fractionation
Western blotting was performed as described elsewhere (6) using antibodies against sGCα1 (Cayman Chemical; catalog no. 160895), CSN4 (Abcam; catalog no. ab12322), CSN5 (Santa Cruz Biotechnology; catalog no. sc-13157), p53 (Santa Cruz Biotechnology; catalog no. sc-263), β-tubulin (Abcam; catalog no. ab6046), retinoic acid receptor-α (Santa Cruz Biotechnology; catalog no. sc-551), β-actin (Abcam; catalog no. ab-6276), CK2 (Thermo Scientific; catalog no. PA1–86381), Survivin (71G4B7), and PARP (Cell Signaling Technology; catalog nos. 2808 and 9242). LNCaP cells treated with CSN5 siRNA were washed with cold PBS and harvested, after which 10% of the cells were saved as Input and the remaining portion was subjected to cytosolic and nuclear cell fractionation using Nuclear/Cytosol Fractionation Kit (MBL International). The fractions were subjected to Western blotting to measure CSN5 and p53 protein levels.
Prostate tissues
Prostate tumors were obtained from the Cooperative Human Tissue Network (CHTN). Protein extracts were prepared by boiling 12 normal and 20 tumor tissues (Gleason scores between 6 and 7) in 3× SDS buffer and then subjected to Western blotting to measure CSN4, sGCα1, and p53 expression levels.
Semiquantitative RT-PCR and quantitative RT-PCR
Total mRNA was isolated from LNCaP and CWR-22Rv1 cells using the Trizol reagent following the manufacturer's protocol (Invitrogen) and subjected to semiquantitative RT-PCR and quantitative RT-PCR analyses as described elsewhere (39). The PCR upstream and downstream primers, respectively, used for each gene were: CSN4, 5′-GTAAG CCTCTGCCTGGACTG-3′ and 5′-AGGAGCAGGTTGCTTCCATA-3′; glyceraldehyde 3-phosphate dehydrogenase, 5′-CGACCACTTTGTCAAGCTCA-3′ and 5′-AGGGGAGATTCAGTGTGGTG-3′; CSN5, 5′-GCCAACCTGTTTTGCATTTT-3′and 5′-TCTGCTGAAGATGGTGATGC-3′; Survivin, 5′-GGACCACCGCATCTCTACAT-3′and 5′-GACAGAAAGGAAAG CGCAAC-3′; p53, 5′-GGCCCACTTCA CCGTACTAA-3′ and 5′-GTGGTTTCAAGG CCAGATGT-3′; sGCα1, 5′-AGCAGTGT GGAGAGCTGGAT-3′ and 5′-CTGATCCAG AGTGCAGTCCA-3′.
Immunocytochemistry
Immunocytochemistry was used to study the subcellular localization of sGCα1 and CSN4 in LNCaP and CWR-22Rv1 cells as described in Reference 38. Reagents used were an anti-CSN4 antibody (1:100 dilution; Abcam) and a fluorescein isothiocyanate-labeled anti-sGCα1 antibody (1:100 dilution; Santa Cruz Biotechnology). Note that all micrographs were taken at the same microscope settings.
Immunoprecipitation
IP experiments were performed as described previously (10, 19). Whole-cell extracts from LNCaP and CWR-22Rv1 cells were subjected to IP using Protein A/G plus Agarose (Santa Cruz). IP antibodies were against sGCα1 (Cayman Chemical), p53 (Santa Cruz Biotechnology), CSN5 (Santa Cruz), CSN4 (Abcam), CK2 (Thermo Scientific), or rabbit or mouse IgG (Santa Cruz) as control.
Mass spectrometry analysis
Whole-cell extracts from LNCaP cells were subjected to an IP experiment, as described previously (10, 19), using an antibody against sGCα1 (Cayman Chemical) or IgG as a negative control. The purified material was run on an SDS-PAGE gel and stained with Coomassie blue. Stained proteins only found in the anti-sGCα1 IP were identified by mass spectrometry analysis (provided by the Mass Spectrometry-based Proteomics Facility at the University of Michigan).
Results
The CSN4 subunit of the CSN signalosome complex is a novel binding partner for sGCα1 in prostate cancer cells
Previously we identified sGCα1 as a novel androgen-regulated gene required for proliferation of both androgen-dependent and castration-resistant, AR-positive prostate cancer cells (6). Furthermore, we reported that sGCα1 expression is significantly elevated in prostate cancer cells and prostate tumors as compared with sGCβ1, with which sGCα1 heterodimerizes and mediates NO signaling. Interestingly the sGCα1 progrowth effect in prostate cancer cells is independent of the NO-signaling pathway (6, 10). Based on these results, we hypothesized that sGCα1 may exist in a protein complex distinct from the heterodimeric complex with sGCβ1, to regulate prostate cancer cell proliferation.
To identify novel sGCα1-interacting proteins, we immunoprecipitated endogenous sGCα1 from LNCaP cells and used mass spectrometric (MS) analysis to identify copurified proteins. This analysis led to the identification of CSN4 (COP9 signalosome subunit 4) as a novel interacting partner for sGCα1. To confirm the MS analysis, immunoprecipitation (IP) was performed in LNCaP cells with endogenous proteins. IP purification of sGCα1 resulted in copurification of endogenous CSN4, which was not seen with the control IgG (Figure 1A). A complementary IP showed that sGCα1 was copurified with endogenous CSN4 (Figure 1B). Collectively, these results strongly suggest that endogenous sGCα1 and CSN4 coassociate in androgen-dependent LNCaP cells. The same interaction was observed in CWR-22Rv1 cells (Figure 1, A and B), a human prostatic carcinoma xenograft cell line that shows androgen-independent proliferation and expresses AR and different AR splice variants, including AR-V7, which play an important role in development of castration resistance in prostate cancer (40, 41). Interestingly, sGCα1 and CSN4 are overexpressed in these cells compared with androgen-dependent LNCaP cells (see Figure 2B).
Figure 1.
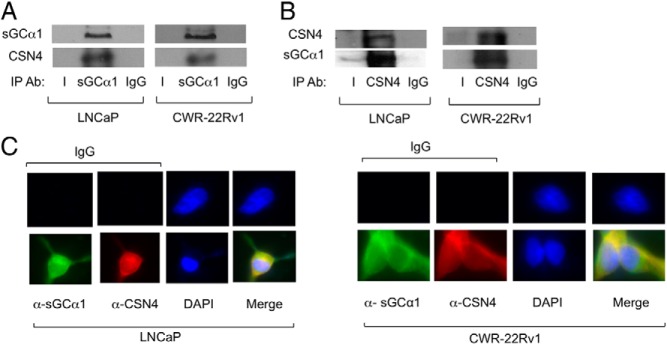
CSN4 is a novel binding partner for sGCα1 in prostate cancer cells. LNCaP and CWR-22Rv1 cell extracts were subjected to IP (panel A) using an (A) anti-sGCα1 antibody or (B) anti-CSN4 antibody. IgG was used as a control. Western blotting was used to measure sGCα1 and CSN4 proteins. C, LNCaP and CWR-22Rv1 cells were subjected to immunocytochemistry using anti-sGCα1 and anti-CSN4 antibodies to measure subcellular localization of endogenous sGCα1 and CSN4. 4′6-diamidino-2-phenylindole (DAPI) stains nuclei. Ab, antibody.
Figure 2.

CSN4 positively regulates sGCα1 stability and is involved in sGCα1-dependent prostate cancer cell growth. A, LNCaP, C81, CWR-22Rv1, and PC-3 cells were transfected with control (Ctrl) siRNA or CSN4 siRNA or (B) LNCaP and CWR-22Rv1 cells were transfected with Ctrl siRNA or CSN4 siRNA with or without CSN4/CMV (cytomegalovirus) expression plasmid. C, LNCaP and CWR-22Rv1 cells were transfected with Ctrl siRNA or CSN4 siRNA and treated with 50 μg/mL cycloheximide (CHX) and 10 μM MG132. CSN4 and sGCα1 levels were measured by Western blotting. Numbers above represent protein quantification levels relative to the first condition, which was set to 1. In panels A, B, and C, β-actin was used as loading control and the molecular weights of the proteins are 77 kDa (sGCα1), 49 kDa (CSN4), and 42 kDa (β-actin). LNCaP and CWR-22Rv1 cells were transfected with (D) Ctrl siRNA or CSN4 siRNA or (E) Ctrl siRNA or CSN4 siRNA with or wtihout sGCα1 expression plasmid. Cell density was measured by methylthiazol tetrazolium assay. Data points in panels D and E represent an average of 3 independent experiments plus SDs. Student's t test was performed to show statistical significance (P < .01), as indicated by asterisks.
To confirm the CSN4-sGCα1 interaction, immunocytochemistry was used in LNCaP and CWR-22RV1 cells to examine the subcellular localization of endogenous CSN4 and sGCα1. As shown in Figure 1C, endogenous CSN4 was localized in cytoplasm and colocalized with sGCα1, suggestive of the existence of a cytoplasmic CSN4-sGCα1 complex. Together, all these results show that endogenous CSN4 and sGCα1 interact in the cytoplasm of both androgen-dependent and castration-resistant prostate cancer cells.
CSN4 promotes prostate cancer cell proliferation by up-regulating sGCα1
The CSN complex shares structural similarities with the proteasome lid subcomplex and can differentially regulate the stability of several target proteins (42). Because CSN4 interacts with sGCα1, we were interested to determine whether this interaction affects the cellular levels of sGCα1. To test our hypothesis, we depleted endogenous level of CSN4 in LNCaP cells by siRNA, which interestingly resulted in markedly reduced levels of sGCα1 protein (Figure 2A). Importantly, when CSN4 was exogenously expressed in CSN4 siRNA-treated cells, sGCα1 protein expression was rescued (Figure 2B), clearly demonstrating that CSN4 is responsible for regulating endogenous sGCα1 protein in LNCaP cells. The same findings were made in C81 cells, an androgen-independent line derived from LNCaP cells (43), and CWR-22Rv1 cells, in which knockdown of CSN4 diminished sGCα1 protein levels (Figure 2A) and exogenous CSN4 rescued these levels (Figure 2B). As shown previously (6), the AR-negative PC-3 cells exhibit very low expression of sGCα1 and CSN4 (Figure 2A). We also observed that sGCα1 mRNA levels remain unaffected in CSN4 knockdown in both LNCaP and CWR-22Rv1 cells (Supplemental Figure 1, A and B), clearly demonstrating that the CSN4 targets the sGCα1 protein.
One possible mechanism of CSN4 regulation of sGCα1 is inhibition of protein degradation. In view of the well-known function of the CSN complex in regulating proteasome-dependent stability of different proteins, we were interested to study a possible role for CSN4 in inhibiting proteasome-dependent degradation of sGCα1. To address this, we measured the time-dependent degradation of endogenous sGCα1 in CSN4 knockdown LNCaP cells, using the translational inhibitor cycloheximide and proteasome inhibitor MG132. The sGCα1 steady-state levels strongly decreased between 0 and 5 hours in CSN4 knockdown LNCaP cells, consistent with down-regulation of sGCα1 in response to reduced CSN4. Interestingly, when MG132 was added to CSN4 siRNA-treated cells, sGCα1 steady state levels were maintained during the first 5 hours, and these levels were comparable to control siRNA-treated cells. The same findings were made in CWR-22Rv1 cells (Figure 2C). Based on these results, we conclude that CSN4 positively regulates endogenous sGCα1 protein stability in both androgen-dependent and castration-resistant prostate cancer cells by preventing its proteasome-dependent degradation.
Previously, we published that reduced sGCα1 expression directly correlates with decreased prostate cancer cell growth (6). Because down-regulation of CSN4 resulted in diminution of sGCα1 protein in prostate cancer cells, we were interested to determine whether CSN4 plays any role in sGCα1-mediated prostate cancer cellular proliferation. To test our hypothesis, endogenous CSN4 expression was reduced in LNCaP cells using CSN4-specific siRNA, and cell growth was measured. As shown in Figure 2D, knockdown of CSN4 resulted in a significant decrease in LNCaP cellular proliferation, whereas control siRNA had no effect. Importantly, the CSN4-knockdown cells were rescued by exogenous overexpression of CSN4 (Supplemental Figure 2A), showing that CSN4 does indeed control prostate cancer cell proliferation. Consistent with the LNCaP cells, C81 (Supplemental Figure 2A) and CWR-22Rv1 (Figure 2D) cells also exhibit significantly reduced growth when in the context of diminution of CSN4 knockdown. Interestingly, CSN4 silencing had no effect on the growth of PC-3 cells (Supplemental Figure 2A), which are deficient in sGCα1 protein and have weak expression of CSN4 compared with LNCaP and CWR-22Rv1 cells (Figure 2A). Based on these results, we conclude that endogenous CSN4 protein levels are directly correlated to prostate cancer cellular proliferation.
In view of the above results, we hypothesized that one mechanism for CSN4 control of cell growth is by regulation of the sGCα1 protein. We tested this hypothesis by overexpressing sGCα1, resulting in significant elevation of sGCα1 protein in the CSN4 siRNA-treated LNCaP cells (Supplemental Figure 2B). Interestingly, overexpression of sGCα1 significantly rescued the growth of CSN4 siRNA-treated cells, both LNCaP and CWR-22Rv1, whereas sGCα1 had only a weakly positive effect in control siRNA-treated cells (Figure 2E). Collectively, these data suggest that sGCα1 down-regulation is responsible for the negative effect of CSN4 siRNA on cell growth.
To determine whether NO signaling is involved in the sGCα1 rescue of CSN4-depleted cells, we used the sGCα1 mutant (D531A), which is deficient in cyclase activity (10). Importantly, this mutant rescued the CSN4-knockdown cells as well as wild-type sGCα1 (Supplemental Figure 3A). Western blotting confirmed equal expression of both sGCα1 proteins in LNCaP cells (Supplemental Figure 3B). In addition, sGCβ1, the binding partner for sGCα1 in NO signaling, was very weakly expressed and remained unaffected in response to CSN4 knockdown in both LNCaP and CWR-22Rv1 cells (Supplemental Figure 3C). Collectively, these data demonstrate that role of sGCα1 in CSN4-mediated growth is independent of NO signaling in prostate cancer cells.
CSN4 down-regulates p53 protein and leads to prostate cancer cell survival and proliferation
Our earlier data showed that overexpression of sGCα1 can significantly, but incompletely, rescue cells with CSN4 knockdown (see Figure 2E). The incomplete rescue suggests that CSN4 knockdown affects additional targets. Because we published earlier that sGCα1 interacts with p53 (10), we examined p53 as an additional target. Interestingly, complementary IP experiments using anti-p53 and anti-sGCα1 antibodies revealed that p53 is associated with CSN4 and sGCα1 prostate cancer cells (Figure 3A). Because CSN4 stabilized the sGCα1 protein, we monitored the p53 protein in the context of altered levels of CSN4. Interestingly, lowering levels of CSN4 resulted in increased p53 protein in LNCaP, C81, and CWR-22Rv1 cells (Figure 3B). Importantly, this positive p53 effect of the CSN4 siRNA was abolished by overexpressed exogenous CSN4 (Figure 3C), clearly demonstrating that CSN4 is indeed regulating p53 protein levels. The mRNA levels of p53 were not affected by CSN4 (Supplemental Figure 1A), showing that CSN4 targets the p53 protein. This was confirmed by performing a protein stability assay using cycloheximide. As shown in Figure 3D, siRNA knockdown of CSN4 greatly enhanced p53 protein stability, mimicking what was observed with the proteasome inhibitor MG132 (Figure 3D).
Figure 3.

CSN4 negatively regulates p53 stability and provides prostate cancer cells enhanced growth. A, CWR-22Rv1 cell extracts were subjected to IP using antibody against p53 or sGCα1. IgG was used as negative control IP. Western blotting was used to measure p53, sGCα1, and CSN4. B, LNCaP, C81, and CWR-22Rv1 cells were transfected with Ctrl siRNA or CSN4 siRNA and subjected to Western blotting to measure CSN4 and p53. C, LNCaP and CWR-22Rv1 cells were transfected with control (Ctrl) siRNA, CSN4 siRNA, and CSN4/CMV (cytomegalovirus) expression plasmid and cell lysates were subjected to Western blotting to measure p53 and CSN4. D, LNCaP cells were treated with vehicle, Ctrl siRNA, and 10 μM MG132, or CSN4 siRNA, in the absence or presence of 50 μg/mL cycloheximide (CHX) for different incubation periods, and Western blotting was used to measure p53 and CSN4. Numbers above represent protein quantification levels relative to the first condition, which was set to 1. In panels B, C, and D, β-actin was used as loading control and the molecular weights of the proteins are 77 kDa (sGCα1), 49 kDa (CSN4), 53 kDa (p53), and 42 kDa (β-actin). E, LNCaP and CWR-22Rv1 cells were transfected with Ctrl siRNA or CSN4 siRNA, with or without p53 siRNA, and cell density was measured by mammalian transient transfection assay. F, LNCaP cells were transfected with Ctrl siRNA or CSN4 siRNA, with or without p53 siRNA or sGCα1 expression plasmid, and cell density was measured by methylthiazol tetrazolium assay. In panels E and F, data points represents an average of 3 independent experiments plus SDs. Student's t test was performed to show statistical significance (P < .03), as indicated by asterisks. Ab, antibody.
Because one important cellular effect of p53 is to shut down cellular proliferation, we tested the possibility that elevated levels of p53 may be involved in the negative effect of CSN4 siRNA on cell growth. To do this, we performed a knockdown of p53 in the background of CSN4 knockdown, which resulted in a significant rescue of the cells (Figure 3E). Yet, like the experiment with sGCα1 overexpression (see Figure 2E), the rescue with p53 knockdown was not complete. Thus, we opted to repeat the rescue experiment with both sGCα1 overexpression and p53 knockdown, resulting in nearly 100% rescue of cells treated with CSN4 siRNA (Figure 3F). Collectively, these results suggest that the CSN4 regulates prostate cancer cell proliferation via the combined effects on the progrowth sGCα1 and the antigrowth p53.
p53 is an important regulator of gene transcription. To begin to study this, we first measured CSN4 effect on p53 transcriptional activity. As measured by a reporter gene assay, endogenous p53 activity was significantly enhanced by CSN4 siRNA, and this was relieved by CSN4 overexpression (Figure 4A). To study an endogenous p53-repressed gene, we chose survivin, which was markedly increased at both the mRNA and protein levels in the context of CSN4 knockdown (Figure 4B), suggesting that CSN4 knockdown leads to p53-dependent apoptosis. Apoptosis was first measured by monitoring Caspase3/7 activity, which was strongly enhanced in response to CSN4 siRNA, comparable to what was observed with Etoposide treatment (Figure 4C). This finding was confirmed using PARP cleavage, which was also greatly enhanced when cells were treated with CSN4 siRNA (Figure 4D). To directly test the role of p53, LNCaP and CWR-22Rv1 cells were transfected with CSN4 siRNA alone or combined with p53 siRNA. Significantly, co-knockdown of p53 markedly reduced PARP cleavage induced by CSN4 knockdown (Figure 4E), demonstrating that apoptosis triggered by CSN4 knockdown is p53 dependent. It is important to mention that p53 siRNA did not influence the negative effect of CSN4 knockdown on sGCα1 (Supplemental Figure 4, A and B). Also, knockdown of endogenous p53 (Supplemental Figure 4D) had no effect on prostate cancer cell proliferation (Supplemental Figure 4C), as reported earlier (10).
Figure 4.

CSN4 affects p53 transcriptional activity and disrupts p53-dependent apoptosis. A, LNCaP cells were transfected with p53-Luc and Ctrl siRNA or CSN4 siRNA, with or without CSN4 expression plasmid, and p53 transcriptional activity was measured by luciferase assay. B, LNCaP and CWR-22Rv1 cells were transfected with Ctrl siRNA or CSN4 siRNA and survivin gene expression was measured by quantitative RT-PCR or Western blotting. LNCaP and C81 cells were transfected with Ctrl siRNA or CSN4 siRNA and apoptosis was measured by (C) Caspase 3/7 activity or (E) Western blotting of cleaved PARP. F, LNCaP and CWR-22Rv1 cells were transfected with control siRNA or CSN4 siRNA, with or without p53 siRNA, and apoptosis was measured by Western blotting of cleaved PARP. In panels B, D, and E, β-actin was used as loading control and the molecular weights of the proteins are 16 kDa (Survivin), 49 kDa (CSN4), 53 kDa (p53), 116 kDa (PARP), 89 kDa (cleaved PARP), and 42 kDa (β-actin). In panels A and C, bar graphs represents an average of 3 independent experiments plus SDs. Student's t test was performed to show statistical significance (P < .005), as indicated by asterisks. Bar graphs represent averages of 3 independent experiments plus SDs.
Expression of CSN4 protein is strongly enhanced in prostate cancer
To obtain evidence for CSN4 regulation of sGCα1 and p53 in prostate cancer, we monitored the expression of all 3 proteins in prostate tumors. The first set of tumors clearly demonstrated high CSN4 protein overexpression as compared with normal (Figure 5A, upper panel). The same finding was made when CSN4 mRNA levels were measured (Supplemental Figure 5A). All 3 proteins were measured in a second set of tumors, which also exhibited a strong overexpression of CSN4 protein in prostate cancer (Figure 5A,). Intriguingly, these cancer tissues also exhibited high levels of sGCα1 and very low levels of p53, whereas the normal prostate tissues expressed little CSN4 and sGCα1, but high levels of p53 (Figure 5B). When protein levels were quantified, we observed a statistically significant direct correlation between CSN4 and sGCα1 proteins and inverse correlation between CSN4 and p53 (Figure 5B, lower). These data are consistent with the activities of CSN4 in prostate cancer cells and make it possible that CSN4 is indeed regulating sGCα1 and p53 protein levels in prostate cancer.
Figure 5.

CSN4 protein is overexpressed in prostate tumors and correlates directly with sGCα1 and inversely with p53 proteins. A, Western blotting was used to measure expression of CSN4, sGCα1, and p53 proteins in normal prostate tissues or prostate tumors. B, Expression of CSN4, sGCα1, and p53 proteins in prostate tumors was quantified and plotted relating CSN4 to either sGCα1 or p53. β-Actin was used as loading control and the molecular weights of the proteins are 77 kDa (sGCα1), 49 kDa (CSN4), 53 kDa (p53), and 42 kDa (β-actin). Linear regression analysis was done to compare protein levels of CSN4, and sGCα1 or CSN4 and p53, with R2 values of 0.73532 and 0.02379 and P values of 0.015 and 0.753, respectively.
CSN5 mediates the CSN4 effects on p53 and sGCα1
CSN4 was identified as a part of the CSN. Another CSN subunit of particular interest here is CSN5 (also known as JAB1), which previously was reported to associate with p53 and mediate its nuclear export (44). Interestingly, siRNA knockdown of CSN4 resulted in markedly reduced levels of CSN5 in both LNCaP and CWR-22Rv1 cells (Figure 6A). The complementary experiment, CSN5 knockdown, did not significantly affect CSN4 protein (Figure 6B), suggesting that CSN5 acts downstream of CSN4.
Figure 6.
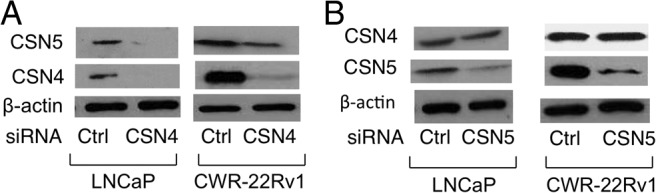
CSN4 regulates CSN5 protein levels in prostate cancer cell lines. LNCaP and CWR-22Rv1 cells were transfected with (A) Ctrl siRNA or CSN4 siRNA or (B) Ctrl siRNA or CSN5 siRNA, and Western blotting was used to measure CSN4 and CSN5. β-Actin was used as loading control and the molecular weights of the proteins are 38 kDa (CSN5), 49 kDa (CSN4), and 42 kDa (β-actin).
Although there was no effect on CSN4 protein levels, CSN5 depletion led to elevated levels of p53 (Figure 7A) and reduced levels of sGCα1 (Figure 7B), mimicking what was observed with the CSN4 knockdown in our earlier experiments (see Figures 2A and 3B). This finding suggests that the effects of CSN4 knockdown on the sGCα1 and p53 proteins are mediated by the elevated levels of CSN5 protein that result from the CSN4 knockdown. IP experiments demonstrated that CSN5 can interact with both p53 (Figure 7C), as shown previously (35), and sGCα1 (Figure 7D and Supplemental Figure 4D), once again duplicating what was observed with CSN4 (see Figure 3A). Indeed, our data showed that CSN5 was found in a single complex with CSN4, p53, and sGCα1 (see Figure 8D). With respect to p53, we confirmed that CSN5 regulates p53 subcellular localization in prostate cancer cells (44), as CSN5 knockdown led to greatly enhanced levels of nuclear p53 (Figure 7E) and increased p53 transcriptional activity (Figure 7F). Because CSN5 regulates the levels of p53 and sGCα1, we expected that CSN5 would act on prostate cancer cell proliferation. Indeed, CSN5 depletion resulted in markedly reduced proliferation of both CWR-22Rv1 (Figure 7G) and LNCaP cells (Figure 7H). These depleted cells were rescued by either siRNA knockdown of p53 (Figure 7H) or sGCα1 overexpression (Figure 7I). Importantly, sGCα1 overexpression had a significantly lower positive effect on cells treated with control siRNA (Figure 7I). Thus, CSN5, like CSN4, regulates prostate cancer cell proliferation by targeting the sGCα1 and p53 proteins.
Figure 7.

CSN5 antagonistically regulates p53 and sGCα1 proteins in prostate cancer. LNCaP and CWR-22Rv1 cells were transfected with Ctrl siRNA or CSN5 siRNA, and Western blotting was used to measure (A) CSN5 and p53, (B) CSN5 and sGCα1, (E) CSN5 and p53 in cytosolic (C) or nuclear (N) fractions, or (F) p53 transcriptional activity using a luciferase assay. β-Actin was used as loading control and the molecular weights of the proteins are 53 kDa (p53), 38 kDa (CSN5), 49 kDa (CSN4), 42 kDa (β-actin), 50 kDa (β-tubulin), and 52 kDa (retinoic acid receptor-α). β-Tubulin and retinoic acid receptor-α were used as markers for cytosolic and nuclear fractions, respectively. LNCaP cell extracts were subjected to IP using antibody against (C) CSN5 or p53 or (D) CSN5 or sGCα1. IgG was used as negative control IP. Western blotting was used to measure CSN5, p53, and sGCα1. CWR-22Rv1 and LNCaP cells were transfected with (G) Ctrl siRNA or CSN5 siRNA, (H) Ctrl siRNA or CSN4 siRNA, with or without p53 siRNA, or (I) Ctrl siRNA or CSN5 siRNA, with or without sGCα1 expression plasmid. Cell density was measured by mammalian transient transfection (MTT) assay. In panels F, G, H, and I, data points represents an average of 3 independent experiments plus SDs. Asterisks indicate statistical significance (P < .004) in panel F and (P < .03) in panels G, H, and I. Student's t test was used to analyze the data. Ab, antibody.
Figure 8.

CK2 associates with and regulates p53 and sGCα1 proteins in prostate cancer cells. LNCaP and CWR-22Rv1 cells were treated with (A) 40 μM Curcumin, (B) 40 μM Emodin, or (C) transfected with Ctrl siRNA or CK2 siRNA, and Western blotting was used to measure CSN4, sGCα1, p53, and CK2. β-Actin was used as loading control. D, LNCaP and CWR-22Rv1 cell extracts were subjected to IP using an antibody against CK2. IgG was used as negative control IP. E, LNCaP cells treated with ethanol (−) or 1 nM R1881 (+) for 24 hours and then subjected to Western blotting to measure CK2, CSN4, CSN5, p53, sGCα1, and AR (110 kDa). The molecular weights of the proteins are 45 kDa (CK2), 53 kDa (p53), 38 kDa (CSN5), 49 kDa (CSN4), 53 kDa (p53), 77 kDa (sGCα1), 110 kDa (kDa), and 42 kDa (β-actin). F, Schematic diagram showing the interaction of the CSN4/5 proteins and CK2 with sGCα1 and p53 and the role of AR. Ab, antibody.
CK2 is involved in the CSN regulation of the p53 and sGCα1 proteins
Previous evidence has shown that caseine kinase-2 (CK2) and protein kinase-D (PKD) are associated with the CSN complex (32). Thus, we tested the possible involvement of these 2 kinases in the CSN4 and CSN5 regulation of sGCα1 and p53 using chemical inhibitors first. Interestingly, treatment of cells with Curcumin, which inhibits both kinases, resulted in reduced sGCα1 and elevated p53 (Figure 8A). To study each kinase individually, we treated cells with CID, a PKD inhibitor, or Emodin, a CK2 inhibitor. Whereas CID had no significant effect (Supplemental Figure 5B), Emodin had a strong negative effect on sGCα1 and a positive effect on p53 (Figure 8B), reproducing the data we observed with CSN4 and CSN5 knockdowns. To confirm these data, CK2 levels were depleted by siRNA, resulting in the same effects on sGCα1 and p53 (Figure 8C) as seen with Emodin treatment (see Figure 8B). Thus, CK2 expression and activity are important in regulating the levels of sGCα1 and p53 proteins.
Because CK2 appears to mediate the antagonistic effects of CSN4 and CSN5 on sGCα1 and p53, it is possible that CK2 may be associated with these proteins. Thus, IP experiments were performed using an anti-CK2 antibody, which yielded copurification of CSN4, CSN5, p53, and sGCα1 in both LNCaP and CWR-22Rv1 cells (Figure 8D), strongly arguing that prostate cancer cells harbor an endogenous complex containing all these proteins.
To monitor the effect of androgens on this complex, LNCaP cells were treated with androgens. Interestingly, only sGCα1 responded (Figure 8E), exhibiting the expected up-regulation (6).
Discussion
The proteins p53 and sGCα1 exhibit an interesting dichotomy in prostate cancer: p53 is widely studied and has antisurvival, antigrowth, and anticancer functions (11), whereas sGCα1 is poorly studied and has prosurvival, progrowth, and procancer functions (6, 10). Moreover, these 2 proteins interact with one another in prostate cancer cells, leading to suppression of p53 activity (10). Now we report that p53 and sGCα1 are part of a larger complex that includes 2 subunits of the CSN complex and the CK2 kinase. The two subunits are CSN4 and CSN5, and together with CK2, they have antagonistic activities on the sGCα1 and p53 proteins: sGCα1 is stabilized whereas p53 is destabilized, providing prostate cancer cells increased capacity to survive and grow. As would be expected, CSN4 protein expression is greatly elevated in prostate tumors, as is sGCα1, whereas p53 expression is down-regulated. Thus, our data suggest that the overexpression of CSN4 in prostate cancer is, at least in part, responsible for the elevated levels of sGCα1 protein (6) and reduced levels of p53 (14).
Our finding that CSN5 regulates p53 is consistent with earlier published work showing that CSN5 mediates p53 nuclear export and protein degradation (44), an effect that we have been able to reproduce in prostate cancer cells. Our finding that CSN4 affects CSN5, leading to sGCα1 stabilization, is completely novel and represents a new target for CSN5. Moreover and more importantly, CSN4 appears to be a master regulator of CSN5 and therefore downstream target of CSN4. Whereas knockdown of CSN5 affects protein levels of both p53 and sGCα1, there is no change in CSN4 protein expression. Yet, knockdown of CSN4 greatly diminished the levels of CSN5 protein, as well as affecting the p53 and sGCα1 proteins. These data suggest that CSN4 is upstream of CSN5 and regulates p53 and sGCα1 via CSN5. There is no published work to date suggesting this hierarchical arrangement of CSN4 and CSN5 in acting on common downstream targets. Although this may be unique to prostate cancer, it is unlikely to be so in view of the ubiquitous expression of the CSN complex and its subunits among different tissues and cells (42).
The CSN complex consists of 8 subunits (45), and our present data show that 2 of these subunits, CSN4 and CSN5, are associated with sGCα1 and p53. Interestingly, our preliminary data suggest that CNS6 and CSN7 may also be part of this complex (data not shown). With respect to this, an earlier study identified a subcomplex of the CSN that consists of CSN4, CSN5, CSN6, and CSN7 (46). The role of this subcomplex and, specifically, the CSN6 and CSN7 subunits in regulating sGCα1 and p53 will be studied in the future.
The CSN complex was previously determined to harbor kinase activity (27), and later work showed that this kinase activity is due to associated CK2 and PKD kinases (32). Our present study found that the kinase activity of CK2, but not PKD, is indeed important in the CSN regulation of sGCα1 and p53. We further showed that CK2 is found in the complex containing sGCα1, p53, CSN4, and CSN5. Thus, we have advanced the earlier study done in erythrocytes (27) to show that a CSN complex containing CK2 and p53 exists in prostate cancer cells and this complex also contains sGCα1. Moreover, both sGCα1 and p53 proteins are targets of regulation within this complex and they are oppositely regulated. Our data also show that CK2 kinase activity is the mediator of sGCα1 and p53 regulation. With respect to p53, it has already been reported that CK2 phosphorylates p53 and thereby mediates its ubiquitin-dependent degradation (32), which is likely occurring in prostate cancer cells. On the other hand, no data have been reported about sGCα1 phosphorylation or interaction with the CSN or CK2. Thus, sGCα1 may be a direct substrate of CK2 kinase activity or an indirect target of this enzyme's activity, with future work directed at addressing this important issue. Published data already have shown that CK2 can phosphorylate CSN2 and CSN7 subunits (32), providing one indirect mechanism for sGCα1 regulation. It is also possible that the multiprotein complex may exist in prostate cancer to regulate CK2 kinase activity and substrate specificity, by bringing p53 and possibly sGCα1 in proximity to CK2. This is another important area of future work. Because p53 mutations are relatively rare in early-stage prostate cancer and become more common in advanced disease (14, 15), the CSN-negative activity on p53 may provide an important mechanism for down-regulating p53 in those prostate tumors that express wild-type p53.
We recently reported that sGCα1 down-regulates p53 activity by mediating its cytoplasmic sequestration via physical interaction in the cytoplasm (10). This current study shows that this cytoplasmic complex of sGCα1 and p53 also contains the CSN subunits and CK2. In view of the CSN5 activity on p53 nuclear export (44), it is possible that CSN5 brings p53 into this multiprotein complex, where CK2 can phosphorylate p53 and mediates its degradation. The sGCα1 protein may be a necessary component of this complex, and its down-regulation may destabilize the complex, thereby preventing p53 phosphorylation and degradation. Thus, it will be important in the future to dissect the cytoplasmic complex to map the interaction partners and determine the necessary components to maintain the stability and function of the complex. Regardless of what this future work will reveal, it is important to emphasize that the CSN activities we observed in prostate cancer cells on sGCα1 and p53 are mimicked in prostate cancer tissues. Intriguingly, CSN4 expression was directly correlated with sGCα1 expression and inversely related to p53 expression in prostate cancer tissues, strongly arguing that CSN4, and likely CSN5 and CK2, are important regulators of the sGCα1 and p53 proteins, providing prostate tumors an enhanced ability to survive and grow. Although survival and growth are important in cancer, there are multiple additional steps that need to be considered in the future to determine the overall importance of our findings to the tumorigenesis process.
Using our data, we provide a model connecting all the proteins (Figure 8F). CSN4 is the upstream player controlling the levels of CSN5, which acts to oppositely regulate the proteasomal degradation of sGCα1 and p53. CK2 enzyme activity is important in this regulation, possibly by affecting CSN4/5. Interestingly, although androgen signaling via AR has the expected positive effect on sGCα1 (6), it has no effect on the other proteins in this complex. Thus, as our data have shown, this endogenous complex exists in both androgen-dependent and -independent prostate cancer cells, and AR may ensure that sGCα1 expression is sufficient to support complex formation. Interestingly, the negative effect of this complex on p53 can lead to enhanced AR activity in prostate cancer, because we have previously reported that p53 disrupts AR transcriptional activity (18). Future work can address this possibility.
Additional material
Supplementary data supplied by authors.
Acknowledgments
We thank Dr Rafael Garcia-Mata for providing the secondary antibody for immunofluorescence and Dr William Taylor for providing the p53 luciferase reporter.
This work was supported by grants from National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AR
- androgen receptor
- CK2
- caseine kinase-2
- CSN4
- COP9 signalosome subunit 4
- IP
- immunoprecipitation
- MS
- mass spectrometric
- NO
- nitric oxide
- PARP
- poly (ADP-ribose) polymerase
- PKD
- protein kinase-D
- SDS
- sodium dodecyl sulfate
- sGCα1
- soluble guanylyl cyclase α1
- siRNA
- small interfering RNA.
References
- 1. Jenster G. The role of the androgen receptor in the development and progression of prostate cancer. Semin Oncol. 1999;26(4):407–421. [PubMed] [Google Scholar]
- 2. Arnold JT, Isaacs JT. Mechanisms involved in the progression of androgen-independent prostate cancers: it is not only the cancer cell's fault. Endocr Relat Cancer. 2002;9(1):61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Han G, Buchanan G, Ittmann M, et al. Mutation of the androgen receptor causes oncogenic transformation of the prostate. Proc Natl Acad Sci USA. 2005;102(4):1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–39. [DOI] [PubMed] [Google Scholar]
- 5. Isaacs JT. Role of androgens in prostatic cancer. Vitam Horm. 1994;49:433–502. [DOI] [PubMed] [Google Scholar]
- 6. Cai C, Chen SY, Zheng Z, et al. Androgen regulation of soluble guanylyl cyclaseα1 mediates prostate cancer cell proliferation. Oncogene. 2007;26(11):1606–1615. [DOI] [PubMed] [Google Scholar]
- 7. Krumenacker JS, Hanafy KA, Murad F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain Res Bull. 2004;62(6):505–515. [DOI] [PubMed] [Google Scholar]
- 8. Sinnaeve P, Chiche JD, Nong Z, et al. Soluble guanylate cyclase α(1) and β(1) gene transfer increases NO responsiveness and reduces neointima formation after balloon injury in rats via antiproliferative and antimigratory effects. Circ Res. 2001;88(1):103–109. [DOI] [PubMed] [Google Scholar]
- 9. Garthwaite G, Bartus K, Malcolm D, et al. Signaling from blood vessels to CNS axons through nitric oxide. J Neurosci. 2006;26(29):7730–7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cai C, Hsieh CL, Gao S, et al. Soluble guanylyl cyclase α1 and p53 cytoplasmic sequestration and down-regulation in prostate cancer. Mol Endocrinol. 2012;26(2):292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 2009;1(5):a001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253(5015):49–53. [DOI] [PubMed] [Google Scholar]
- 13. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54(18):4855–4878. [PubMed] [Google Scholar]
- 14. Qian J, Hirasawa K, Bostwick DG, et al. Loss of p53 and c-myc overrepresentation in stage T(2–3)N(1–3)M(0) prostate cancer are potential markers for cancer progression. Mod Pathol. 2002;15(1):35–44. [DOI] [PubMed] [Google Scholar]
- 15. Navone NM, Labate ME, Troncoso P, et al. p53 mutations in prostate cancer bone metastases suggest that selected p53 mutants in the primary site define foci with metastatic potential. J Urol. 1999;161(1):304–308. [PubMed] [Google Scholar]
- 16. Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget. 2012;3(3):228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roemer K. Notch and the p53 clan of transcription factors. Adv Exp Med Biol. 2012;727:223–240. [DOI] [PubMed] [Google Scholar]
- 18. Shenk JL, Fisher CJ, Chen SY, Zhou XF, Tillman K, Shemshedini L. p53 represses androgen-induced transactivation of prostate-specific antigen by disrupting hAR amino- to carboxyl-terminal interaction. J Biol Chem. 2001;276(42):38472–38479. [DOI] [PubMed] [Google Scholar]
- 19. Gao S, Hsieh CL, Bhansali M, Kannan A, Shemshedini L. A peptide against soluble guanylyl cyclase α1: a new approach to treating prostate cancer. PLoS One. 2013;8(5):e64189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei N, Deng XW. Making sense of the COP9 signalosome. A regulatory protein complex conserved from Arabidopsis to human. Trends Genet. 1999;15(3):98–103. [DOI] [PubMed] [Google Scholar]
- 21. Wei N, Deng XW. The COP9 signalosome. Annu Rev Cell Dev Biol. 2003;19:261–286. [DOI] [PubMed] [Google Scholar]
- 22. Schwechheimer C, Isono E. The COP9 signalosome and its role in plant development. Eur J Cell Biol. 2010;89(2–3):157–162. [DOI] [PubMed] [Google Scholar]
- 23. Lykke-Andersen K, Schaefer L, Menon S, Deng XW, Miller JB, Wei N. Disruption of the COP9 signalosome Csn2 subunit in mice causes deficient cell proliferation, accumulation of p53 and cyclin E, and early embryonic death. Mol Cell Biol. 2003;23(19):6790–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Menon S, Chi H, Zhang H, Deng XW, Flavell RA, Wei N. COP9 signalosome subunit 8 is essential for peripheral T cell homeostasis and antigen receptor-induced entry into the cell cycle from quiescence. Nat Immunol. 2007;8(11):1236–1245. [DOI] [PubMed] [Google Scholar]
- 25. Tomoda K, Yoneda-Kato N, Fukumoto A, Yamanaka S, Kato JY. Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J Biol Chem. 2004;279(41):43013–43018. [DOI] [PubMed] [Google Scholar]
- 26. Scheel H, Hofmann K. Prediction of a common structural scaffold for proteasome lid, COP9-signalosome and eIF3 complexes. BMC Bioinformatics. 2005;6:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bech-Otschir D, Seeger M, Dubiel W. The COP9 signalosome: at the interface between signal transduction and ubiquitin-dependent proteolysis. J Cell Sci. 2002;115(Pt 3):467–473. [DOI] [PubMed] [Google Scholar]
- 28. Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114(6):663–671. [DOI] [PubMed] [Google Scholar]
- 29. Richardson KS, Zundel W. The emerging role of the COP9 signalosome in cancer. Mol Cancer Res. 2005;3(12):645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao R, Yeung SC, Chen J, et al. Subunit 6 of the COP9 signalosome promotes tumorigenesis in mice through stabilization of MDM2 and is upregulated in human cancers. J Clin Invest. 2011;121(3):851–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398(6723):160–165. [DOI] [PubMed] [Google Scholar]
- 32. Uhle S, Medalia O, Waldron R, et al. Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. EMBO J. 2003;22(6):1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harari-Steinberg O, Chamovitz DA. The COP9 signalosome: mediating between kinase signaling and protein degradation. Curr Protein Pept Sci. 2004;5(3):185–189. [DOI] [PubMed] [Google Scholar]
- 34. Henke W, Ferrell K, Bech-Otschir D, et al. Comparison of human COP9 signalsome and 26S proteasome lid'. Mol Biol Rep. 1999;26(1–2):29–34. [DOI] [PubMed] [Google Scholar]
- 35. Bech-Otschir D, Kraft R, Huang X, et al. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 2001;20(7):1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kapelari B, Bech-Otschir D, Hegerl R, Schade R, Dumdey R, Dubiel W. Electron microscopy and subunit-subunit interaction studies reveal a first architecture of COP9 signalosome. J Mol Biol. 2000;300(5):1169–1178. [DOI] [PubMed] [Google Scholar]
- 37. Granata A, Koo SJ, Haucke V, Schiavo G, Warner TT. CSN complex controls the stability of selected synaptic proteins via a torsinA-dependent process. EMBO J. 2011;30(1):181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cai C, Hsieh CL, Shemshedini L. c-Jun has multiple enhancing activities in the novel cross talk between the androgen receptor and Ets variant gene 1 in prostate cancer. Mol Cancer Res. 2007;5(7):725–735. [DOI] [PubMed] [Google Scholar]
- 39. Cai C, Hsieh CL, Omwancha J, et al. ETV1 is a novel androgen receptor-regulated gene that mediates prostate cancer cell invasion. Mol Endocrinol. 2007;21(8):1835–1846. [DOI] [PubMed] [Google Scholar]
- 40. Tepper CG, Boucher DL, Ryan PE, et al. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62(22):6606–6614. [PubMed] [Google Scholar]
- 41. Nadiminty N, Tummala R, Liu C, et al. NF-κB2/p52 induces resistance to enzalutamide in prostate cancer: role of androgen receptor and its variants. Mol Cancer Ther. 2013;12(8):1629–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schwechheimer C. The COP9 signalosome (CSN): an evolutionary conserved proteolysis regulator in eukaryotic development. Biochim Biophys Acta. 2004;1695(1–3):45–54. [DOI] [PubMed] [Google Scholar]
- 43. Igawa T, Lin FF, Lee MS, Karan D, Batra SK, Lin MF. Establishment and characterization of androgen-independent human prostate cancer LNCaP cell model. Prostate. 2002;50(4):222–235. [DOI] [PubMed] [Google Scholar]
- 44. Oh W, Lee EW, Sung YH, et al. Jab1 induces the cytoplasmic localization and degradation of p53 in coordination with Hdm2. J Biol Chem. 2006;281(25):17457–17465. [DOI] [PubMed] [Google Scholar]
- 45. Kato JY, Yoneda-Kato N. Mammalian COP9 signalosome. Genes Cells. 2009;14(11):1209–1225. [DOI] [PubMed] [Google Scholar]
- 46. Kotiguda GG, Weinberg D, Dessau M, et al. The organization of a CSN5-containing subcomplex of the COP9 signalosome. J Biol Chem. 2012;287(50):42031–42041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.