

Abstract
There has been much evidence demonstrating the involvement of oxidative stress in the pathology of neurological disorders. Moreover, the vulnerability of the central nervous system to reactive oxygen species mediated injury is well established since neurons consume large amounts of oxygen, the brain has many areas containing high iron content, and neuronal mitochondria generate large amounts of hydrogen peroxide. Furthermore, neuronal membranes are rich in polyunsaturated fatty acids, which are particularly susceptible to oxidative stress. Recently, the biological roles of products produced by lipid peroxidation have received much attention, not only for their pathological mechanisms associated with neurological disorders, but also for their practical clinical applications as biomarkers. Here, we discuss the production mechanisms of reactive oxygen species in some neurological disorders, including Alzheimer’s disease, Down syndrome, Parkinson’s disease, and stroke. We also describe lipid peroxidation biomarkers for evaluating oxidative stress.
Keywords: lipid peroxidation, neurological disorder, Alzheimer’s disease, Down syndrome, Parkinson’s disease, stroke
Introduction
Redox homeostasis is maintained in vivo as an equilibrium between oxidant and antioxidant levels. An imbalance in these molecules in favor of oxidants results in oxidative stress.(1) Increasing evidence has pointed out the involvement of in vivo oxidation as an underlying mechanism in several disorders and diseases, such as diabetes,(2) cardiovascular diseases,(3) cancer, neurodegenerative diseases,(4) and aging.(5) Recently, the role of oxidative stress in neurodegenerative diseases has received much attention.(6)
The brain is extremely sensitive to oxidative damage for several reasons. For one, the brain consumes an inordinate amount of oxygen (around 20%), particularly when considering the fact that the brain accounts for only 2% of body weight.(4) A major reason for the high O2 uptake is the vast amounts of ATP needed to maintain neuronal intracellular ion homeostasis in the face of ion channels need for action potentials and neurosecretion. Additionally, several brain areas, including the substantia nigra, caudate nucleus, putamen, and globus pallidus, are rich in iron content.(7) It is generally accepted that iron accumulates in the brain of older individuals, and iron ions that are released following brain damage can catalyze free radical reactions. The brain is a source of reactive oxygen species (ROS). Complex I-dependent hydrogen peroxide generation in brain mitochondria is greater than that in skeletal muscle mitochondria.(8) Lastly, neuronal membranes are rich in polyunsaturated fatty acids (PUFAs), particularly arachidonic acid (AA), docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA).(9) These PUFAs are particularly vulnerable to oxidative stress due to their possession of unsaturated double bonds. For these reasons, neural cells are more susceptible to oxidative damage when compared to other body tissues.
Lipid Peroxidation
It has been well documented that lipid peroxidation comprises of three distinct mechanisms: free radical-mediated oxidation, free radical-independent, nonenzymatic oxidation, and enzymatic oxidation.(10) Free radical-mediated lipid peroxidation proceeds by a chain mechanism, that is, one initiating free radical can oxidize both lipid molecules in biological membranes and low density lipoproteins. The primary reactions of lipid peroxidation include hydrogen-atom abstraction by peroxyl or alkoxyl radicals (initiation); oxygen addition to carbon radicals (propagation); peroxyl radical fragmentation or rearrangement; peroxyl radical addition to carbon-carbon double bonds cyclization; and peroxyl-peroxyl termination. Vitamin E (α-tocopherol) acts as a “chain-breaking” antioxidant and can terminate the propagation steps of lipid peroxidation. Lipid oxidization by nonradical mechanisms can be accomplished by singlet oxygen and ozone.(11) Moreover, singlet oxygen may be used in photodynamic therapy or can cause deleterious damage such as a disease porphyria on the skin. Myeloperoxidase, a heme protein secreted by activated phagocytes, reacts with hydrogen peroxide in the presence of chloride and bromide to yield hypochlorous acid and hypobromous acid, HOCl and HOBr, respectively. These acids are strong oxidants and can oxidize biological molecules via either free radical or non-radical pathways.(12) The third mechanism of oxidation, enzymatic oxidation, can be illustrated by lipoxygenase and cyclooxygenase, which have both been reported to oxidize arachidonic acid into hydroperoxyeicosatetraenoic aicd (HPETE), prostaglandins, prostacyclin, thromboxane, and leukotrienes. Moreover, lipoxygenase and cyclooxygenase can oxidize lipids regio-, stereo-, and enantiospecifically.(13)
Lipid peroxidation has been shown to induce disturbance to membrane organization and functional loss/modification of proteins and DNA. However, lipid peroxidation products also act as redox signaling mediators.(14,15)
Lipid Peroxidation Biomarkers for Neurological Dysfunction
A number of studies have been performed to measure lipid oxidation products in humans; the biomarkers measuring the degree of lipid peroxidation are discussed below.
Lipid peroxidation products from linoleic acid
Linoleic acid (LA) is the most abundant PUFA in vivo, and its oxidation is initiated by a straightforward mechanism that yields much simpler products than AA. As shown in Fig. 1, hydroperoxyoctadecadienoic acids (HPODEs) that are formed by radical-mediated oxidation consist of four isomers: 13-hydroperoxy-9(Z),11(E)-octadecadienoic acid [13-(Z,E)-HPODE], 13-hydroperoxy-9(E), 11(E)-octadecadienoic acid [13-(E,E)-HPODE], 9-hydroperoxy-10(E), 12(Z)-octadecadienoic acid [9-(E,Z)-HPODE], and 9-hydroperoxy-10(E), 12(E)-octadecadienoic acid [9-(E,Z)-HPODE]. 13-(Z,E)-HPODE is also formed by enzymatic oxidation via lipoxygenase as enantio-, regio-, and stereo-specific products, while 9- and 13-(E,E)-HPODE are specific products of radical-mediated oxidation. HPODEs are then reduced to hydroxyoctadecanoic acids (HODEs) by a variety of enzymes, such as glutathione peroxidase.
Fig. 1.
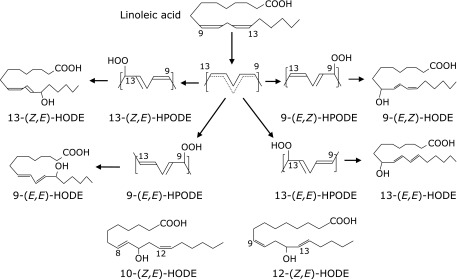
Structures of HODE formation.
Singlet oxygen has been shown to oxidize LA by a non-radical mechanism to form 13-(Z,E)-HPODE, [9-(E,Z)-HPODE], 10-hydroperoxy-8(E), 12(Z)-octadecadienoic acid [10-(E,Z)-HPODE] and 12-hydroperoxy-9(Z), 13(E)-octadecadienoic acid [12-(Z,E)-HPODE]. In this case, 10- and 12-(Z,E)-HPODEs are specific oxidation products produced by singlet oxygen. We recently found that the fasting levels of [10- and 12-(Z,E)-HODE]/LA increase significantly with increasing levels of HbA1c and glucose during oral glucose tolerance tests.(16)
We propose that measurement of total hydroxyoctadecanoic acid (tHODE) could be an effective biomarker of oxidative stress in vivo.(17) In this method, biological samples such as plasma, erythrocytes, urine, and tissues are reduced followed by saponification. Hydroperoxides, as well as hydroxides of both free and ester forms of LA, are then measured as tHODE.
Lipid peroxidation products from arachidonic acid
Arachidonic acid is more susceptible to oxidation than LA because it has three bisallylic positions that serve as possible sites for initial hydrogen atom abstraction. In the presence of hydrogen atom donors, six major hydroperoxide, HPETEs, are produced (Fig. 2a).(18) Furthermore, lipoxygenases oxidize AA to 5-, 8-, 12-, and 15-HPETE.(19) HPETEs are then further reduced to hydroxyeicosatetraenoic acids (HETEs).
Fig. 2a.
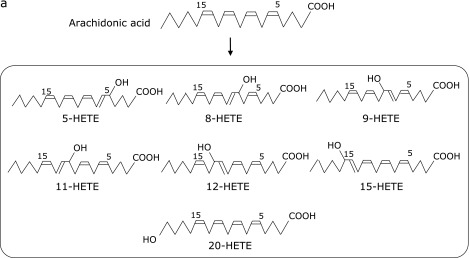
Structures of HETE formation.
Recently, elevated levels of 20-HETE have been reported in cerebrospinal fluid (CSF) and plasma in patients with subarachnoid hemorrhage (SAH).(20,21) It has also been reported that an inhibitor of cytochrome P450 oxidoreductase, which catalyzes AA to 20-HETE, can reduce infarct size in rats after cerebral ischemia.(22)
F2-isoprostanes (F2-IsoPs) are products of free radical-induced peroxidation of AA and are currently thought to be the most reliable markers of oxidative damage in humans.(23–25) As shown in Fig. 2b, F2-IsoPs comprise of many isomers through various reactions. For example, prostaglandin H2-like bicyclic endoperoxide (H2-isoprostanes) intermediates, which are formed by autoxidation of AA, are reduced to form F-ring IsoPs. Additionally, three arachidonyl radicals give rise to the formation of four F2-IsoP regioisomers, 5-, 12-, 8-, and 15-series, each of which contain eight racemic diastereomers for a total of 64 compounds.(23) F2-IsoPs have been shown to increase in the CSF of patients with AD (26) and Huntington’s disease.(27)
Fig. 2b.

Structures of isoprostane (IsoP) and isofuran (IsoF) formation.
Isofurans (IsoFs), which have a substituted tetrahydrofuran ring structure, are generated by the peroxidation of AA as well as F2-IsoPs. Interestingly, IsoFs levels in the substantia nigra have been reported to be elevated in patients with Parkinson’s disease (PD), whereas levels of F2-IsoPs were not.(28) The formation of IsoFs and F2-IsoPs occur via similar mechanisms and share a carbon-centered radical as an intermediate. However, IsoFs are generated by a reaction with molecular oxygen. Milne et al.(29) used gas chromatography-mass spectrometry (GC-MS) to measure F2-IsoPs and IsoFs and showed that normal levels of IsoFs in human plasma (0.071 ± 0.010 ng/ml) were higher than F2-IsoPs (15-F2t-IsoP) (0.035 ± 0.006 ng/ml), although it is unclear what kind of IsoF they measured.
Lipid peroxidation products from docosahexaenoic acid
DHA and AA are highly enriched in the central nervous system. Since DHA has a higher number of double bonds compared with AA, DHA is more susceptible to free radical-mediated oxidation. Oxidation of DHA leads to the formation of an F2-IsoPs-like compound, neuroprostane (NP), which is highly concentrated in neuronal membranes.(30) Endoperoxide intermediates, which are formed by autoxidation of DHA, are reduced to form F-ring NPs (F4-NPs) which have eight regioisomers (Fig. 3). These F4-NP regioisomers (4-, 7-, 10-, 11-, 13-, 14-, 17-, and 20-series) further comprise of eight racemic diastereomers for a total of 128 compounds. It has been reported that F4-NP levels are higher in the CSF of patients with aneurysmal subarachnoid hemorrhage(31) and in the plasma of patients with Rett syndrome.(32)
Fig. 3.

Structures of neuroprostanes (NPs) and neurofurans (NFs).
Recently, it was shown that a novel class of IsoFs-like compounds, neurofurans (NFs), are formed from DHA (Fig. 3).(33) Both NPs and NFs are considered sensitive and specific markers of neuronal oxidative damage. In a case of basal levels, Solberg et al.(34) demonstrated that concentrations of IsoPs and IsoFs were similar to levels of NPs and NFs in the brain tissue of newborn piglets (about 10 ng/g of brain tissue).
Lipid peroxidation-derived short-chain aldehydes
PUFAs are finally broken down to short-chain aldehydes by peroxidation.(35) These short-chain aldehydes are mainly classified into three families: 2-alkenals, 4-hydroxy-2-alkenals, and ketoaldehydes (Fig. 4). These products are generated from the oxidation of free fatty acids including AA and LA. Acrolein, the most potent electrophilic 2-alkenal, has been detected in automobile emissions, cigarette smoke, and other products of thermal degradation.(36) One type of 4-hydroxy-2-alkenal, 4-hydroxy-2-nonenal (HNE), is known to be a major aldehyde produced during peroxidation of ω6-PUFAs, such as LA and AA. Lastly, malondialdehyde (MDA) is known as the most abundant lipid peroxidation-specific aldehyde. In order to identify lipid peroxidation, the thiobarbituric acid (TBA) assay has been widely used to measure red pigment formation which is adduct of TBA with aldehydes, MDA, alkenals, and alkadienals.
Fig. 4.

General structure of short-chain aldehydes.
These aldehydes are highly reactive with proteins, DNA, and phospholipids, and cause deleterious effects. HNE- and acrolein-protein adducts are considered to be good biomarkers of lipid peroxidation in vivo and have been widely applied by using antibodies directed against them.(37) In many clinical reports, these antibodies were used not only for measuring biological samples, but also for immunohistochemical staining of brain sections.(38)
Lipid peroxidation products from cholesterol
Cholesterol is present in all cells and regulates the fluidity of lipid bilayers. Cholesterol oxidation products, oxysterols, have received increasing attention as messengers for cell signaling and cholesterol transport.(39) Cholesterol is also oxidized by both enzymatic and non-enzymatic mechanisms (Fig. 5). The free radical-mediated oxidation of cholesterol yields 7α- and 7β-hydroperoxycholesterol (7α-OOHCh and 7β-OOHCh), 7α- and 7β-hydroxycholesterol (7α-OHCh and 7β-OHCh) and 7-ketocholesterol (7-KCh) as major products.(39) 7β-OHCh is regarded as a marker of free radical-mediated oxidation, and s. Singlet oxygen oxidizes cholesterol to 5α-OOHCh.
Fig. 5.
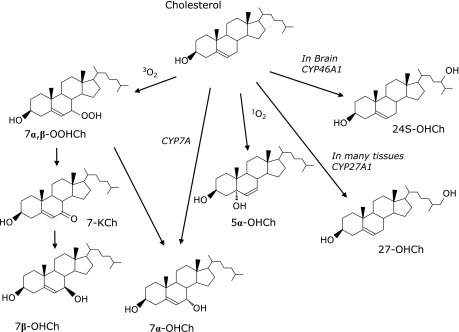
Mechanisms for the formation of cholesterol oxidation.
Various enzymes oxidize cholesterol to yield specific hydroxycholesterols. For example, the mitochondrial enzyme, CYP27A1, produces 27-hydoroxycholesterol (27-OHCh). Furthermore, 24-hydroxylase (CYP46A1), an enzyme found exclusively in the brain and retina, yields 24(S)-hydroxycholesterol [24(S)-OHCh] as a specific product,(40) which has been reported to play an important role in affecting cholesterol metabolism in the brain.(41) Because almost all plasma 24(S)-OHCh seem to generate in brain, plasma levels of 24(S)-OHCh are considered to be reflective of the number of metabolically-active neurons.(42) Consistent with this, Leoni and Cacciam reported reduced levels of 24(S)-OHCh in neurodegenerative diseases, including Alzheimer’s disease (AD), and suggested that the reduction of 24(S)-OHCh was related to the degree of atrophy in the brain.(41) In contrast, slightly increased 24(S)-OHCh levels have been observed in cases of mild cognitive impairment and in patients with AD.(43)
Lipid peroxidation products from α-tocopherol
α-Tocopherol is the highest bioavailable form of vitamin E, the major lipid-soluble chain-breaking antioxidant. α-Tocopherol reacts with peroxyl radicals to form α-tocopheroxyl radicals, which are further oxidized into α-tocopheryl quinone (αTQ) (Fig. 6).(44) Once α-tocopheroxyl radical is formed, it can be converted back to α-tocopherol by the action of other antioxidants, including ascorbic acid and coenzyme Q10.(45) This reversal mechanism allows for the maintenance of adequate concentrations of the active form of α-tocopherol to protect from oxidative damage that is induced by highly-reactive free radicals. The degree of oxidative injury can be evaluated by measurements of the αTQ and α-tocopherol levels. However, there have only been a few studies examining αTQ in biological samples. Ravaglia et al.(46) reported that plasma concentration of αTQ was associated with cognitive impairment in elderly individuals. Additionally, Kanazawa et al.(47) reported that the ratios of αTQ/α-tocopherol were increased in several tissue types by hyperoxia while Murphy et al.(48) reported that the plasma concentration of αTQ increased in patients who had undergone an aortic cross-clamping operation. We reported that αTQ was significantly higher in the plasma and hippocampus of a Ts65Dn mouse model of Down syndrome.(49) Recently, we also demonstrated an increase of plasma αTQ is correlated with fatigue induced by overnight deskwork without sleep.(50) This suggests that plasma levels of αTQ could serve as a potential objective marker of fatigue.
Fig. 6.

Mechanisms for the formation of α-tocopherol oxidation.
Mechanisms of Free Radical Production in Neurological Disorders
The mechanism of ROS production differs in each neurological disorder. Here, we discuss the lipid peroxidation products that relate to each neurological disorder as well as possible therapeutic interventions with antioxidants.
Alzheimer’s disease
The aggregation of Aβ (1-42) is proposed to play a central role in the pathogenesis of AD which is the most common cause of dementia. Aβ are small oligomers that can insert into the lipid bilayer and generate hydrogen peroxide.(51) In the presence of redox-active transition metal ions, such as Fe and Cu, hydrogen peroxide leads to the formation of hydroxyl radicals via the Fenton reaction. This reaction then initiates lipid peroxidation, and causes oxidative damage to proteins and other biomolecules.(52) On the other hand, using an animal model of AD it has been shown that oxidative stress and lipid peroxidation can induce Aβ accumulation during the progression of AD.(53)
Many studies have demonstrated increased levels of oxidative stress markers, such as F2-isoPs and HNE, in the biological fluids of patients with AD.(54–57) Through Michael addition and Schiff base formation, HNE binds to proteins and modifies enzymatic activities(56) and the conformation of tau (τ), which is associated with neurofibrillary tangles.(58)
We previously reported significantly elevated levels of tHODE and oxidatively modified peroxiredoxin (oxPrx)-2 and oxPrx-6 in plasma and erythrocytes of patients with AD.(59) Furthermore, the tHODE levels were shown to increase with increasing clinical dementia ratings.
The level of plasma 24(S)-OHCh has also been reported to be higher in patients with AD and vascular dementia(43); however, these levels are elevated only in the early stages and have been shown to be lower in long-term cases of AD.(60,61) Associations between plasma 24(S)-OHCh levels and AD have been inconsistent in many reports.
As far as treatment strategies for this neurodegenerative disease, vitamin E seems to be a promising candidate since it acts as a “chain-breaking” antioxidant that can terminate the propagation steps of lipid peroxidation. However, Petersen et al.(62) reported that vitamin E given to 769 patients with mild cognitive impairment did nothing to improve their symptoms. Moreover, significant effects of vitamin E have also not been observed in treatment for AD.(63,64)
Down syndrome
Down syndrome (DS) is considered to be the most common genetic cause of mental retardation. DS is caused by total or partial trisomy of chromosome 21. Cu, Zn superoxide dismutase (Cu,Zn-SOD) has been proposed as a potential gene candidate closely implicated in oxidative stress.(65) Furthermore, overexpression of Cu,Zn-SOD in DS has been shown to result in the excessive production of hydrogen peroxide. However, other antioxidant enzymes, such as catalase and glutathione peroxidase, which can catalyze hydrogen peroxide to H2O are not coded in chromosome 21. The imbalance between Cu, Zn SOD and these enzymes has been suggested to induce oxidative damage in patients with DS. More specifically, a three- to four-fold increase in intracellular ROS and elevated levels of lipid peroxidation markers have been revealed in the brains of patients with DS.(66) Interestingly, the concentration of IsoPs in the amniotic fluid of mothers who were pregnant with fetuses with DS has been reported to be 9-fold higher than in pregnancies with normal fetuses.(67) The levels of HNE, protein carbonyl (2,4-dinitrophenylhydrazine), and advanced glycation end products have all been found to be increased in the brains of fetuses with DS (at 18–20 weeks of gestation) in comparison with matched controls.(68) These results provide evidence that accelerated brain oxidation occurs early in the life of individuals with DS.
We previously reported elevated levels of IsoPs (8-iso-prostaglandin F2α) in brain tissue and tHODE and 7-OHCh in the plasma Ts65Dn mice, a mouse model of Down syndrome, than those of control mice.(49) Ts65Dn mice are the most widely used animal model of DS, since these mice carry a segmental trisomy of mouse chromosome 16 in which many of the genes in human chromosome 21 are conserved.(69) We found that acrolein was stained in the dentate gyrus region of Ts65Dn mice. Furthermore, we have also demonstrated that αTQ was significantly higher in the plasma and hippocampus of Ts65Dn mice than in control mice.
There is no clear clinical evidence that antioxidant supplementation is an effective treatment for DS(70); however, in trials that have investigated this, antioxidant vitamin supplements were begun at 7.5 months of age or later, which possibly account for the lack of efficacy observed. We conducted an experiment to test whether chronic administration of vitamin E could reverse the cognitive deficit found in Ts65Dn mice.(49) α-Tocopherol was administered to pregnant Ts65Dn females from the day of conception throughout the pregnancy, and to Ts65Dn and control pus from birth until the end of the behavioral testing period. We found that administration of α-tocopherol attenuated cognitive impairment and decreased the levels of lipid peroxidation products in Ts65Dn mice. The results of animal experiments cannot be easily extrapolated to humans; however, this study implies a potential benefit of early supplementation of vitamin E in patients with DS.
Parkinson’s disease
PD clinically manifests as resting tremors, slowness of movement, rigidity, and postural instability. PD is pathologically defined by the loss of neurons in the substantia nigra (SN) and by the presence of cytoplasmic protein inclusions named Lewy bodies and neuritis.(71) A small synaptic protein, α-synuclein, is recognized to be the main component of Lewy bodies.(72) The loss of SN cells lead to striatal dopamine deficiency and are related to the clinical severity of PD. Three independent mutations in α-synuclein, including A53T, A30P, and E46K, are involved in the development of familial PD. Recombinant synuclein is reported to produce hydrogen peroxide by Electron Spin Resonance (ESR),(73) and hydrogen peroxide exposure induces α-synuclein fragmentation and accumulation in the nucleus.(74)
It has been reported that the lipid peroxidation adducts, HNE and Nε-(carboxymethyl) lysine, are localized in Lewy bodies in post-mortem PD brain tissue.(75) HNE modification of α-synuclein results in conformational changes and oligomerization. Furthermore, HNE-modified oligomers are potentially toxic and could contribute to the demise of neurons subjected to oxidative damage.(76) The levels of plasma F2-IsoPs, HETEs, 7β-and 27-OHCh, 7-KCh, and NPs have all been shown to be elevated in patients with PD compared to controls.(77) Furthermore, the levels of plasma F2-IsoPs and HETEs have been reported to be higher in the early stages of PD. Recently, IsoFs have been shown to be increased in the SN of patients with PD.(28) It was also reported that glutathione, catalase, and glutathione peroxidase are reduced in PD.(78,79)
We recently developed specific antibodies against oxidized DJ-1(80) since DJ-1 has recently been found to be a causative gene for a familial form of PD. A cysteine residue at position 106 (Cys-106) in DJ-1 was found to be preferentially oxidized under oxidative stress.(81) We observed higher levels of oxidized DJ-1 in the erythrocytes of un-medicated patients with PD than in healthy subjects and medicated patients with PD.(80) Immunohistochemical analysis also revealed that the number of oxidized DJ-1 antibody-positive cells in the SN of MPTP-treated mice increase in a dose-dependent manner.(82) These results suggest that oxidized DJ-1 might be a useful biomarker for the detection and diagnosis of PD at early-stages.
As far as studies investigating the use of antioxidants in PD, Zhang et al.(83) reported that vitamin E can reduce the risk of developing PD; however, other studies have shown contradictory results about the dietary intake of antioxidants, and their efficacy for preventing PD progression.(84,85)
Stroke
Stroke is characterized by a block of blood flow to the brain. Important risk factors for stroke are hypertension, diabetes, hypercholesterolemia, smoking, and old age.(86) These risk factors are associated with the endothelial dysfunction and lead to atherogenesis and cerebral ischemic injury. With cerebral reperfusion by thrombolytic therapy, for example tissue plasminogen activator (tPA), ROS production is stimulated and causes cytotoxicity through oxidation of lipids, proteins and DNA.(87,88) Moreover, elevated plasma levels of IsoPs and 20-HETE have been observed in acute patients with acute ischemic stroke.(21)
In a previous study, we found changes in lipid peroxidation products in the CSF of patients with or without symptomatic vasospasm (SVS) after subarachnoid hemorrhage (SAH).(89) One of the most important complications of SAH is ischemia. Ischemia after SAH is hypothesized to be caused by decreased cerebral perfusion induced by SVS. SVS occurs in about 30% of patients surviving SAH, mostly between days four and 10 after SAH. We found that the levels of free IsoPs (8-iso-PGF2α) and HODE in CSF and plasma activity of platelet-activating factor-acetylhydrolase (PAF-AH) were higher in patients without SVS than in patients with SVS. These findings lead to the speculation that plasma PAF-AH can hydrolyze oxidized phospholipids and attenuate the spreading of lipid peroxidation.
It has been suggested that a combination of antioxidants with thrombolytic therapy might offer advantages, such as improving clinical outcome after cerebral ischemia, or extending the treatment window for tPA.(90) The effect of vitamin E is difficult to assess, since stroke is an emergency disease. However, edaravone has been effectively used as a neuroprotective agent for the treatment of acute cerebral infarction in Japan.
Conclusions
As discussed, lipid peroxidation is involved in neurological disorders, including AD, PD, stroke, and DS. There are few clinical reports about the efficacy of antioxidants for neurological disorders. It is considered that the timing to start antioxidants therapy is important. As our report about the effectiveness of α-tocopherol administration to the fetuses of DS mouse,(49) antioxidants therapy before appearance of symptoms may be effective against these neurological disorders. In order to start a treatment in early stages, biomarkers which can be used to diagnose in early stages of each neurological disorders are required. The use of biomarkers to diagnose each neurological disorder has been the focus of intense study. It should be noted that there is no specific lipid peroxidation marker for each disease; however, it has been suggested that the peroxidation products from AA and DHA, particularly NPs and NFs, may serve as potential markers for neurological disorder, since these compounds may exist in central nervous system. The development of technology that can measure infinitesimal content of NPs and NFs will be necessary in future studies.
Acknowledgments
This study was supported in part by a Grants-in-Aid for Young Scientists (A) No. 22680051 from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Conflict of Interest
No potential conflicts of interest were disclosed.
References
- 1.Sies H. Oxidative stress: oxidants and antioxidants. Exp physiol. 1997;82:291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 2.Miyamoto S, Kawano H, Hokamaki J, et al. Increased plasma levels of thioredoxin in patients with glucose intolerance. Intern Med. 2005;44:1127–1132. doi: 10.2169/internalmedicine.44.1127. [DOI] [PubMed] [Google Scholar]
- 3.Cracowski JL. Isoprostanes: an emerging role in vascular physiology and disease? Chem Phys Lipids. 2004;128:75–83. doi: 10.1016/j.chemphyslip.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 5.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 6.Reed TT. Lipid peroxidation and neurodegenerative disease. Free Radic Biol Med. 2011;51:1302–1319. doi: 10.1016/j.freeradbiomed.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 7.Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 8.Malinska D, Kudin AP, Debska-Vielhaber G, Vielhaber S, Kunz WS. Chapter 23 Quantification of superoxide production by mouse brain and skeletal muscle mitochondria. Methods Enzymol. 2009;456:419–437. doi: 10.1016/S0076-6879(08)04423-6. [DOI] [PubMed] [Google Scholar]
- 9.Chen CT, Green JT, Orr SK, Bazinet RP. Regulation of brain polyunsaturated fatty acid uptake and turnover. Prostaglandins Leukot Essent Fatty Acids. 2008;79:85–91. doi: 10.1016/j.plefa.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Niki E, Yoshida Y, Saito Y, Noguchi N. Lipid peroxidation: mechanisms, inhibition, and biological effects. Biochem Biophys Res Commun. 2005;338:668–676. doi: 10.1016/j.bbrc.2005.08.072. [DOI] [PubMed] [Google Scholar]
- 11.Pryor WA, Houk KN, Foote CS, et al. Free radical biology and medicine: it’s a gas, man! Am J Physiol Regul Integr Comp Physiol. 2006;291:R491–R511. doi: 10.1152/ajpregu.00614.2005. [DOI] [PubMed] [Google Scholar]
- 12.Panasenko OM, Vakhrusheva TV, Vlasova , II , Chekanov AV, Baranov YV, Sergienko VI. Role of myeloperoxidase-mediated modification of human blood lipoproteins in atherosclerosis development. Bull Exp Biol Med. 2007;144:428–431. doi: 10.1007/s10517-007-0346-x. [DOI] [PubMed] [Google Scholar]
- 13.Schneider C, Pratt DA, Porter NA, Brash AR. Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem Biol. 2007;14:473–488. doi: 10.1016/j.chembiol.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ceaser EK, Moellering DR, Shiva S, et al. Mechanisms of signal transduction mediated by oxidized lipids: the role of the electrophile-responsive proteome. Biochem Soc Trans. 2004;32:151–155. doi: 10.1042/bst0320151. [DOI] [PubMed] [Google Scholar]
- 15.Zmijewski JW, Landar A, Watanabe N, Dickinson DA, Noguchi N, Darley-Usmar VM. Cell signalling by oxidized lipids and the role of reactive oxygen species in the endothelium. Biochem Soc Trans. 2005;33:1385–1389. doi: 10.1042/BST20051385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umeno A, Shichiri M, Ishida N, et al. Singlet oxygen induced products of linoleates, 10- and 12-(Z,E)-hydroxyoctadecadienoic acids (HODE), can be potential biomarkers for early detection of type 2 diabetes. PLoS one. 2013;8:e63542. doi: 10.1371/journal.pone.0063542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshida Y, Shichiri M. Quantitative analysis of lipid peroxidation products using mass spectrometry. In: Ivanov AR, Lazarev AV, editors. Sample Preparation in Biological Mass Spectrometry. Springer; 2011. pp. 877–884. [Google Scholar]
- 18.Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111:5944–5972. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto S. Mammalian lipoxygenases: molecular structures and functions. Biochim Biophys Acta. 1992;1128:117–131. doi: 10.1016/0005-2760(92)90297-9. [DOI] [PubMed] [Google Scholar]
- 20.Roman RJ, Renic M, Dunn KM, Takeuchi K, Hacein-Bey L. Evidence that 20-HETE contributes to the development of acute and delayed cerebral vasospasm. Neurol Res. 2006;28:738–749. doi: 10.1179/016164106X152016. [DOI] [PubMed] [Google Scholar]
- 21.Ward NC, Croft KD, Blacker D, et al. Cytochrome P450 metabolites of arachidonic acid are elevated in stroke patients compared with healthy controls. Clin Sci. 2011;121:501–507. doi: 10.1042/CS20110215. [DOI] [PubMed] [Google Scholar]
- 22.Renic M, Klaus JA, Omura T, et al. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2009;29:629–639. doi: 10.1038/jcbfm.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., 2nd A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halliwell B, Lee CY. Using isoprostanes as biomarkers of oxidative stress: some rarely considered issues. Antioxid Redox Signal. 2010;13:145–156. doi: 10.1089/ars.2009.2934. [DOI] [PubMed] [Google Scholar]
- 25.Roberts LJ,, 2nd, Fessel JP. The biochemistry of the isoprostane, neuroprostane, and isofuran pathways of lipid peroxidation. Chem Phys Lipids. 2004;128:173–186. doi: 10.1016/j.chemphyslip.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 26.Montine TJ, Beal MF, Cudkowicz ME, et al. Increased CSF F2-isoprostane concentration in probable AD. Neurology. 1999;52:562–565. doi: 10.1212/wnl.52.3.562. [DOI] [PubMed] [Google Scholar]
- 27.Montine TJ, Beal MF, Robertson D, et al. Cerebrospinal fluid F2-isoprostanes are elevated in Huntington’s disease. Neurology. 1999;52:1104–1105. doi: 10.1212/wnl.52.5.1104. [DOI] [PubMed] [Google Scholar]
- 28.Fessel JP, Hulette C, Powell S, Roberts LJ, 2nd, Zhang J. Isofurans, but not F2-isoprostanes, are increased in the substantia nigra of patients with Parkinson’s disease and with dementia with Lewy body disease. J Neurochem. 2003;85:645–650. doi: 10.1046/j.1471-4159.2003.01709.x. [DOI] [PubMed] [Google Scholar]
- 29.Milne GL, Gao B, Terry ES, Zackert WE, Sanchez SC. Measurement of F2- isoprostanes and isofurans using gas chromatography-mass spectrometry. Free Radic Biol Med. 2013;59:36–44. doi: 10.1016/j.freeradbiomed.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts LJ,, 2nd, Montine TJ, Markesbery WR, et al. Formation of isoprostane-like compounds (neuroprostanes) in vivo from docosahexaenoic acid. J Biol Chem. 1998;273:13605–13612. doi: 10.1074/jbc.273.22.13605. [DOI] [PubMed] [Google Scholar]
- 31.Hsieh YP, Lin CL, Shiue AL, et al. Correlation of F4-neuroprostanes levels in cerebrospinal fluid with outcome of aneurysmal subarachnoid hemorrhage in humans. Free Radic Biol Med. 2009;47:814–824. doi: 10.1016/j.freeradbiomed.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 32.Signorini C, De Felice C, Leoncini S, et al. F4-neuroprostanes mediate neurological severity in Rett syndrome. Clin Chim Acta. 2011;412:1399–1406. doi: 10.1016/j.cca.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 33.Song WL, Lawson JA, Reilly D, et al. Neurofurans, novel indices of oxidant stress derived from docosahexaenoic acid. J Biol Chem. 2008;283:6–16. doi: 10.1074/jbc.M706124200. [DOI] [PubMed] [Google Scholar]
- 34.Solberg R, Longini M, Proietti F, Vezzosi P, Saugstad OD, Buonocore G. Resuscitation with supplementary oxygen induces oxidative injury in the cerebral cortex. Free Radic Biol Med. 2012;53:1061–1067. doi: 10.1016/j.freeradbiomed.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 35.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 36.Ghilarducci DP, Tjeerdema RS. Fate and effects of acrolein. Rev Environ Contam Toxicol. 1995;144:95–146. doi: 10.1007/978-1-4612-2550-8_2. [DOI] [PubMed] [Google Scholar]
- 37.Toyokuni S, Miyake N, Hiai H, et al. The monoclonal antibody specific for the 4-hydroxy-2-nonenal histidine adduct. FEBS Lett. 1995;359:189–191. doi: 10.1016/0014-5793(95)00033-6. [DOI] [PubMed] [Google Scholar]
- 38.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 39.Diczfalusy U. Analysis of cholesterol oxidation products in biological samples. J AOAC Int. 2004;87:467–473. [PubMed] [Google Scholar]
- 40.Lund EG, Guileyardo JM, Russell DW. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S A. 1999;96:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leoni V, Caccia C. 24S-hydroxycholesterol in plasma: a marker of cholesterol turnover in neurodegenerative diseases. Biochimie. 2013;95:595–612. doi: 10.1016/j.biochi.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 42.Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260:493–508. doi: 10.1111/j.1365-2796.2006.01725.x. [DOI] [PubMed] [Google Scholar]
- 43.Lutjohann D, Papassotiropoulos A, Bjorkhem I, et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. 2000;41:195–198. [PubMed] [Google Scholar]
- 44.Niki E. Tocopherylquinone and tocopherylhydroquinone. Redox Rep. 2007;12:204–210. doi: 10.1179/135100007X200353. [DOI] [PubMed] [Google Scholar]
- 45.Stoyanovsky DA, Osipov AN, Quinn PJ, Kagan VE. Ubiquinone-dependent recycling of vitamin E radicals by superoxide. Arch Biochem Biophys. 1995;323:343–351. doi: 10.1006/abbi.1995.9955. [DOI] [PubMed] [Google Scholar]
- 46.Ravaglia G, Forti P, Lucicesare A, et al. Plasma tocopherols and risk of cognitive impairment in an elderly Italian cohort. Am J Clin Nutr. 2008;87:1306–1313. doi: 10.1093/ajcn/87.5.1306. [DOI] [PubMed] [Google Scholar]
- 47.Kanazawa H, Miyata C, Nagata Y, Urano S, Matsushima Y. Determination of alpha-tocopherol and alpha-tocopherylquinone in rat tissues and plasma by high-performance liquid chromatography with electrochemical detection. Chem Pharm Bull (Tokyo) 2000;48:1462–1466. doi: 10.1248/cpb.48.1462. [DOI] [PubMed] [Google Scholar]
- 48.Murphy ME, Kolvenbach R, Aleksis M, Hansen R, Sies H. Antioxidant depletion in aortic crossclamping ischemia: increase of the plasma alpha-tocopheryl quinone/alpha-tocopherol ratio. Free Radic Biol Med. 1992;13:95–100. doi: 10.1016/0891-5849(92)90069-s. [DOI] [PubMed] [Google Scholar]
- 49.Shichiri M, Yoshida Y, Ishida N, et al. α-Tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic Biol Med. 2011;50:1801–1811. doi: 10.1016/j.freeradbiomed.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 50.Shichiri M, Harada N, Ishida N, et al. Oxidative stress is involved in fatigue induced by overnight deskwork as assessed by increase in plasma tocopherylhydroqinone and hydroxycholesterol. Biol Psychol. 2013;94:527–533. doi: 10.1016/j.biopsycho.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Huang X, Atwood CS, Hartshorn MA, et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 52.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 53.Praticò D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol. 2005;58:730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- 55.Montine TJ, Montine KS, McMahan W, Markesbery WR, Quinn JF, Morrow JD. F2-isoprostanes in Alzheimer and other neurodegenerative diseases. Antioxid Redox Signal. 2005;7:269–275. doi: 10.1089/ars.2005.7.269. [DOI] [PubMed] [Google Scholar]
- 56.Reed TT, Pierce WM, Markesbery WR, Butterfield DA. Proteomic identification of HNE-bound proteins in early Alzheimer disease: insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009;1274:66–76. doi: 10.1016/j.brainres.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 57.Volkel W, Sicilia T, Pähler A, et al. Increased brain levels of 4-hydroxy-2-nonenal glutathione conjugates in severe Alzheimer’s disease. Neurochem Int. 2006;48:679–686. doi: 10.1016/j.neuint.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 58.Liu Q, Smith MA, Avilá J, et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic Biol Med. 2005;38:746–754. doi: 10.1016/j.freeradbiomed.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 59.Yoshida Y, Yoshikawa A, Kinumi T, et al. Hydroxyoctadecadienoic acid and oxidatively modified peroxiredoxins in the blood of Alzheimer’s disease patients and their potential as biomarkers. Neurobiol Aging. 2009;30:174–185. doi: 10.1016/j.neurobiolaging.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 60.Bretillon L, Sidén A, Wahlund LO, et al. Plasma levels of 24S-hydroxycholesterol in patients with neurological diseases. Neurosci Lett. 2000;293:87–90. doi: 10.1016/s0304-3940(00)01466-x. [DOI] [PubMed] [Google Scholar]
- 61.Papassotiropoulos A, Lütjohann D, Bagli M, et al. Plasma 24S-hydroxycholesterol: a peripheral indicator of neuronal degeneration and potential state marker for Alzheimer’s disease. Neuroreport. 2000;11:1959–1962. doi: 10.1097/00001756-200006260-00030. [DOI] [PubMed] [Google Scholar]
- 62.Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 63.Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 64.Lloret A, Badía MC, Mora NJ, Pallardó FV, Alonso MD, Viña J. Vitamin E paradox in Alzheimer’s disease: it does not prevent loss of cognition and may even be detrimental. J Alzheimers Dis. 2009;17:143–149. doi: 10.3233/JAD-2009-1033. [DOI] [PubMed] [Google Scholar]
- 65.Groner Y, Elroy-Stein O, Avraham KB, et al. Cell damage by excess CuZnSOD and Down’s syndrome. Biomed Pharmacother. 1994;48:231–240. doi: 10.1016/0753-3322(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 66.Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature. 1995;378:776–779. doi: 10.1038/378776a0. [DOI] [PubMed] [Google Scholar]
- 67.Perrone S, Longini M, Bellieni CV, et al. Early oxidative stress in amniotic fluid of pregnancies with Down syndrome. Clin Biochem. 2007;40:177–180. doi: 10.1016/j.clinbiochem.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 68.Odetti P, Angelini G, Dapino D, et al. Early glycoxidation damage in brains from Down’s syndrome. Biochem Biophys Res Commun. 1998;243:849–851. doi: 10.1006/bbrc.1998.8186. [DOI] [PubMed] [Google Scholar]
- 69.Moldrich RX, Dauphinot L, Laffaire J, Rossier J, Potier MC. Down syndrome gene dosage imbalance on cerebellum development. Prog Neurobiol. 2007;82:87–94. doi: 10.1016/j.pneurobio.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 70.Ellis JM, Tan HK, Gilbert RE, et al. Supplementation with antioxidants and folinic acid for children with Down’s syndrome: randomised controlled trial. BMJ. 2008;336:594–597. doi: 10.1136/bmj.39465.544028.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jellinger KA. Recent developments in the pathology of Parkinson’s disease. J Neural Transm Suppl. 2002:347–376. doi: 10.1007/978-3-7091-6139-5_33. [DOI] [PubMed] [Google Scholar]
- 72.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 73.Turnbull S, Tabner BJ, El-Agnaf OM, Moore S, Davies Y, Allsop D. alpha-Synuclein implicated in Parkinson’s disease catalyses the formation of hydrogen peroxide in vitro. Free Radic Biol Med. 2001;30:1163–1170. doi: 10.1016/s0891-5849(01)00513-5. [DOI] [PubMed] [Google Scholar]
- 74.Xu S, Zhou M, Yu S, et al. Oxidative stress induces nuclear translocation of C-terminus of alpha-synuclein in dopaminergic cells. Biochem Biophys Res Commun. 2006;342:330–335. doi: 10.1016/j.bbrc.2006.01.148. [DOI] [PubMed] [Google Scholar]
- 75.Castellani RJ, Perry G, Siedlak SL, et al. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett. 2002;319:25–28. doi: 10.1016/s0304-3940(01)02514-9. [DOI] [PubMed] [Google Scholar]
- 76.Qin Z, Hu D, Han S, Reaney SH, Di Monte DA, Fink AL. Effect of 4-hydroxy-2-nonenal modification on alpha-synuclein aggregation. J Biol Chem. 2007;282:5862–5870. doi: 10.1074/jbc.M608126200. [DOI] [PubMed] [Google Scholar]
- 77.Seet RC, Lee CY, Lim EC, et al. Oxidative damage in Parkinson disease: measurement using accurate biomarkers. Free Radic Biol Med. 2010;48:560–566. doi: 10.1016/j.freeradbiomed.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 78.Sian J, Dexter DT, Lees AJ, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 79.Ambani LM, Van Woert MH, Murphy S. Brain peroxidase and catalase in Parkinson disease. Arch Neurol. 1975;32:114–118. doi: 10.1001/archneur.1975.00490440064010. [DOI] [PubMed] [Google Scholar]
- 80.Saito Y, Hamakubo T, Yoshida Y, et al. Preparation and application of monoclonal antibodies against oxidized DJ-1. Significant elevation of oxidized DJ-1 in erythrocytes of early-stage Parkinson disease patients. Neurosci Lett. 2009;465:1–5. doi: 10.1016/j.neulet.2009.08.074. [DOI] [PubMed] [Google Scholar]
- 81.Kinumi T, Kimata J, Taira T, Ariga H, Niki E. Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;317:722–728. doi: 10.1016/j.bbrc.2004.03.110. [DOI] [PubMed] [Google Scholar]
- 82.Akazawa YO, Saito Y, Hamakubo T, et al. Elevation of oxidized DJ-1 in the brain and erythrocytes of Parkinson disease model animals. Neurosci Lett. 2010;483:201–205. doi: 10.1016/j.neulet.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 83.Zhang SM, Hernán MA, Chen H, Spiegelman D, Willett WC, Ascherio A. Intakes of vitamins E and C, carotenoids, vitamin supplements, and PD risk. Neurology. 2002;59:1161–1169. doi: 10.1212/01.wnl.0000028688.75881.12. [DOI] [PubMed] [Google Scholar]
- 84.Etminan M, Gill SS, Samii A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: a meta-analysis. Lancet Neurol. 2005;4:362–365. doi: 10.1016/S1474-4422(05)70097-1. [DOI] [PubMed] [Google Scholar]
- 85.Weber CA, Ernst ME. Antioxidants, supplements, and Parkinson’s disease. Ann Pharmacother. 2006;40:935–938. doi: 10.1345/aph.1G551. [DOI] [PubMed] [Google Scholar]
- 86.Feigin VL. Stroke epidemiology in the developing world. Lancet. 2005;365:2160–2161. doi: 10.1016/S0140-6736(05)66755-4. [DOI] [PubMed] [Google Scholar]
- 87.Crack PJ, Taylor JM. Reactive oxygen species and the modulation of stroke. Free Radic Biol Med. 2005;38:1433–1444. doi: 10.1016/j.freeradbiomed.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 88.Gursoy-Ozdemir Y, Can A, Dalkara T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke. 2004;35:1449–1453. doi: 10.1161/01.STR.0000126044.83777.f4. [DOI] [PubMed] [Google Scholar]
- 89.Hirashima Y, Doshi M, Hayashi N, et al. Plasma platelet-activating factor-acetyl hydrolase activity and the levels of free forms of biomarker of lipid peroxidation in cerebrospinal fluid of patients with aneurysmal subarachnoid hemorrhage. Neurosurgery. 2012;70:602–609. doi: 10.1227/NEU.0b013e3182333c69. [DOI] [PubMed] [Google Scholar]
- 90.Green AR. Pharmacological approaches to acute ischaemic stroke: reperfusion certainly, neuroprotection possibly. Br J Pharmacol. 2008;153:S325–S338. doi: 10.1038/sj.bjp.0707594. [DOI] [PMC free article] [PubMed] [Google Scholar]