

Abstract
Testicular nuclear receptor 4 (TR4), also known as NR2C2, belongs to the nuclear receptor superfamily and shares high homology with the testicular nuclear receptor 2. The natural ligands of TR4 remained unclear until the recent discoveries of several energy/lipid sensors including the polyunsaturated fatty acid metabolites, 13-hydroxyoctadecadienoic acid and 15-hydroxyeicosatetraenoic acid, and their synthetic ligands, thiazolidinediones, used for treatment of diabetes. TR4 is widely expressed throughout the body and particularly concentrated in the testis, prostate, cerebellum, and hippocampus. It has been shown to play important roles in cerebellar development, forebrain myelination, folliculogenesis, gluconeogenesis, lipogenesis, muscle development, bone development, and prostate cancer progression. Here we provide a comprehensive summary of TR4 signaling including its upstream ligands/activators/suppressors, transcriptional coactivators/repressors, downstream targets, and their in vivo functions with potential impacts on TR4-related diseases. Importantly, TR4 shares similar ligands/activators with another key nuclear receptor, peroxisome proliferator-activated receptor γ, which raised several interesting questions about how these 2 nuclear receptors may collaborate with or counteract each other's function in their related diseases. Clear dissection of such molecular mechanisms and their differential roles in various diseases may help researchers to design new potential drugs with better efficacy and fewer side effects to battle TR4 and peroxisome proliferator-activated receptor γ involved diseases.
Testicular nuclear receptor 4 (TR4), also known as NR2C2, was first cloned from human and rat hypothalamus, prostate, and testes libraries in 1994 (1). The open reading frame contains 1848 nucleotides in human and 1791 nucleotides in rat, which encode 615 and 596 amino acids, respectively (1). TR4 is widely expressed throughout the body, and its expression is particularly high in testis, prostate, ovary, cerebellum, and hippocampus (2). From functional analyses based on the signaling involved, TR4 was assigned to the gene cluster related to the central nervous system (CNS), circadian, and basal metabolic functions (2, 3).
The human TR4 gene was mapped to chromosome 3 at band p24.3. Interestingly, several other nuclear receptors (NRs) have also been mapped close to this TR4 gene region, including the human peroxisome proliferator–activated receptor γ (PPARγ), retinoic acid receptor (RAR) β, and thyroid hormone receptor β on chromosomes 3p25, 3p24.2–p24.3, and 3p22–p24.1, respectively (4).
TR4, like other NRs, has a common modular organization with a variable N-terminal domain, a conserved DNA-binding domain (DBD), and a ligand-binding domain (LBD) that activates gene transcription upon ligand binding. The DBD is composed of a highly conserved 66-amino acid core domain located centrally in each NR, together with a short, nonconserved extension into the hinge region of the receptor (5). Its modular design is composed of 2 zinc-binding loops and a pair of α helices (6).
TR4 and some other NRs, including testicular nuclear receptor 2 (TR2), 9-cis-retinoic acid receptor (retinoid X receptor [RXR]), RAR, thyroid hormone receptor, vitamin D receptor (VDR), and PPAR (7–9), can bind to consensus hormone response elements (HREs) that contain direct repeats (DRs) via one of the α helices. DRs are composed of 2 pieces of the AGGTCA sequence with 1 to 5 nucleotides between them (DR1–DR5) (10). Human TR4 binds specifically to DR1 and DR5 HREs (11). The human TR2, one of the first orphan receptors identified, shares structural homology with members of the NR superfamily (12, 13) and binds to DRs in the following order: DR1 > DR2 > DR5 = DR4 > DR3 (9). PPARγ binding sites (peroxisome proliferator–activated receptor response elements [PPREs]) are generally DR1 (14–17). Thyroid hormone receptor and VDRs preferably bind to DR4 and DR3, respectively (10, 18).
TR4 Upstream and Downstream Signaling Pathways
TR4 upstream signaling pathways with natural and synthetic ligands/activators
TR4 has been an orphan NR without an identified ligand since it was cloned in 1994 (1). Recently, Xie et al (19) found that polyunsaturated fatty acid (PUFA) metabolites, 13-hydroxyoctadecadienoic acid (13-HODE) and 15-hydroxyeicosatetraenoic acid (15-HETE), and the thiazolidinedione (TZD) family of antidiabetic drugs could transactivate TR4 to a degree similar to their ability to transactivate PPARγ. This discovery was first inspired by the phenotypes found in mice lacking TR4 (TR4−/−), which demonstrated decreased expression of key genes involved in the lipid metabolism and insulin sensitivity, including CD36, stearoyl-CoA desaturase 1 (SCD1), and phosphoenolpyruvate carboxykinase (PEPCK) (19–22). Among the inducers/activators of CD36, Xie et al (19) found that 13-HODE and 15-HETE could induce CD36 expression via transactivation of TR4. Other studies also confirmed these findings, showing that the PUFA, γ-linoleic acid, could induce the TR4 target genes, including apolipoprotein E (ApoE) and PEPCK (23), via induction of TR4 transactivation (23, 24).
In addition to the natural ligands, the TZD rosiglitazone has been identified as a TR4 ligand/activator (19). TZDs, including rosiglitazone, pioglitazone, ciglitazone, and troglitazone, have been used widely as antidiabetic drugs (25). However, side effects including hepatitis, cardiovascular diseases, and bladder cancer have resulted in restrictions for their use in the United States (26–28). Early studies showed that TZD has protective effects in lung cancer, but not in colorectal cancer, breast cancer, or prostate cancer (PCa) (29, 30). Other reports suggested that continued TZD treatment in some diabetic patients might lead to increased bladder cancer risk (28). Interestingly, other reports also indicated that TZDs might be able to reduce PCa cell growth (31, 32). The discrepancy between clinical studies and in vitro cell line studies led to the speculation that the effects of TZDs may be different under different microenvironments. The finding that TZDs can transactivate both PPARγ and TR4 (19) has raised the interesting question of whether TZDs may function differentially, depending on the relative expression of these two NRs in the same cell type. We will discuss this in more detail in a later section.
TR4 upstream signaling pathways with kinases and other modulators
Early studies concluded that kinases/phosphatases could induce transactivation of NRs by altering their conformation via phosphorylation or dephosphorylation (33). This observation was especially important for the study of orphan NRs before their ligands were found.
Through peptide mapping and motif scanning by Scansite (http://scansite.mit.edu), Kim et al (22) found that the TR4 serine 351 (S351), conserved in human, mouse, rat, and chicken, was a potential target for AMP activated protein kinase (AMPK) (Figure 1). The in vitro phosphorylation assay proved that TR4 could be phosphorylated by AMPK in the presence of AMP, and mutation of S351 completely abolished this phosphorylation (22). Suppression of AMPK by the inhibitor, compound C, enhanced TR4 transactivation, and activation of AMPK by the drug, AICAR (5-aminoimidazole-4-carboxamide ribonucleotide), suppressed TR4 transactivation. Importantly, they also found that metformin, an antidiabetic drug that could activate AMPK (34), could also suppress TR4 function via alteration of AMPK-mediated TR4 phosphorylation (22).
Figure 1.
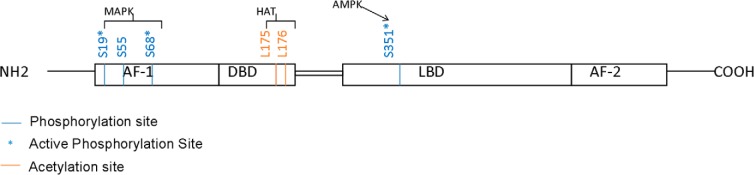
TR4 organization and sites of modulation by phosphorylation and acetylation.
Another report found that TR4 could be phosphorylated by MAPK at its AF-1 domain in the absence of specific ligands, and activation of MAPK by anisomycin could suppress TR4 transactivation. In contrast, inhibition of MAPK by PD98059 significantly increased TR4 transactivation (35). A mass spectrometric analysis of TR4 revealed 3 MAPK phosphorylation sites at Ser19, Ser55, and Ser68 in its AF-1 domain, and site-directed mutagenesis studies demonstrated the functionality of phosphorylation on Ser19 and Ser68, but not Ser55 (Figure 1). Further mechanism dissection indicated that MAPK-mediated phosphorylation of the AF-1 domain could make TR4 function as a repressor via recruitment of the corepressor receptor-interacting protein 140 (RIP140) (35). In contrast, dephosphorylation of its AF-1 made TR4 function as an activator via recruitment of the coactivator p300/CREB-binding protein-associated factor (PCAF) (Figure 2) (35).
Figure 2.
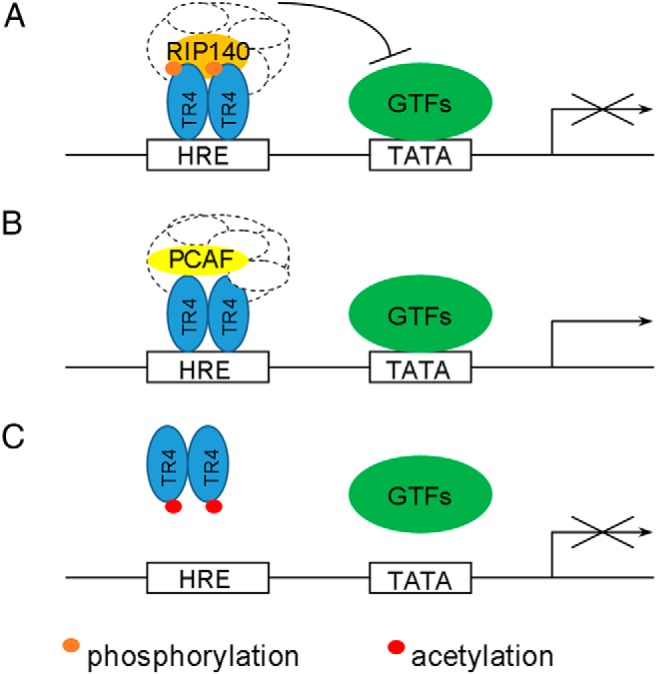
Mechanisms of modulation of TR4 activity by phosphorylation and acetylation. A, Phosphorylation of the TR4 AF-1 domain leads to recruitment of the corepressor complex that inhibits transcription initiation. B, Dephosphorylated TR4 recruits coactivator complex that promote transcription initiation. C, Acetylation of TR4 at the DBD prevents TR4-binding to HRE and inhibits transcription initiation.
Liu et al (21) also found that cAMP/protein kinase A (PKA) signaling could induce TR4 expression by triggering CCAAT/enhancer-binding protein (C/EBP) α- and C/EBPβ-binding to the selective cAMP response element (CRE) located on the TR4 promoter that led to the modulation of gluconeogenesis.
Taken together, results from the above studies suggest that phosphorylation or dephosphorylation via various kinases including AMPK, MAPK, or PKA might be able to modulate TR4 function via transactivation of TR4.
TR4 can also be acetylated at lysine-175 and lysine-176 to alter its transactivation via attenuation of the ability of TR4 to bind to its target genes (Figure 1) (36). Interestingly, unlike histone acetylation to induce its downstream target genes (37), TR4 acetylation via histone acetyltransferase (HAT) actually suppressed the TR4 downstream targets, and this acetylation could be enhanced via association with the coregulator ARA55 involving recruitment of the HAT enzyme CREB-binding protein (36). Such suppression of TR4 transactivation via acetylation might then alter expression of the TR4 downstream target CD36 (36).
TR4 downstream signaling pathways involving heterodimerization with other NRs
TR4 can form heterodimers with various NRs to modulate its downstream target genes. TR4 forms heterodimers preferentially with TR2 to bind at DR5, and 3 leucine residues on helix 10 of TR2 are critical for this dimerization (38). Coexpression of TR4 and TR2 resulted in much stronger repression of a DR5-containing reporter than expression of either receptor alone (38).
TR4 can also interact with the androgen receptor (AR). The heterodimerization of TR4-AR may prevent TR4-binding to its target DNA and thus suppress TR4 target gene expression (39). Interestingly, the heterodimerization of TR4-AR also suppresses AR target gene expression (39).
TR4 can also repress estrogen receptor (ER)–mediated transactivation through a direct protein-protein interaction. The interaction between TR4 and ER suppresses the ER-ER interaction and prevents ER from binding to the estrogen response element, which results in the suppression of ER-mediated cell proliferation in MCF-7 cells (40).
TR4 binds specifically to DR response elements, which are also the response elements for other NRs including thyroid hormone receptor, VDR, RAR, RXR, and PPAR (11, 41, 42). Interestingly, via competing with other NRs binding to their response elements, TR4 can either increase or decrease the number of their downstream targets. For example, TR4 increases the transcriptional activity of a thyroid hormone receptor response element (TRE) containing reporter gene (41, 42). Electrophoretic mobility shift assay (EMSA) and Scatchard analysis indicated a strong binding affinity between TR4 and DR4. Furthermore, this transcriptional activation by TR4 is dose dependent, and that for DR4 is sequence dependent (41).
In contrast, TR4 suppresses VDR downstream targets by competing with the DR3–vitamin D response element (VDRE). Cell transfection and in situ hybridization analyses demonstrated that the expression of the vitamin D3 target gene, 25-hydroxyvitamin D3 24-hydroxylase, can be suppressed by TR4 through high-affinity binding to the VDRE (42). EMSA and proteolytic analysis showed that TR4 adopts different conformations once bound to DR3-VDRE or DR4–TRE (42). These different TR4-DR3-VDRE and TR4-DR4-TRE conformations may allow TR4 to recruit different coregulators (42).
TR4 also suppresses retinoic acid–induced transactivation in a dose-dependent manner via binding to the DR1/DR5-retinoic acid receptor response element/RXR response element (11). These results indicate that retinoid signaling can be suppressed by TR4 to maintain homeostasis. In the PPAR family, TR4 plays opposite roles in terms of regulating downstream genes, ie, suppresses PPARα targets (43), but enhances PPARγ targets (19). TR4 binds to the DR1-PPRE and represses the PPARα target genes rat enoyl-coenzyme A (CoA) hydratase and peroxisomal fatty acetyl-CoA oxidase (43) in a dose-dependent manner. In contrast, TR4 binds to the DR1-PPRE and enhances the PPARγ target CD36 in a dose-dependent manner (19).
TR4 downstream signaling pathways involving the interaction with other coregulators
To date, only one coactivator, PCAF (35), has been identified as being able to induce TR4 transactivation. As a HAT, PCAF can also acetylate nonhistone transcription–related proteins such as p53 (44). This TR4-PCAF interaction may then turn on its natural target gene, apoE.
TIP27 has 2 zinc finger motifs and can function as a TR4 corepressor via interaction of TR4 with its region between Asp39 and Lys79, which suppresses TR4-mediated DR1-dependent transactivation. This repression does not involve the inhibition of TR4 homodimerization or DR1-binding but may be due to an interruption between TR4 and coactivator interaction (45). Further studies have to be done to determine the physiological function of TIP27 in the repression of gene transcription by TR4.
TR4 associated protein (TRA16) is another identified corepressor that can suppress TR4 activity via inhibition of TR4-binding to the TR4 response element (TR4RE) and/or via blocking the interaction between TR4-DBD and TR4-LBD. These unique suppression mechanisms identify TRA16 as a selective TR4 corepressor. TRA16 expression in the H1299 human lung cancer cell line was found to be higher than that in normal human lung tissue. This TR4-TRA16 interaction may then lead to lung cancer transformation, suggesting that TRA16 could play roles in some kinds of cancer tissues (46).
RIP140 has been shown to be a common coregulator of many NRs (47, 48). Huq et al (35) demonstrated that MAPK-mediated phosphorylation of the AF-1 domain rendered TR4 a repressor, mediated through the preferential recruitment of the corepressor RIP140. Kim et al (49) also reported TR4-induced Bcl-2 expression can be suppressed by cotransfection with RIP140 in a dose-dependent manner.
TR4 In Vivo Roles in Aging and Cancer
TR4 in aging
Collins et al (50) first found that almost all mice lacking TR4 (TR4−/−) developed cachexia by 1 month, characterized by severe weight loss, prominent reduction of fat tissue, and weakness. Cachexia in the TR4−/− mice was progressive and heterogeneous in severity and was followed by premature death. Generally, TR4−/− mice had a much shorter life span than wild-type (TR4+/+) and heterozygous (TR4+/−) mice, with 70% of TR4−/− mice dying before 4 months of age and >90% (72 of 77) not living longer than 1 year (51). More than 90% of TR4+/+ and TR4+/− mice survived over this period of study, and the average life span of dams and sires in this wild-type C57BL/6J background exceeds 2 years (51). Besides the shorter life span, TR4−/− mice also acquired an aged appearance at an earlier stage. By 6 months if age, most TR4−/− mice had gray hair and an extramedullary hematopoiesis in the liver that is often found in aged mice. Furthermore, examination of bones for age-associated changes in TR4+/+ and TR4−/− mice showed that by 6 months of age, TR4−/− mice developed severe kyphosis, a hallmark for aged bone. A reduction in bone mineral density was also found in spines of 6- to 7-month-old TR4−/− mice (51).
TR4−/− mice also exhibited premature aging phenotypes that are associated with increased oxidative stress and genome instability (51). At the cellular level, a rapid cellular growth arrest accompanied by elevated intracellular reactive oxygen species and more DNA damage were observed in TR4−/− mouse embryonic fibroblasts (MEFs). Restoring TR4 to TR4−/− MEFs reduced the reactive oxygen species levels and DNA damage, which resulted in slowed cellular growth arrest. The data suggest that acceleration of aging in TR4−/− mice results from oxidative DNA damage via loss of function in the DNA repair systems mediated by TR4 (51).
TR4 in DNA damage/repair and apoptosis
The discovery of the linkage of TR4 to aging suggested that TR4 might play important roles in the DNA damage system (51). Liu et al (52) also found that TR4 could modulate UV-induced DNA damage by promoting the transcription-coupled nucleotide excision repair (TC-NER) DNA repair pathway through transcriptional regulation of Cockayne syndrome B protein (CSB), a member of the TC-NER pathway (52). They found that UV treatment could modulate DNA repair specifically through the TC-NER pathway but not the global genomic NER pathway. The level of CSB is 10-fold reduced in tissues of TR4−/− mice, and TR4 directly regulates CSB at the transcriptional level. Moreover, restored CSB expression decreases UV hypersensitivity of TR4−/− cells. Taken together, these results indicate that TR4 modulates UV sensitivity by promoting the TC-NER DNA repair pathway through transcriptional regulation of CSB. These results support the previous finding that lack of TR4 will cause premature aging (51).
The consequences of DNA damage may then induce apoptosis, which can also contribute to the aging phenotype. Kim et al (49) reported that MEFs prepared from TR4−/− mice are more susceptible to UV irradiation–mediated apoptosis than MEFs from TR4+/+ littermates (49). TR4−/− MEFs showed more apoptosis upon UV irradiation caused by down-regulation of Bcl-2 RNA and protein expression and consequently increased caspase-3 activity. Conversely, this TR4-induced Bcl-2 expression can be suppressed by cotransfection with TR4 coregulators, such as AR and RIP140 in a dose-dependent manner. Taken together, these results demonstrate that TR4 might function as an apoptosis modulator through induction of Bcl-2 expression (49).
Interestingly, ionizing radiation (IR) can also induce DNA damage–dependent apoptosis that is linked to TR4 function. Yan et al (53) found that knockdown of TR4 by TR4 siRNA sensitized the cells to IR-induced apoptosis, whereas IR exposure increased TR4 expression in the scramble siRNA control cells. Examination of IR-responsive molecules showed that expression of the growth arrest and DNA damage response gene (Gadd45a) was dramatically decreased in tissues of TR4−/− mice and, as a result, failed to respond to IR. Interestingly, luciferase reporter and chromatin immunoprecipitation (ChIP) assays indicated that TR4 regulates Gadd45a expression through the TR4RE located in intron 3. Taken together, these results show that TR4 can protect genomic integrity through up-regulation of Gadd45a, which in turn repairs DNA damage and prolongs the life of the cells (53).
While studying upstream TR4 regulation under the condition of oxidative stress, Li et al (54) identified the transcription factor forkhead box O3a (FOXO3a), a key stress-responsive factor, which could regulate TR4 expression through a daf-16 family protein-binding element (5′-TGTTTAC-3′) in the TR4 promoter. The binding of FOXO3a to the daf-16 family protein-binding element on TR4 was confirmed using EMSA and ChiP. In contrast, suppression of FOXO3a by siRNA could reduce oxidative stress-induced TR4 expression. These authors also showed that cells with reduced FOXO3a were less resistant to oxidative stress. In contrast, addition of functional TR4 into these cells could restore the stress resistance. Taken together, the results unveil the new pathway: oxidative stress → FOXO3a → TR4 signaling → cell survival (54).
TR4 in prostate cancer
The linkage of TR4 to DNA damage/repair → oxidative stress–resistance → antiapoptosis → cell survival indicates that TR4 might play important roles in tumor progression. Lin et al (55) found that TR4 might act as a caretaker tumor suppressor to suppress PCa initiation through promotion of DNA repair and maintenance of genome integrity. They found from a prostate histology survey that TR4−/− mice and not their TR4+/+ littermates developed prostatic intraepithelial neoplasia (PIN) lesions. In another PCa mouse model, PTEN+/−/TR4+/− mice but not their PTEN+/−/TR4+/+ littermates not only formed PIN lesions as expected but also formed prostate tumors at 15 months of age. Furthermore, in the TRAMP mouse model, TRAMP/TR4+/− mice formed severe PIN lesions at 24 weeks, whereas TRAMP/TR4+/+ littermates only formed minor PIN lesions (55).
Mechanism dissection indicated that loss of TR4 led to increased DNA damage with decreased levels of the DNA repair gene ATM. Luciferase reporter and ChIP assays further revealed that TR4 could modulate ATM gene expression at the transcriptional level, and human clinical sample surveys also confirmed that the expression of ATM was decreased with decreased TR4 expression (55). The above results suggest that TR4 is a tumor suppressor gene in the early development of PCa (55).
Interestingly, in vitro migration/invasion assays and in vivo mouse studies showed that TR4 promoted various PCa cells migration/invasion via modulation of the CCL2/CCR2 axis, suggesting that TR4 can enhance PCa metastasis (Ding X.F., D.R. Yang, S.O. Lee, Y.L. Chen, L.Q. Xia, S.J. Lin, S.C. Yu, Y.J. Niu, G.H. Li, and C. Chang, submitted for publication). These conflicting findings indicate that TR4, like AR, may have distinct and opposite functions in influencing PCa progression (56–60). Whereas AR can function as a stimulator to promote PCa initiation and development at early stages, it can also function as a suppressor to inhibit PCa metastasis at later stages (56–60). In contrast, TR4 may function as a suppressor to inhibit PCa initiation and development at early stages yet function as a stimulator to promote PCa metastasis at later stages.
Cancer stem cells are known to have higher chemoresistance than nonstem cancer cells (61), but the underlying molecular mechanisms remain unclear. Yang et al (62) found that the expression of TR4 was significantly higher in the PCa CD133+ stem/progenitor cells than in CD133− nonstem/progenitor cells. Knockdown of TR4 in the CD133+ population of the C4–2 PCa cell line led to increased sensitivity to docetaxel and etoposide, drugs used in chemotherapy (62). Mechanism dissection showed that knocking down TR4 in these CD133+ cells led to down-regulation of Oct4 expression, which, in turn, down-regulated IL-1 receptor antagonist (IL1Ra) expression. Adding back Oct4 and IL1Ra into the TR4 knockdown CD133+ cells restored the chemoresistance, suggesting that TR4 may function through Oct4 and IL1Ra to modulate chemoresistance. Taken together, these studies suggest that targeting of TR4 may improve the chemotherapy sensitivity of PCa stem/progenitor cells.
In addition to directly modulating PCa progression, TR4 may also be able to cooperate with other NRs to influence PCa progression. Via interaction with AR, Lee et al (39) found that TR4 could suppress both AR and TR4 downstream target genes. They also found that the expression of prostate-specific antigen (an androgen target widely used as a marker for PCa progression) in LNCaP cells was clearly suppressed by the addition of TR4 in the absence or presence of 10 nM dihydrotestosterone, suggesting that TR4 could suppress AR function in PCa cells (39). These results further strengthen the observation that TR4 has different roles as either a suppressor or promotor of PCa progression at different stages.
TR4 in liver cancer
Through the interaction with other NRs, TR4 can also have effects on other cancers. EMSA demonstrated that TR4 and thyroid hormone receptor could co-occupy the same response element. The interaction of TR4 with the thyroid hormone receptor may then enhance the thyroid hormone receptor downstream target genes including fibrinogen, cdk2, and p21 (63). Interestingly, it was found that TR4 alone could also modulate these downstream genes, which might then lead to increased cell migration in liver cancer HepG2-TR cells. However, addition of TR4 in these HepG2-TR cells also suppressed cell proliferation, suggesting that TR4, like its bifunctional roles in PCa, also functions oppositely in liver cancer as a suppressor to inhibit liver cancer initiation and as a stimulator to promote liver cancer metastasis.
Importantly, TR4 may also be able to function through modulation of hepatitis B virus (HBV) (64) expression to influence HBV-induced liver cancer (65). Using EMSA, Lin et al (65) found that TR4 could bind to the DR1 on the HBV core promoter, which might then suppress HBV-mediated transactivation, suggesting that TR4 might be able to function as a suppressor in the transcriptional regulation of the HBV core gene expression.
Interestingly, in addition to the interaction with thyroid hormone receptor to influence liver cancer progression, Lin et al (66) found that TR4 could also interact with another NR, TR2, to significantly enhance its transcriptional suppression of HBV expression via binding to the DR6 response element in the enhancer.
Taken together, these results suggest that TR4 can differentially influence liver cancer progression via interaction with various NRs.
TR4 in Breast Cancer
TR4 can also interact with ER to influence breast cancer progression (40). EMSA and pull-down assays clearly demonstrated that the direct interaction between TR4 and ER would suppress the homodimerization of ER and interrupt/prevent ER-binding to the ERE (40). This interaction might then suppress the ER targets, including cyclin D1 and pS2, and inhibit ER-mediated cell proliferation in the breast cancer MCF-7 cells (40).
TR4 in Cushing's disease and pituitary corticotroph tumors
Cushing's disease is a life-threatening disorder attributed to excess ACTH secretion caused by pituitary corticotroph tumors. Du et al (67) first revealed that TR4 is overexpressed in human pituitary corticotroph tumors as well as in human and mouse pituitary corticotroph tumor cell lines. Overexpression of TR4 in both human and mouse tumor cells increases ACTH precursor polypeptide proopiomelanocortin (POMC) transcription, ACTH secretion, cellular proliferation and tumor invasion (67). In contrast, knocking down TR4 reverses all these phenotypes. Similar results were also obtained from in vivo mouse studies. Mechanistically, they showed that TR4 could transcriptionally activate POMC expression through binding to the DR1 response element in the promoter, and POMC expression was enhanced by MAPK-mediated TR4 phosphorylation (67). Taken together, their findings indicate that TR4 can promote pituitary corticotroph tumor progression, which is consistent with the roles of TR4 in PCa (Ding X.F., D.R. Yang, S.O. Lee, Y.L. Chen, L.Q. Xia, S.J. Lin, S.C. Yu, Y.J. Niu, G.H. Li, and C. Chang, submitted for publication).
TR4 In Vivo Roles in Metabolic Syndrome and Cardiovascular Diseases
TR4 in metabolic syndrome
Liu et al (20) identified TR4 as a key gluconeogenic regulator via transactivation of PEPCK (20). The PEPCK promoter contains the TR4RE located at −451 to −439. Luciferase reporter assays, EMSA, and ChIP proved that TR4 can regulate PEPCK through promoter binding. Eliminating TR4 in hepatocytes with TR4 small interfering RNA (siRNA) or knocking out TR4 in mice significantly reduced PEPCK expression and glucose production in response to glucose depletion. In contrast, overexpression of TR4 increased PEPCK expression and hepatic glucose production in human and mouse hepatocytes. These in vitro and in vivo data suggest a new pathway of TR4 → PEPCK → gluconeogenesis that may be able to control blood glucose.
Liu et al (21) also found that cAMP/PKA, the mediator of fasting and glucagon, could function through induction of TR4 → PEPCK signaling to modulate gluconeogenesis (21). Mechanism dissection in hepatocytes demonstrated that cAMP/PKA might trigger C/EBPα/β binding to the selective CRE located at the TR4 promoter to induce TR4 transcription. They also demonstrated that the binding activity of C/EBPs to the TR4 promoter is increased in response to cAMP treatment. Taken together, these results identify a new signaling pathway from the fasting signal → glucagon → cAMP/PKA activation → C/EBPα/β activation → TR4 function increase to modulate the gluconeogenesis (21).
In contrast, Kim et al (22) found that TR4 transactivation is suppressed via phosphorylation by metformin-induced AMPK at Ser351, which results in the suppression of SCD1, the rate-limiting enzyme in the biosynthesis of monounsaturated fatty acids (22), which plays key roles in obesity, diabetes, lipogenesis, β-oxidation, and insulin sensitivity (68, 69). This pathway was further examined in TR4 knockout mice and primary hepatocytes with either knocked-down or overexpressed TR4, and results showed that TR4 could alter expression of the lipogenic gene SCD1 at the transcriptional level to influence lipid storage and insulin sensitivity.
TR4 could also facilitate fatty acid uptake into 3T3-L1 adipocytes via up-regulation of the FATP1 gene, which might then result in lipid accumulation in adipocytes (70), suggesting that TR4 might function as an important modulator to control lipid anabolism, and the antidiabetic drug metformin could suppress TR4 to reduce lipid synthesis and increase insulin sensitivity (22).
Kang et al (71) also reported that TR4−/− mice showed significantly lower hepatic triglyceride levels with reduced lipid accumulation in adipose tissue than TR4+/+ mice. Mechanism dissection showed that loss of TR4 decreased expression levels of several genes involved in lipid uptake and triglyceride synthesis/storage in hepatocytes, including Cidea, Cidec, Mogat1, and CD36. Moreover, TR4−/− mice exhibited reduced infiltrated inflammatory cells and were resistant to the development of glucose intolerance and insulin resistance.
Taken together, these results suggest that TR4 is an important modulator in insulin sensitivity and glucose/lipid metabolism. However, two newly identified signaling pathways involving TR4 (1, the fasting signal → glucagon → cAMP/PKA activation → C/EBPα/β activation → TR4 function increase → gluconeogenesis increase in hepatocytes vs 2, starvation → AMP increase → AMPK activation → TR4 function decrease/inactivation → SCD1 decrease → β-oxidation increase/lipogenesis decrease → insulin sensitivity increase) (22) suggest the starvation can either increase TR4 function through cAMP/PKA or decrease TR4 function through AMPK. It will be important to further dissect the detailed mechanisms by which the human body can differentially use these opposite roles of TR4 upon facing starvation.
Moreover, TZDs were initially identified via activation of PPARγ as increasing insulin sensitivity (25) but were also identified as the ligands/activators of TR4 modulating PEPCK and/or SCD-1 expression to alter the systemic lipid and glucose metabolism for the reduction of insulin sensitivity. It will be interesting in the future to screen new types of TZDs that can distinguish between the activation of PPARγ vs TR4.
TR4 in cardiovascular diseases
Xie et al (19) found reduced CD36 expression with reduced foam cell formation in TR4−/− mice. Mechanism dissection suggests that TR4 induces CD36 expression via a transcriptional regulation. Knocking down TR4 via TR4 siRNA resulted in decreased CD36 expression and foam cell formation, whereas addition of functional TR4 cDNA resulted in increased CD36 expression and foam cell formation in the macrophage RAW264.7 cells (19). Restoring functional CD36 in the TR4 knocked-down RAW264.7 cells reversed the decreased foam cell formation (19). Importantly, in vitro studies revealed that TZDs could also induce CD36 expression via TR4-binding to the DNA response element located on the CD36 5′ promoter (19). Early studies found that PPARγ could transactivate CD36 expression via binding to the same DNA response element, suggesting that TZDs could modulate atherosclerosis via activation of the two different NRs, TR4 and PPARγ.
Interestingly, TR4 may also be involved in pathogen infection via modulation of CD36 expression. Mahajan et al (24) reported that TR4 could interact with Mycobacterium tuberculosis macrophage lipids to ensure survival of the pathogen. TR4 might create a foamy niche within the macrophages by modulating oxidized low-density lipoprotein receptor CD36, suggesting that targeting of TR4 may eliminate the tuberculosis infection.
The ApoE protein is synthesized mainly in the liver where it can redistribute cholesterol and other lipids to peripheral tissues. Kim et al (72) demonstrated that the TR4 can induce ApoE expression in HepG2 cells, and mice lacking TR4 had significant suppressed ApoE at both protein and ApoE mRNA levels. Mechanism dissection showed that TR4 could modulate ApoE at the transcriptional level.
Loss of TR4 in TR4−/− mice led to altered expression of the ApoC-I and ApoC-II genes (72), and TR4 could also form a complex with Sp1 to synergistically induce ApoE expression (73). γ-Linoleic acid could function through induction of TR4 activity to promote the expression of ApoE and PEPCK (23), suggesting that TR4 can function through modulation of the expression of the ApoE/C-I/C-II gene cluster in liver cells to influence lipoprotein metabolism (72).
TR4 In Vivo Roles in Reproduction and Male/Female Fertility
Collins et al (50) first generated TR4−/− mice by mating TR4−/+ mice and found that the number of TR4−/− pups generated was well under that predicted by the normal Mendelian ratio. They also found that TR4−/− mice have high rates of early postnatal mortality with significant growth retardation (50). Interestingly, TR4−/− females show defects in reproduction and maternal behavior. This in vivo evidence suggests that TR4 may play important roles in embryonic and early postnatal pup survival, growth, female reproductive function, and maternal behavior (50).
Chen et al (74) found that TR4−/− female mice displayed subfertility and irregular estrous cycles, reduced size, reduced weight of ovaries, and more intensive granulosa apoptosis, as well as fewer oocytes, preovulatory follicles, and corpora lutea. They concluded that instead of a deficiency in pituitary gonadotropins, TR4−/− mice subfertility was due to ovarian defects with impaired folliculogenesis. Mechanism dissection showed that TR4 might function through transcriptional regulation to reduce LH receptor expression that might then result in reduced gonadal sex hormones (74).
In addition to influencing female fertility, TR4 can also affect male fertility. Mu et al (75) found that sperm production in TR4−/− mice was reduced because of the delay and disruption in spermatogenesis, but not sperm morphology or motility, at stages X to XII, during which late meiotic prophase and meiotic divisions take place. TR4−/− mice also showed delayed and decreased expression of sperm 1 and cyclin A1 at the late meiotic prophase. Mechanistically, TR4 is specifically and stage dependently expressed in late-stage pachytene spermatocytes and round spermatids. In the developing mouse testes, the highest expression of TR4 can be detected at postnatal days 16 to 21 when the first wave of spermatogenesis progresses to the late meiotic prophase. Analysis of the first wave of spermatogenesis in TR4−/− mice shows that the delayed spermatogenesis can be due to a delay and disruption at the end of the late meiotic prophase and subsequent meiotic divisions. Histological examination of testes sections from TR4−/− mice shows degenerated primary spermatocytes and some necrotic tubules (75).
Mu et al (76) also reported that testosterone withdrawal led to inhibited TR4 expression in rat testes, indicating that the TR4 expressed in rat testis to control spermatogenesis is both stage dependent and androgen inductive.
Interestingly, Collins et al (77) found that TR4−/− males have penile dysfunction with priapism. Mechanism dissection revealed that TR4 could modulate the key enzyme, neuronal nitric oxide synthase, which regulates penile erection, at the transcriptional level. Loss of TR4 results in neuronal nitric oxide synthase reduction at both mRNA and protein levels, which consequently leads to erectile dysfunction. TR4−/− males also displayed defects in sexual behavior, such as mounting, intromission, and ejaculation (77). Taken together, these results indicate that TR4 may function through internal spermatogenesis and external penile physiology to control male fertility.
TR4 In Vivo Roles in Neuron and Brain
In 1995, Lopes da Silva et al (78) performed a survey of the NR superfamily and reported that TR4 was expressed in rat supraoptic nucleus of the hypothalamo-neurohypophyseal system. Dot-blot screening of amplified gene fragment analysis showed that TR4 was one of the most abundant factors expressed in the supraoptic nucleus region. Two years later, Young et al (79) also proved that abundant TR4 transcripts were detected in brain, spinal cord, ganglia (sympathetic and sensory), and neuronal epithelia (retinal, otic, olfactory, and gustatory). In 2000, van Schaick et al (80) further found that, at embryonic days 14.5 and 19.5, higher expression of TR4 was detected in the CNS, and TR4 remained highly expressed in the hippocampus and cerebellum in postnatal rats. In the adult brain, TR4 was predominantly expressed in granule cells of both hippocampus and cerebellum (80). Young et al (79) also found striking similarities in the expression patterns of TR4 and ciliary neurotrophic factor receptor (CNTFR) α in the developing and postnatal nervous systems.
Mechanism dissection indicated that TR4 could transactivate CNTFRα gene expression at the transcriptional level via binding to a DR1-TR4RE on the promoter region of the CNTFRα gene (79). Interestingly, via a feedback mechanism, Young et al (81) also demonstrated that CNTF could increase TR4 expression and enhance the DNA-binding capacity of TR4.
Throughout postnatal development, Chen et al (82–84) and Kim et al (85) found that TR4−/− mice exhibited behavioral deficits in motor coordination, ie, unsteady gait, an increase in startle reactivity, and involuntary postural and kinetic movements, and displayed a plateau in performance on the running wheel, indicating that impaired cerebellar function was caused by loss of TR4 in these mice. In the TR4−/− brain, cerebellar-restricted hypoplasia was severe, and cerebellar vermal lobules VI and VII were underdeveloped. Histological analysis of the TR4−/− cerebellar cortex revealed reductions in granule cell density, as well as fewer parallel fiber boutons that were enlarged in size (83). Further analyses demonstrated that the laminations of the TR4−/− cerebellar cortex were changed, including reductions in the thickness of the molecular layer and the internal granule layer, as well as delayed disappearance of the external granule cell layer. These lamination irregularities may result from interference with granule cell proliferation within the external granule cell layer, delayed inward migration of postmitotic granule cells, and a higher incidence of apoptosis (82). In addition, abnormal development of Purkinje cells was observed in the postnatal TR4−/− cerebellum, as evidenced by aberrant dendritic arborization and reduced calbindin staining intensity (82). At postnatal day 21, Golgi-positive Purkinje cells in TR4−/− mice displayed a smaller soma (18%) and a shorter distance to the first branch point (35%) (85). Furthermore, reduction of PAX6, sonic hedgehog (Shh), astrotactin (Astn), reelin, CDK-5, and astrocyte-specific glutamate transporter (GLAST) gene expressions, which are correlated with the morphological development of the cerebellum, were observed in the cerebellum of TR4−/− mice (82, 85).
Chen et al (83) also found that the behavioral phenotype of TR4−/− mice may result from disrupted inhibitory pathways in the cerebellum. The levels of the major inhibitory neurotransmitter γ-aminobutyric acid and the synthetic enzyme for γ-aminobutyric acid, glutamic acid decarboxylase, were decreased in both Purkinje cells and interneurons of the TR4−/− cerebellum, suggesting that the inhibitory circuits signaling within and from the cerebellum may be perturbed (83). No progressive atrophy was observed at various adult stages in the TR4−/− brain, suggesting that the disturbances most likely originate from a failure to establish proper connections between principal neurons in the cerebellum during development (83).
Taken together, these findings from various TR4−/− mice suggest that TR4 is required for normal cerebellar development and that failure to establish proper cytoarchitecture results in dysfunction of the cerebellum and leads to abnormal behavior (82–85).
Zhang et al (86) found reduced myelination in TR4−/− mice, particularly in the forebrain during early developmental stages, and further analysis revealed that oligodendrocytes, which generate the myelin sheath, were decreased. However, the number and proliferation rate of oligodendrocyte precursor cells remained unaffected in forebrains, suggesting that loss of TR4 interrupts oligodendrocyte differentiation. They also found higher Jagged1 expression in axon fiber–enriched regions of TR4−/− forebrains, suggesting a more activated Notch signaling in these regions, which correlates with a previous report showing that Notch activation inhibits oligodendrocyte differentiation (87).
Mechanism dissection suggested that the increased Jagged1-Notch signaling in TR4−/− forebrain inhibited oligodendrocyte differentiation, which in turn contributed to the reduction of myelination in the forebrain (86). Taken together, these results suggest that TR4 is required for proper myelination in the CNS.
Finally, Wang et al (88) reported that loss of TR4 in the CNS resulted in loss of some excitatory interneurons in the superficial dorsal horn associated with a near complete absence of supraspinally integrated pain and itch behaviors. The reflex responsiveness to noxious heat– and the tissue injury–induced heat and mechanical hypersensitivity were intact, whereas nerve injury–induced mechanical hypersensitivity was severely compromised (88).
TR4 In Vivo Roles in Bone, Muscle, and Mitochondria Diseases
Lin et al (89) found that mice lacking TR4 might develop osteoporosis. Using alkaline phosphatase and alizarin red staining to assay bone differentiation, they found that osteoblast progenitor cells isolated from bone marrow of TR4−/− mice displayed reduced osteoblast differentiation capacity and calcification. Furthermore, higher proliferation rates with a lower osteoblast differentiation ability were detected in TR4−/− mice. Mechanism dissection indicated that the expression levels of osteoblast marker genes, including ALP, type I collagen-α1, osteocalcin, PTH, and PTH receptor, were dramatically reduced in osteoblasts from TR4−/− mice. Luciferase reporter and ChIP assays further demonstrated that TR4 could bind directly to the promoter region of the osteocalcin gene to induce its gene expression at the transcriptional level in a dose-dependent manner (89).
Interestingly, in addition to osteoporosis, Lee et al (51) found that 6-month-old TR4−/− mice had severe kyphosis (curvature of the spine) with reduced spine bone mineral density by dual-energy x-ray absorptiometry scanning. Some TR4−/− mice at 6 months of age also started to develop signs of osteoarthritis with increasing superficial zone cells and disruption of the articular surface.
Taken together, the results from bone studies suggest that TR4 may play important roles in bone development and maintenance, and loss of TR4 may lead to development of bone disease, including osteoporosis, kyphosis, and osteoarthritis.
Liu et al (90) showed that mice lacking TR4 may have mitochondrial myopathy with muscle weakness. They found that TR4−/− mice had exercise intolerance with abnormal mitochondrial accumulation with ragged red fibers. In addition, increased serum lactate levels, decreased mitochondrial ATP production, and decreased electron transport chain complex I activity were also found in TR4−/− mouse skeletal muscle tissues (90). Restoration of TR4 in the TR4−/− mice rescued the mitochondrial ATP generation capacity and complex I activity in primary cultured myoblasts, and mechanism dissection revealed that TR4 could modulate NDUFAF1 (the complex I assembly factor) gene expression at the transcriptional level. Restoration of NDUFAF1 rescued mitochondrial ATP generation capacity and complex I activity (90).
Taken together, these results suggest that TR4 may play important roles in maintaining the normal function of bone, muscle, and mitochondria.
TR4 In Vivo Roles in Sickle Cell Diseases
Tanabe et al (91) reported that a erythroid ϵ-globin gene repressor, direct repeat erythroid-definitive, a complex containing the TR2/TR4 heterodimer, could bind to the human embryonic ϵ-globin and fetal γ-globin gene promoters at their DR1 sites. Cui et al (92) also identified DNMT1, NuRD, and LSD1/CoREST repressor complexes plus HDAC3 and TIF1β as potential corepressors of TR2/TR4, and using ChIP assays they found that TR2/TR4 might be able to recruit these transcriptional corepressors to elicit adult stage–specific silencing of the embryonic β-type globin genes through coordinated epigenetic chromatin modifications. Reduced TR2/TR4 binding to the DR1 mutant site may then lead to elevated γ-globin transcription in human hereditary persistence of fetal hemoglobin syndrome (91).
TR2−/− and TR4−/− mice showed delayed silencing of both embryonic and fetal β-type globin genes in definitive erythroid cells. In transgenic mice that express dominant-negative TR4, human embryonic ϵ-globin is activated in both primitive and definitive erythroid cells. In contrast, human fetal γ-globin is activated by dominant-negative TR4 in definitive, but not in primitive, erythroid cells, indicating that TR2/TR4 may function as a stage-selective repressor. Overexpression of TR2 and TR4 leads to repression of ϵ-globin but induction of γ-globin in definitive erythroid cells. These temporally specific and gene-selective alterations in ϵ-globin and γ-globin gene expression via alterations in TR2/TR4 functions provide the first genetic evidence showing that TR2/TR4 may play roles in sequential and stage-specific repression of the human ϵ-globin and γ-globin during development (93).
The embryos of mice with overexpressed TR2 and TR4 in the erythroid cells exhibit a transient midgestational anemia as a consequence of a reduction in the number of primitive erythroid cells. Overexpression of TR2 and TR4 in CD34+ erythroid progenitor cells also reduces GATA1 mRNA expression via alteration of the binding to a DR element in the GATA1 hematopoietic enhancer. Mutation of the DR element with reduced TR2/TR4-binding led to elevated GATA1 promoter activity, suggesting that TR2/TR4 may directly repress GATA1 transcription in erythroid progenitor cells through the DR binding site on a tissue-specific GATA1 enhancer, thereby providing a mechanism by which GATA1 can be directly silenced during terminal erythroid maturation (94).
Sickle cell disease (SCD) is a hematologic disorder caused by a missense mutation in the adult β-globin gene. Higher fetal hemoglobin (HbF) levels in red blood cells of patients with SCD have been shown to decrease morbidity and mortality. Because overexpression of TR2/TR4 in murine adult erythroid cells paradoxically enhanced fetal γ-globin gene expression in transgenic mice, Campbell et al (95) studied whether forced TR2/TR4 expression in a SCD model mouse would result in elevated HbF synthesis and thereby alleviate the disease phenotype. In a “humanized” sickle cell model mouse, TR2/TR4 overexpression increased HbF abundance from 7.6% of total hemoglobin to 18.6%, accompanied by increased hematocrit from 23% to 34% and reticulocyte reduction from 61% to 18%, indicating a significant reduction in hemolysis. Moreover, forced TR2/TR4 expression reduced hepatosplenomegaly and liver parenchymal necrosis and inflammation in the SCD mice, indicating alleviation of the usual pathophysiological characteristics.
Taken together, these results suggest that TR2/TR4 may play important roles in the SCD, and targeting of TR2/TR4 may represent a promising therapeutic approach to induce persistent HbF accumulation for treatment of the disease.
Differential TR4 and PPARγ In Vivo Roles in the TR4-Related Diseases and Their Impacts on Drug Development
TR4 and PPARγ are 2 members of the NR superfamily sharing similar ligands/activators yet having distinct pathophysiological functions that may affect their application to drug development to treat their related diseases.
Both TR4 and PPARγ could be activated by several ligands/activators including PUFAs, 13-HODE, and 15-HETE, as well as some antidiabetic drugs, the TZDs (19). However, 4 differential phenotypes were observed as the consequences of TR4 and PPARγ activation. The first phenotype was activation of PPARγ increasing insulin sensitivity, yet activation of TR4 decreasing the insulin sensitivity (22, 96, 97). The second phenotype was suppression by PPARγ, yet induction by TR4 of cardiovascular diseases with atherosclerosis, a lipid disorder disease in which TZD treatment can improve the lipid profiles of these patients (19, 98). The third phenotype was the differential modulation of these two NRs in cancer progression. PPARγ was believed to act as a tumor suppressor in tumors of colon, prostate, breast, thyroid, and lung by inducing apoptosis (99–101), yet TR4 can be either a suppressor for PCa development or enhancer for PCa metastasis (55 and Ding X.F., D.R. Yang, S.O. Lee, Y.L. Chen, L.Q. Xia, S.J. Lin, S.C. Yu, Y.J. Niu, G.H. Li, and C. Chang, submitted for publication). Furthermore, TR4 also plays dual roles in liver cancer: it functions as a suppressor to inhibit liver cancer initiation and also functions as a stimulator to promote liver cancer metastasis (63). In breast cancer, TR4 can suppress cell growth by interacting with ER (40) and can also promote pituitary corticotroph tumor progression by increasing transcription of the ACTH precursor polypeptide POMC (67). The fourth phenotype was the differential roles of TR4 and PPARγ in bone formation: TR4 can promote bone formation, whereas PPARγ decreases bone formation. The following subsections will further review the detailed mechanistic differences between the differential TR4 vs PPARγ phenotypes.
TR4 and PPARγ in metabolic syndromes
The first phenotype is the opposite functions of these two NRs in their differential modulation of insulin sensitivity: loss of PPARγ reduced insulin sensitivity (96, 97) and, in contrast, loss of TR4 increased insulin sensitivity (22).
The development of insulin resistance is an early event in the onset and progression of type 2 diabetes mellitus, and PPARγ ligand TZDs have been used in type 2 diabetes mellitus as insulin sensitizers with adipose tissue as the primary target tissue where PPARγ is predominantly expressed. In contrast, reduced fat mass and smaller adipocyte size, low glucose levels at birth and in fasting conditions, and improved insulin sensitivity in TR4−/− mice suggest that loss of TR4 increases insulin sensitivity.
In addition to glucose metabolism, TR4 also plays an important role in lipid metabolism by directly regulating expression of SCD-1 (22), which is the rate-limiting enzyme in the biosynthesis of monounsaturated fatty acid. SCD-1 activity has been implicated in obesity, diabetes, lipogenesis, β-oxidation, and insulin sensitivity (68, 69). TR4−/− mice showed reduced fat volume and triglyceride deposition and improved insulin sensitivity that may be partially due to reduced SCD-1 expression (22).
TR4 and PPARγ in cardiovascular diseases
The second phenotype is suppression by PPARγ (98) and induction by TR4 (19) of cardiovascular diseases with atherosclerosis, in which TZD treatment can improve patient lipid profiles.
Xie et al (19) found that CD36 is a TR4 target gene, and thus TR4 might have direct and indirect influences on atherosclerosis via regulation of CD36 (19). TR4−/− mice have reduced foam cell formation with reduced CD36 gene expression and TR4 could function as a transcription factor to directly regulate CD36 gene expression in macrophage cells. Overexpression of CD36 can rescue impaired foam cell formation in macrophages due to reduced expression of TR4, suggesting that CD36 is involved in TR4-mediated foam cell formation (19). In vitro studies showed that TZDs could activate the CD36 promoter through TR4 via binding to the same response element as PPARγ.
These results suggested that TZDs might be able to modulate atherosclerosis via activation of two different NRs, TR4 and PPARγ, that might then lead to differential influence on atherosclerosis. Revealing details of the mechanisms by which these two NRs respond to TZDs in mediating lipid metabolism will be beneficial to our understanding of the complexity of TZDs activity and specificity.
TR4 and PPARγ interplay in PCa initiation
The third phenotype is the differential modulation of these two NRs on cancer development, especially in PCa. Lin et al (Lin S.J., D.R. Yang, H. Miyamoto, M. Jiang, L. Li, N. Wang, G. Li, and C. Chang, submitted for publication) analyzed PPARγ deletion in 69 human PCa samples using fluorescence in situ hybridization assays. Results showed that 9% of PCa samples have a 1-allele deletion of PPARγ compared with no PPARγ deletion in their normal prostate compartment, suggesting that the deletion of PPARγ may have some linkage to PCa development. Similarly, Jiang et al (102) reported disruption of PPAR signaling results in mouse prostatic intraepithelial neoplasia involving active autophagy. In the earlier section, we discussed that Lin et al (55) reported TR4 plays a protective role in PCa iniation. Now they were interested to see whether TR4 might exert different effects in prostate epithelial cells with PPAR deletion (mPrE−/−). Thus, they knocked down TR4 and treated mPrE−/− cells with the carcinogen nitrosomethylurea (100 μg/mL) to induce cell transformation, and results showed suppressed cell proliferation. In contrast, overexpression of TR4 and treatment with nitrosomethylurea in mPrE−/− cells promoted cell proliferation (Lin S.J., D.R. Yang, H. Miyamoto, M. Jiang, L. Li, N. Wang, G. Li, and C. Chang, submitted for publication). However, in the mPrE PPARγ intact cells, they found that knocking down TR4 promoted cell proliferation and overexpression of TR4 suppressed cell proliferation (55).
Using cell transformation with colony formation assays, they also found that knocking down TR4 suppressed, while overexpressing TR4 promoted PCa tumorigenesis in mPrE−/− cells (Lin S.J., D.R. Yang, H. Miyamoto, M. Jiang, L. Li, N. Wang, G. Li, and C. Chang, submitted for publication). In contrast, knocking down TR4 in PPARγ intact prostate epithelial cell lines mPrE and RWPE1 promote PCa tumorigenesis (55). In vivo mouse studies via xenografted mPrE−/− cells with either knocked down TR4 or overexpressed TR4 also led to similar conclusions, showing that differential TR4 effects on PCa cell proliferation and PCa initiation were dependent on the deletion status of PPARγ (Lin S.J., D.R. Yang, H. Miyamoto, M. Jiang, L. Li, N. Wang, G. Li, and C. Chang, submitted for publication). Molecular mechanism dissection revealed that overexpressed TR4 in mPrE−/− cells enhanced the expression of CD44 and Scal1 compared with knocking down TR4, which suppressed the expression of CD44 and Scal1. Furthermore, the epithelial-mesenchymal transition is also increased with over-expressing TR4 in mPrE−/− cells (Lin S.J., D.R. Yang, H. Miyamoto, M. Jiang, L. Li, N. Wang, G. Li, and C. Chang, submitted for publication).
Another similar approach to suppress PPARs is to apply PPARγ antagonist(s). Lin et al (Lin S.J., D.R. Yang, H. Miyamoto, M. Jiang, L. Li, N. Wang, G. Li, and C. Chang, submitted for publication) treated mPrE−/− cells with PPARγ antagonist and assayed cell proliferation. They found the results similar to those in the PPARγ knockout cell line.
Taken together, results of the two approaches (knockout of PPARγ and treatment with an antagonist of PPARγ) show that TR4 might exert differential effects on prostate cell proliferation and PCa initiation, depending on whether PPARγ is either deleted or suppressed by its antagonist (Lin S.J., D.R. Yang, H. Miyamoto, M. Jiang, L. Li, N. Wang, G. Li, and C. Chang, submitted for publication).
TR4 and PPARγ interplay in PCa progression
Interestingly, Lin et al (Lin S.J., C.Y. Lin, K. Izumi, E. Yan, X. Niu, D.R. Yang, N. Wang, G. Li, and C. Chang, submitted for publication) found that TZD (rosiglitazone) treatment of PCa cells with or without TR4 knocked down also led to differential effects. They found that rosiglitazone could promote CWR22RV1 cell growth significantly compared with that for the vehicle control when TR4 was knocked down by siRNA (CWR22RV1-siTR4). In contrast, rosiglitazone showed no effect on CWR22RV1 cell growth compared with that for the vehicle control when cells were transfected with scramble siRNA (CWR22RV1-scr) (Lin S.J., C.Y. Lin, K. Izumi, E. Yan, X. Niu, D.R. Yang, N. Wang, G. Li, and C. Chang, submitted for publication). Other assays including the colony formation assay, migration assay, and invasion assay and in vivo mice studies with xenografted CWR22RV1 cells showed results similar to those for in vitro growth assays. Molecular mechanism dissection revealed increased mRNA expression of HRAS, one of the key metastasis-related genes, in both PCa cell lines (CWR22RV1-siTR4 and C4-2-siTR4) treated with a TZD but decreases in the scramble controls treated with a TZD (Lin S.J., C.Y. Lin, K. Izumi, E. Yan, X. Niu, D.R. Yang, N. Wang, G. Li, and C. Chang, submitted for publication).
Taken together, the results suggest that the TZD rosiglitazone may have differential effects on PCa growth and metastasis that depend on the TR4 expression status, ie TZD may have adverse effects on PCa progression in diabetic PCa patients who lost 1 allele of TR4 (identified in 9% of PCa patients, Lin S.J., C.Y. Lin, K. Izumi, E. Yan, X. Niu, D.R. Yang, N. Wang, G. Li, and C. Chang, submitted for publication).
TR4 and PPARγ in PCa summary
The interaction between TR4 and PPARγ in PCa is complicated. In the absence of PPARγ, TR4 may become an oncogene instead of a tumor suppressor gene. Reciprocally, in the absence of TR4, PPARγ may become an oncogene instead of a tumor suppressor gene through TZD signalings, but the detailed mechanisms are still unclear. Importantly, for the 2 major TZD antidiabetic drugs, rosiglitazone (Avandia) and pioglitazone (Actos), the Food and Drug Administration has recently been withdrawn and retained warnings, respectively. The major warning against Actos is that long-term treatment may increase bladder cancer risk. Because 38% of patients with bladder cancer have PPARγ gene amplification (103), the imbalance between TR4 and PPARγ might contribute to the Actos increased bladder cancer risk (103). Furthermore, Lin et al (Lin S.J., C.Y. Lin, K. Izumi, E. Yan, X. Niu, D.R. Yang, N. Wang, G. Li, and C. Chang, submitted for publication) found that TZDs could increase mRNA expression of HRAS, an oncogene that enhances cancer initiation and metastasis, only when TR4 is reduced. In contrast, TZDs decreases the HRAS mRNA level when TR4 expression is normal.
Taken together, these results indicate that TR4 may be a key regulator to prevent TZDs from being oncogenic, and patients with a 1-allele TR4 deletion need to be warned about taking TZDs.
TR4 and PPARγ in bone physiology
Lin et al (89) were the first to find that mice lacking TR4 develop osteoporosis. Mechanism dissection revealed that TR4 could regulate bone differentiation by directly binding to the promoter region of the osteocalcin gene and induce its expression at the transcriptional level in a dose-dependent manner, suggesting that TR4 may function as a novel transcription factor to play pathophysiological roles in maintaining normal osteoblast activity during bone development and remodeling and that disruption of TR4 function may result in multiple skeletal abnormalities. In contrast, Marie and Kaabeche (104) showed that PPARγ negatively regulates osteoblast differentiation, resulting in bone loss. The opposite roles in bone formation between TR4 and PPARγ open the possibility that manipulation of these 2 NRs may correct the balance between osteoporosis and osteosclerosis, which may have potential implications in the treatment of bone diseases in clinical conditions.
Starvation leads to differential TR4 regulation
In the section, TR4 In Vivo Roles in Metabolic Syndrome and Cardiovascular Diseases, we discussed the differential regulation of TR4 by the same fasting stage (fasting signal → glucagon → cAMP/PKA activation → C/EBPα/β activation → TR4 function increase → gluconeogenesis increase in hepatocytes vs starvation → AMP increase → AMPK activation → TR4 function decrease/inactivation → SCD1 decrease → β-oxidation increase/lipogenesis decrease → insulin sensitivity increase) (22), suggesting that starvation can either increase TR4 function through cAMP/PKA or decrease TR4 function through AMPK. These contrasting findings raise the interesting question of whether to target TR4 to alter starvation-related responses and illnesses. Further dissection of the detailed mechanisms of how our body can differentially utilize the opposite roles of TR4 during starvation may help us to solve this puzzle.
Summary and Future Perspectives
Previous in vitro and in vivo studies suggested that TR4 plays essential roles in aging, cancer, metabolic syndrome, cardiovascular diseases, male/female fertility, neuron and brain development, bone/muscle/mitochondria, and SCD. TR4 has long been viewed as an orphan receptor until the recent discovery that the PPARγ ligands/activators, such as PUFAs and its metabolites, 15-HETE and 13-HODE, as well as the TZD rosiglitazone, could also transactivate TR4 to a degree similar to the activation of PPARγ (Figure 3) (19).
Figure 3.

Scheme of TR4 upstream regulators and downstream targets.
Although sharing similar ligands/activators, TR4 and PPARγ can trigger similar or distinct downstream signals, depending on the context. Whereas TZDs improve insulin sensitivity through the activation of PPARγ and its downstream pathways in adipose tissues, they might trigger some side effects in other tissues by the unexpected activation of TR4. Thus, it is extremely important to fully understand details about the spatial and temporal action of TZDs on TR4 and PPARγ, the individual resulting downstream events, and the interaction between these 2 networks. With this knowledge, the drug industry might be able to improve drug tissue specificity and reduce the side effects of the present TZD drugs by limiting the “bad” effects of TR4, and they might be able to develop new therapeutic approaches for metabolic and other diseases by selectively targeting TR4.
Acknowledgments
We thank Karen Wolf for help in the preparation of this manuscript.
This work was supported by the National Institutes of Health (Grants CA156700 and DK73414), the George Whipple Professorship Endowment; and the Taiwan Department of Health Clinical Trial, Research Center of Excellence (Grant DOH99-TD-B-111-004 to China Medical University, Taichung, Taiwan).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AMPK
- AMP activated protein kinase
- Apo
- apolipoprotein
- AR
- androgen receptor
- C/EBP
- CCAAT/enhancer-binding protein
- ChIP
- chromatin immunoprecipitation
- CNS
- central nervous system
- CNTFR
- ciliary neurotrophic factor receptor
- CoA
- coenzyme A
- CRE
- cAMP response element
- CSB
- Cockayne syndrome B protein
- DBD
- DNA-binding domain
- DR
- direct repeat
- EMSA
- electrophoretic mobility shift assay
- ER
- estrogen receptor
- FOXO3a
- forkhead box O3a
- HAT
- histone acetyltransferase
- HbF
- fetal hemoglobin
- HBV
- hepatitis B virus
- 15-HETE
- 15-hydroxyeicosatetraenoic acid
- 13-HODE
- 13-hydroxyoctadecadienoic acid
- HRE
- hormone response element
- IL-1Ra
- IL-1 receptor antagonist
- IR
- ionizing radiation
- LBD
- ligand-binding domain
- MEF
- mouse embryonic fibroblast
- NR
- nuclear receptor
- PCa
- prostate cancer
- PCAF
- p300/CREB-binding protein-associated factor
- PEPCK
- phosphoenolpyruvate carboxykinase
- PIN
- prostatic intraepithelial neoplasia
- PKA
- protein kinase A
- POMC
- polypeptide proopiomelanocortin
- PPARγ
- peroxisome proliferator–activated receptor γ
- PPRE
- peroxisome proliferator–activated receptor response element
- PUFA
- polyunsaturated fatty acid
- RAR
- retinoic acid receptor
- RIP140
- receptor-interacting protein 140
- RXR
- retinoid X receptor
- SCD
- sickle cell disease
- SCD1
- stearoyl-CoA desaturase 1
- siRNA
- small interfering RNA
- TC-NER
- transcription-coupled nucleotide excision repair
- TR2
- testicular nuclear receptor 2
- TR4
- testicular nuclear receptor 4
- TR4RE
- testicular nuclear receptor 4 response element
- TRA16
- testicular nuclear receptor 4–associated protein 16
- TRE
- thyroid hormone response element
- TZD
- thiazolidinedione
- VDR
- vitamin D receptor
- VDRE
- vitamin D response element.
References
- 1. Chang C, Da Silva SL, Ideta R, Lee Y, Yeh S, Burbach JP. Human and rat TR4 orphan receptors specify a subclass of the steroid receptor superfamily. Proc Natl Acad Sci USA 1994;91:6040–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang X, Downes M, Yu RT, et al. Nuclear receptor expression links the circadian clock to metabolism. Cell. 2006;126:801–810 [DOI] [PubMed] [Google Scholar]
- 4. Yoshikawa T, DuPont BR, Leach RJ, Detera-Wadleigh SD. New variants of the human and rat nuclear hormone receptor, TR4: expression and chromosomal localization of the human gene. Genomics. 1996;35:361–366 [DOI] [PubMed] [Google Scholar]
- 5. Wilson TE, Paulsen RE, Padgett KA, Milbrandt J. Participation of non-zinc finger residues in DNA binding by two nuclear orphan receptors. Science. 1992;256:107–110 [DOI] [PubMed] [Google Scholar]
- 6. Luisi BF, Xu WX, Otwinowski Z, Freedman LP, Yamamoto KR, Sigler PB. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature. 1991;352:497–505 [DOI] [PubMed] [Google Scholar]
- 7. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850 [DOI] [PubMed] [Google Scholar]
- 8. Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin TM, Young WJ, Chang C. Multiple functions of the TR2–11 orphan receptor in modulating activation of two key cis-acting elements involved in the retinoic acid signal transduction system. J Biol Chem. 1995;270:30121–30128 [PubMed] [Google Scholar]
- 10. Rastinejad F, Perlmann T, Evans RM, Sigler PB. Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature. 1995;375:203–211 [DOI] [PubMed] [Google Scholar]
- 11. Lee YF, Young WJ, Burbach JP, Chang C. Negative feedback control of the retinoid-retinoic acid/retinoid X receptor pathway by the human TR4 orphan receptor, a member of the steroid receptor superfamily. J Biol Chem. 1998;273:13437–13443 [DOI] [PubMed] [Google Scholar]
- 12. Chang C, Kokontis J. Identification of a new member of the steroid receptor super-family by cloning and sequence analysis. Biochem Biophys Res Commun. 1988;155:971–977 [DOI] [PubMed] [Google Scholar]
- 13. Chang C, Kokontis J, Acakpo-Satchivi L, Liao S, Takeda H, Chang Y. Molecular cloning of new human TR2 receptors: a class of steroid receptor with multiple ligand-binding domains. Biochem Biophys Res Commun. 1989;165:735–741 [DOI] [PubMed] [Google Scholar]
- 14. Tontonoz P, Hu E, Devine J, Beale EG, Spiegelman BM. PPAR gamma 2 regulates adipose expression of the phosphoenolpyruvate carboxykinase gene. Mol Cell Biol. 1995;15:351–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yue L, Rasouli N, Ranganathan G, Kern PA, Mazzone T. Divergent effects of peroxisome proliferator-activated receptor γ agonists and tumor necrosis factor α on adipocyte ApoE expression. J Biol Chem. 2004;279:47626–47632 [DOI] [PubMed] [Google Scholar]
- 16. Semple RK, Chatterjee VK, O'Rahilly S. PPARγ and human metabolic disease. J Clin Invest. 2006;116:581–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiol Rev. 1998;78:783–809 [DOI] [PubMed] [Google Scholar]
- 18. Rastinejad F. Retinoid X receptor and its partners in the nuclear receptor family. Curr Opin Struct Biol. 2001;11:33–38 [DOI] [PubMed] [Google Scholar]
- 19. Xie S, Lee YF, Kim E, et al. TR4 nuclear receptor functions as a fatty acid sensor to modulate CD36 expression and foam cell formation. Proc Natl Acad Sci USA 2009;106:13353–13358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu NC, Lin WJ, Kim E, et al. Loss of TR4 orphan nuclear receptor reduces phosphoenolpyruvate carboxykinase-mediated gluconeogenesis. Diabetes. 2007;56:2901–2909 [DOI] [PubMed] [Google Scholar]
- 21. Liu NC, Lin WJ, Yu IC, et al. Activation of TR4 orphan nuclear receptor gene promoter by cAMP/PKA and C/EBP signaling. Endocrine. 2009;36:211–217 [DOI] [PubMed] [Google Scholar]
- 22. Kim E, Liu NC, Yu IC, et al. Metformin inhibits nuclear receptor TR4-mediated hepatic stearoyl-CoA desaturase 1 gene expression with altered insulin sensitivity. Diabetes. 2011;60:1493–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsai NP, Huq M, Gupta P, Yamamoto K, Kagechika H, Wei LN. Activation of testicular orphan receptor 4 by fatty acids. Biochim Biophys Acta. 2009;1789:734–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mahajan S, Dkhar HK, Chandra V, et al. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARγ and TR4 for survival. J Immunol. 2012;188:5593–5603 [DOI] [PubMed] [Google Scholar]
- 25. Quinn CE, Hamilton PK, Lockhart CJ, McVeigh GE. Thiazolidinediones: effects on insulin resistance and the cardiovascular system. Br J Pharmacol. 2008;153:636–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shibuya A, Watanabe M, Fujita Y, et al. An autopsy case of troglitazone-induced fulminant hepatitis. Diabetes Care. 1998;21:2140–2143 [DOI] [PubMed] [Google Scholar]
- 27. Ziyadeh N, McAfee AT, Koro C, Landon J, Arnold Chan K. The thiazolidinediones rosiglitazone and pioglitazone and the risk of coronary heart disease: a retrospective cohort study using a US health insurance database. Clin Ther. 2009;31:2665–2677 [DOI] [PubMed] [Google Scholar]
- 28. Lewis JD, Ferrara A, Peng T, et al. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care. 2011;34:916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Govindarajan R, Ratnasinghe L, Simmons DL, et al. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J Clin Oncol. 2007;25:1476–1481 [DOI] [PubMed] [Google Scholar]
- 30. Koro C, Barrett S, Qizilbash N. Cancer risks in thiazolidinedione users compared to other anti-diabetic agents. Pharmacoepidemiol Drug Saf. 2007;16:485–492 [DOI] [PubMed] [Google Scholar]
- 31. Bolden A, Bernard L, Jones D, Akinyeke T, Stewart LV. The PPAR gamma agonist troglitazone regulates Erk 1/2 phosphorylation via a PPARγ-independent, MEK-dependent pathway in human prostate cancer cells. PPAR Res. 2012;2012:929052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lyles BE, Akinyeke TO, Moss PE, Stewart LV. Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARγ-dependent and PPARγ-independent pathways. Cell Cycle. 2009;8:268–277 [DOI] [PubMed] [Google Scholar]
- 33. Jiang G, Hunter T. Receptor signaling: when dimerization is not enough. Curr Biol. 1999;9:R568–R571 [DOI] [PubMed] [Google Scholar]
- 34. Santomauro Júnior AC, Ugolini MR, Santomauro AT, Souto RP. Metformin and AMPK: an old drug and a new enzyme in the context of metabolic syndrome (in Portuguese). Arq Bras Endocrinol Metabol. 2008;52:120–125 [DOI] [PubMed] [Google Scholar]
- 35. Huq MD, Gupta P, Tsai NP, Wei LN. Modulation of testicular receptor 4 activity by mitogen-activated protein kinase-mediated phosphorylation. Mol Cell Proteomics. 2006;5:2072–2082 [DOI] [PubMed] [Google Scholar]
- 36. Xie S, Ni J, Lee YF, et al. Increased acetylation in the DNA-binding domain of TR4 nuclear receptor by the coregulator ARA55 leads to suppression of TR4 transactivation. J Biol Chem. 2011;286:21129–21136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–295 [DOI] [PubMed] [Google Scholar]
- 38. Lee CH, Chinpaisal C, Wei LN. A novel nuclear receptor heterodimerization pathway mediated by orphan receptors TR2 and TR4. J Biol Chem. 1998;273:25209–25215 [DOI] [PubMed] [Google Scholar]
- 39. Lee YF, Shyr CR, Thin TH, Lin WJ, Chang C. Convergence of two repressors through heterodimer formation of androgen receptor and testicular orphan receptor-4: a unique signaling pathway in the steroid receptor superfamily. Proc Natl Acad Sci USA 1999;96:14724–14729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shyr CR, Hu YC, Kim E, Chang C. Modulation of estrogen receptor-mediated transactivation by orphan receptor TR4 in MCF-7 cells. J Biol Chem. 2002;277:14622–14628 [DOI] [PubMed] [Google Scholar]
- 41. Lee YF, Pan HJ, Burbach JP, Morkin E, Chang C. Identification of direct repeat 4 as a positive regulatory element for the human TR4 orphan receptor. A modulator for the thyroid hormone target genes. J Biol Chem. 1997;272:12215–12220 [DOI] [PubMed] [Google Scholar]
- 42. Lee YF, Young WJ, Lin WJ, Shyr CR, Chang C. Differential regulation of direct repeat 3 vitamin D3 and direct repeat 4 thyroid hormone signaling pathways by the human TR4 orphan receptor. J Biol Chem. 1999;274:16198–16205 [DOI] [PubMed] [Google Scholar]
- 43. Yan ZH, Karam WG, Staudinger JL, Medvedev A, Ghanayem BI, Jetten AM. Regulation of peroxisome proliferator-activated receptor alpha-induced transactivation by the nuclear orphan receptor TAK1/TR4. J Biol Chem. 1998;273:10948–10957 [DOI] [PubMed] [Google Scholar]
- 44. Schiltz RL, Nakatani Y. The PCAF acetylase complex as a potential tumor suppressor. Biochim Biophys Acta. 2000;1470:M37–M53 [DOI] [PubMed] [Google Scholar]
- 45. Nakajima T, Fujino S, Nakanishi G, Kim YS, Jetten AM. TIP27: a novel repressor of the nuclear orphan receptor TAK1/TR4. Nucl Acids Res. 2004;32:4194–4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang Y, Wang X, Dong T, Kim E, Lin WJ, Chang C. Identification of a novel testicular orphan receptor-4 (TR4)-associated protein as repressor for the selective suppression of TR4-mediated transactivation. J Biol Chem. 2003;278:7709–7717 [DOI] [PubMed] [Google Scholar]
- 47. Cavaillès V, Dauvois S, L'Horset F, et al. Nuclear factor RIP140 modulates transcriptional activation by the estrogen receptor. EMBO J. 1995;14:3741–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Treuter E, Albrektsen T, Johansson L, Leers J, Gustafsson JA. A regulatory role for RIP140 in nuclear receptor activation. Mol Endocrinol. 1998;12:864–881 [DOI] [PubMed] [Google Scholar]
- 49. Kim E, Ma WL, Lin DL, Inui S, Chen YL, Chang C. TR4 orphan nuclear receptor functions as an apoptosis modulator via regulation of Bcl-2 gene expression. Biochem Biophys Res Commun. 2007;361:323–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Collins LL, Lee YF, Heinlein CA, et al. Growth retardation and abnormal maternal behavior in mice lacking testicular orphan nuclear receptor 4. Proc Natl Acad Sci USA 2004;101:15058–15063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee YF, Liu S, Liu NC, et al. Premature aging with impaired oxidative stress defense in mice lacking TR4. Am J Physiol Endocrinol Metab. 2011;301:E91–E98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu S, Yan SJ, Lee YF, et al. Testicular nuclear receptor 4 (TR4) regulates UV light-induced responses via Cockayne syndrome B protein-mediated transcription-coupled DNA repair. J Biol Chem. 2011;286:38103–38108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yan SJ, Lee YF, Ting HJ, et al. Deficiency in TR4 nuclear receptor abrogates Gadd45a expression and increases cytotoxicity induced by ionizing radiation. Cell Mol Biol Lett. 2012;17:309–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li G, Lee YF, Liu S, et al. Oxidative stress stimulates testicular orphan receptor 4 through forkhead transcription factor forkhead box O3a. Endocrinology. 2008;149:3490–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lin SJ, Lee SO, Lee YF, et al. TR4 nuclear receptor functions as a tumor suppressor for prostate tumorigenesis via modulation of DNA damage/repair system [published online February 28, 2014]. Carcinogenesis. doi: 10.1093/carcin/bgu052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Niu Y, Altuwaijri S, Lai KP, et al. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci USA. 2008;105(34):12182–12187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Niu Y, Chang TM, Yeh S, Ma WL, Wang YZ, Chang C. Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene. 2010;29:3593–3604 [DOI] [PubMed] [Google Scholar]
- 58. Chang C, Lee SO, Yeh S, Chang TM. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver [published online ahead of print July 22, 2013]. Oncogene. doi: 10.1038/onc.2013.274 [DOI] [PubMed] [Google Scholar]
- 59. Lin TH, Lee SO, Niu Y, et al. Differential androgen deprivation therapies with anti-androgens Casodex/bicalutamide or MDV3100/enzalutamide versus anti-androgen receptor ASC-J9(R) Lead to promotion versus suppression of prostate cancer metastasis. J Biol Chem. 2013;288:19359–19369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin TH, Izumi K, Lee SO, Lin WJ, Yeh S, Chang C. Anti-androgen receptor ASC-J9 versus anti-androgens MDV3100 (enzalutamide) or Casodex (bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3-CCL2 signaling. Cell Death Dis. 2013;4:e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Crea F, Danesi R, Farrar WL. Cancer stem cell epigenetics and chemoresistance. Epigenomics. 2009;1:63–79 [DOI] [PubMed] [Google Scholar]
- 62. Yang DR, Ding XF, Luo J, et al. Increased chemosensitivity via targeting testicular nuclear receptor 4 (TR4)-Oct4-interleukin 1 receptor antagonist (IL1Ra) axis in prostate cancer CD133+ stem/progenitor cells to battle prostate cancer. J Biol Chem. 2013;288:16476–16483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huang YH, Liao CH, Chen RN, Liao CJ, Lin KH. Human testicular orphan receptor 4 enhances thyroid hormone receptor signaling. J Cell Physiol. 2010;222:347–356 [DOI] [PubMed] [Google Scholar]
- 64. Tiollais P, Pourcel C, Dejean A. The hepatitis B virus. Nature. 1985;317:489–495 [DOI] [PubMed] [Google Scholar]
- 65. Lin WJ, Li J, Lee YF, et al. Suppression of hepatitis B virus core promoter by the nuclear orphan receptor TR4. J Biol Chem. 2003;278:9353–9360 [DOI] [PubMed] [Google Scholar]
- 66. Lin TJ, Yang RY, Lee HJ. Collective repression of the hepatitis B virus enhancer II by human TR4 and TR2 orphan receptors. Hepatol Res. 2008;38:79–84 [DOI] [PubMed] [Google Scholar]
- 67. Du L, Bergsneider M, Mirsadraei L, et al. Evidence for orphan nuclear receptor TR4 in the etiology of Cushing disease. Proc Natl Acad Sci USA 2013;110:8555–8560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cohen P, Ntambi JM, Friedman JM. Stearoyl-CoA desaturase-1 and the metabolic syndrome. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3:271–280 [DOI] [PubMed] [Google Scholar]
- 69. Cohen P, Friedman JM. Leptin and the control of metabolism: role for stearoyl-CoA desaturase-1 (SCD-1). J Nutr. 2004;134:2455S–2463S [DOI] [PubMed] [Google Scholar]
- 70. Choi H, Kim SJ, Park SS, Chang C, Kim E. TR4 activates FATP1 gene expression to promote lipid accumulation in 3T3–L1 adipocytes. FEBS Lett. 2011;585:2763–2767 [DOI] [PubMed] [Google Scholar]
- 71. Kang HS, Okamoto K, Kim YS, et al. Nuclear orphan receptor TAK1/TR4-deficient mice are protected against obesity-linked inflammation, hepatic steatosis, and insulin resistance. Diabetes. 2011;60:177–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim E, Xie S, Yeh SD, et al. Disruption of TR4 orphan nuclear receptor reduces the expression of liver apolipoprotein E/C-I/C-II gene cluster. J Biol Chem. 2003;278:46919–46926 [DOI] [PubMed] [Google Scholar]
- 73. Kim E, Yang Z, Liu NC, Chang C. Induction of apolipoprotein E expression by TR4 orphan nuclear receptor via 5′ proximal promoter region. Biochem Biophys Res Commun. 2005;328:85–90 [DOI] [PubMed] [Google Scholar]
- 74. Chen LM, Wang RS, Lee YF, et al. Subfertility with defective folliculogenesis in female mice lacking testicular orphan nuclear receptor 4. Mol Endocrinol. 2008;22:858–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mu X, Lee YF, Liu NC, et al. Targeted inactivation of testicular nuclear orphan receptor 4 delays and disrupts late meiotic prophase and subsequent meiotic divisions of spermatogenesis. Mol Cell Biol. 2004;24:5887–5899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mu X, Yang L, Chang C. Stage dependent and androgen inductive expression of orphan receptor TR4 in rat testis. Biochem Biophys Res Commun. 2006;341:464–469 [DOI] [PubMed] [Google Scholar]
- 77. Collins LL, Lee YF, Ting HJ, et al. The roles of testicular nuclear receptor 4 (TR4) in male fertility-priapism and sexual behavior defects in TR4 knockout mice. Reprod Biol Endocrinol. 2011;9:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lopes da Silva S, Van Horssen AM, Chang C, Burbach JP. Expression of nuclear hormone receptors in the rat supraoptic nucleus. Endocrinology. 1995;136:2276–2283 [DOI] [PubMed] [Google Scholar]
- 79. Young WJ, Smith SM, Chang C. Induction of the intronic enhancer of the human ciliary neurotrophic factor receptor (CNTFRα) gene by the TR4 orphan receptor. A member of steroid receptor superfamily. J Biol Chem. 1997;272:3109–3116 [DOI] [PubMed] [Google Scholar]
- 80. van Schaick HS, Rosmalen JG, Lopes da Silva S, Chang C, Burbach JP. Expression of the orphan receptor TR4 during brain development of the rat. Brain Res Mol Brain Res. 2000;77:104–110 [DOI] [PubMed] [Google Scholar]
- 81. Young WJ, Lee YF, Smith SM, Chang C. A bidirectional regulation between the TR2/TR4 orphan receptors (TR2/TR4) and the ciliary neurotrophic factor (CNTF) signaling pathway. J Biol Chem. 1998;273:20877–20885 [DOI] [PubMed] [Google Scholar]
- 82. Chen YT, Collins LL, Uno H, Chang C. Deficits in motor coordination with aberrant cerebellar development in mice lacking testicular orphan nuclear receptor 4. Mol Cell Biol. 2005;25:2722–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen YT, Collins LL, Uno H, et al. Abnormal cerebellar cytoarchitecture and impaired inhibitory signaling in adult mice lacking TR4 orphan nuclear receptor. Brain Res. 2007;1168:72–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chen YT, Collins LL, Chang SS, Chang C. The roles of testicular orphan nuclear receptor 4 (TR4) in cerebellar development. Cerebellum. 2008;7:9–17 [DOI] [PubMed] [Google Scholar]
- 85. Kim YS, Harry GJ, Kang HS, et al. Altered cerebellar development in nuclear receptor TAK1/TR4 null mice is associated with deficits in GLAST+ glia, alterations in social behavior, motor learning, startle reactivity, and microglia. Cerebellum. 2010;9:310–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhang Y, Chen YT, Xie S, et al. Loss of testicular orphan receptor 4 impairs normal myelination in mouse forebrain. Mol Endocrinol. 2007;21:908–920 [DOI] [PubMed] [Google Scholar]
- 87. Wang S, Sdrulla AD, diSibio G, et al. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 1998;21:63–75 [DOI] [PubMed] [Google Scholar]
- 88. Wang X, Zhang J, Eberhart D, et al. Excitatory superficial dorsal horn interneurons are functionally heterogeneous and required for the full behavioral expression of pain and itch. Neuron. 2013;78:312–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lin SJ, Ho HC, Lee YF, et al. Reduced osteoblast activity in the mice lacking TR4 nuclear receptor leads to osteoporosis. Reprod Biol Endocrinol. 2012;10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu S, Lee YF, Chou S, et al. Mice lacking TR4 nuclear receptor develop mitochondrial myopathy with deficiency in complex I. Mol Endocrinol. 2011;25:1301–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tanabe O, Katsuoka F, Campbell AD, et al. An embryonic/fetal beta-type globin gene repressor contains a nuclear receptor TR2/TR4 heterodimer. EMBO J. 2002;21:3434–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cui S, Kolodziej KE, Obara N, et al. Nuclear receptors TR2 and TR4 recruit multiple epigenetic transcriptional corepressors that associate specifically with the embryonic β-type globin promoters in differentiated adult erythroid cells. Mol Cell Biol. 2011;31:3298–3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tanabe O, McPhee D, Kobayashi S, et al. Embryonic and fetal beta-globin gene repression by the orphan nuclear receptors, TR2 and TR4. EMBO J. 2007;26:2295–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tanabe O, Shen Y, Liu Q, et al. The TR2 and TR4 orphan nuclear receptors repress Gata1 transcription. Genes Dev. 2007;21:2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Campbell AD, Cui S, Shi L, et al. Forced TR2/TR4 expression in sickle cell disease mice confers enhanced fetal hemoglobin synthesis and alleviated disease phenotypes. Proc Natl Acad Sci USA 2011;108:18808–18813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Olefsky JM, Saltiel AR. PPAR gamma and the treatment of insulin resistance. Trends Endocrinol Metab. 2000;11(9):362–368 [DOI] [PubMed] [Google Scholar]
- 97. Hevener AL, He W, Barak Y, et al. Muscle-specific Pparg deletion causes insulin resistance. Nat Med. 2003;9(12):1491–1497 [DOI] [PubMed] [Google Scholar]
- 98. Subramanian V, Golledge J, Ijaz T, Bruemmer D, Daugherty A. Pioglitazone-induced reductions in atherosclerosis occur via smooth muscle cell-specific interaction with PPARγ. Circ Res. 2010;107(8):953–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Koeffler HP. Peroxisome proliferator-activated receptor γ and cancers. Clin Cancer Res. 2003;9:1–9 [PubMed] [Google Scholar]
- 100. Philips JC, Petite C, Willi JP, Buchegger F, Meier CA. Effect of peroxisome proliferator-activated receptor gamma agonist, rosiglitazone, on dedifferentiated thyroid cancers. Nucl Med Commun. 2004;25:1183–1186 [DOI] [PubMed] [Google Scholar]
- 101. Kim HJ, Hwang JY, Choi WS, et al. Expression of a peroxisome proliferator-activated receptor gamma 1 splice variant that was identified in human lung cancers suppresses cell death induced by cisplatin and oxidative stress. Clin Cancer Res. 2007;13:2577–2583 [DOI] [PubMed] [Google Scholar]
- 102. Jiang M, Fernandez S, Jerome WG, et al. Disruption of PPARγ signaling results in mouse prostatic intraepithelial neoplasia involving active autophagy. Cell Death Differ. 2010;17(3):469–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yang DR, Lin SJ, Ding XF, et al. Higher expression of peroxisome proliferator-activated receptor gamma or its activation by agonist thiazolidinedione-rosiglitazone promotes bladder cancer cell migration and invasion. Urology. 2013:81(5):1109 e1101–e1106 [DOI] [PubMed] [Google Scholar]
- 104. Marie PJ, Kaabeche K. PPARγ activity and control of bone mass in skeletal unloading. PPAR Res. 2006;2006:64807. [DOI] [PMC free article] [PubMed] [Google Scholar]