

Abstract
Mesenchymal stromal cells (MSCs) or their media (MSC-M) were reported to reverse acute lung injury (ALI)-induced decrease of alveolar fluid clearance. To determine the mechanisms by which MSC-M exert their beneficial effects, an in vitro model of alveolar epithelial injury was created by exposing primary rat alveolar epithelial cells (AECs) to hypoxia (3% O2) plus cytomix, a combination of IL-1β, TNF-α, and IFN-γ. MSC-M were collected from human MSCs exposed for 12 h to either normoxia (MSC-M) or to hypoxia plus cytomix (HCYT-MSC-M). This latter condition was used to model the effect of alveolar inflammation and hypoxia on paracrine secretion of MSCs in the injured lung. Comparison of paracrine soluble factors in MSC media showed that the IL-1 receptor antagonist and prostaglandin E2 were markedly increased while keratinocyte growth factor (KGF) was twofold lower in HCYT-MSC-M compared with MSC-M. In AECs, hypoxia plus cytomix increased protein permeability, reduced amiloride-sensitive short-circuit current (AS-Isc), and also decreased the number of α-epithelial sodium channel (α-ENaC) subunits in the apical membrane. To test the effects of MSC media, MSC-M and HCYT-MSC-M were added for an additional 12 h to AECs exposed to hypoxia plus cytomix. MSC-M and HCYT-MSC-M completely restored epithelial permeability to normal. MSC-M, but not HCYT-MSC-M, significantly prevented the hypoxia plus cytomix-induced decrease of ENaC activity and restored apical α-ENaC channels. Interestingly, KGF-deprived MSC-M were unable to restore amiloride-sensitive sodium transport, indicating a possible role for KGF in the beneficial effect of MSC-M. These results indicate that MSC-M may be a preferable therapeutic option for ALI.
Keywords: growth factors, membrane transport, hypoxia, inflammation, mesenchymal stem cells, sodium channels
acute lung injury (ALI) is a major clinical syndrome manifested by an inflammatory response with injury to the lung endothelium and the alveolar epithelium that leads to acute respiratory failure (53). Although mortality has declined with improved supportive care, especially lung protective ventilation and a conservative fluid strategy, the overall mortality of ALI remains high, ∼25–40% (51). No pharmacological therapies have yet been found to improve clinical outcomes in ALI (8), thus underscoring the need for novel therapeutic strategies (40).
There has been a rapidly growing interest in the potential value of stem cell therapies as therapeutic approaches in multiple clinical disorders, including myocardial infarction, diabetes, and hepatic and acute renal failure (34). Moreover, recent studies indicate that bone marrow-derived, multipotent mesenchymal (stem) stromal cells (MSCs) have therapeutic efficacy in experimental models of ALI in adult animals. Intravenous or intra-alveolar administration of MSCs attenuates the severity of lung damage, reduces lung inflammation and fibrosis, and increases survival of rats after bleomycin, endotoxin-induced lung injury, and live bacteria or ventilator-induced injury (34, 41, 44). The potential mechanisms through which MSC therapy improves lung function include lung engraftment, anti-inflammatory, or immunomodulatory functions, and antiapoptotic effects (1, 19, 27, 28). However, although MSCs delivered by the intravenous route engraft in mice lungs after bleomycin injury (44), other reports have shown that engraftment of MSCs in the lung is very low (<5%) (9, 21). Interestingly, most studies reported striking protective effects of MSC therapy despite low engraftment rates, supporting the concept that the protective effects may be largely mediated through production of paracrine mediators that constitute the MSC secretome (29, 33, 43). This assumption has been confirmed by another recent study reporting that the cultured medium of MSC themselves can reverse hyperoxia-induced lung injury in a perinatal mouse model (4).
The direct delivery of MSCs for treating acute respiratory distress syndrome (ARDS) is reasonable to test for safety and efficacy in the clinical setting, but if the cultured media of MSCs were as effective as the MSCs themselves, it would be a simpler treatment that might have fewer potential side effects that may be associated with cell-based therapy. Moreover, in the context of ALI, these cells are exposed to acute inflammation and hypoxic conditions with unknown consequences on their secretome. Under resting conditions, MSCs secrete several soluble factors that may reduce ALI (11, 23, 33). Based on previous studies, we and others identified potential soluble factors that could be responsible for the observed beneficial effects, keratinocyte growth factor (KGF) (23, 32), IL-1 receptor antagonist (IL-1ra) (43), and prostaglandin E2 (PGE2) (42). These three paracrine products of MSCs modulate alveolar ion and fluid transport and lung barrier permeability or acute lung inflammation, important pathways of lung injury (17, 34, 42). The two objectives of this study were 1) to test the impact of the alveolar environment of acute inflammation and hypoxia on the phenotype of MSCs in terms of apoptosis and secretion of three mediators, IL-1ra, PGE2, and KGF; and 2) to assess the therapeutic efficacy of MSC-conditioned media on the injured cultured alveolar epithelial cells (AECs) and determine the mechanisms that explain how the conditioned media restored the vectorial sodium transport in an in vitro model of acute alveolar injury.
METHODS
Extraction, Culture, and Conditioning of Human MSCs
Bone marrow samples were harvested from washed filters used during bone marrow graft processing for allogeneic transplantation after healthy donor informed consent according to approved institutional guidelines (Assistance Publique–Hôpitaux de Paris, Paris, France). MSCs were cultured as previously described (3). Briefly, healthy donors bone marrrow cells obtained after Ficoll (Invitrogen, Cergy-Pontoise, France) were cultured at an initial density of 5 × 104 cells/cm2 in minimum essential medium-α (Invitrogen), supplemented with 10% defined fetal calf serum (DFBS; HyClone, Logan, UT), 2 mM l-glutamine (Invitrogen), 1 ng/ml basic fibroblast growth factor (bFGF; R&D Systems, Lille, France), and antibiotic/antimycotic (Invitrogen). After 24–48 h, nonadherent cells were removed and the medium was changed. The media were changed every 2 or 3 days until confluence. Adherent cells were then trypsinized, harvested, and cultured by seeding 0.5 × 103 cells/cm2.
At confluence, the MSC medium was changed and replaced by DMEM medium containing 10% fetal bovine serum. To test the effect of inflammation on MSC secretion, MSCs were exposed to hypoxia (3% O2) plus 20 ng/ml of cytomix. Cytomix is a mixture of IL-1β, TNF-α, and INF-γ (R&D Systems), which are the major proinflammatory cytokines in the pulmonary edema fluid from patients with ARDS (31). After 12 h, the media of MSCs exposed to cytomix plus hypoxia (HCYT-MSC-M) or the media of control MSC (MSC-M) were harvested and rapidly centrifuged (10 × 103 rpm). The aliquots were stored at −80°C.
Measurements of Soluble Mediator Concentrations in MSC Supernatants
MSCs between passages 2 and 5 were cultured on sixwell plates until 85–90% confluence was achieved. After exposure to experimental conditions, the supernatants were harvested and immediately centrifuged. The concentrations of soluble mediators, IL-1ra, PGE2, and KGF were measured by ELISA (R&D Systems). KGF (FGF-7) was measured with a twofold sample dilution factor, PGE2 was measured with a threefold sample dilution factor, and IL-1ra was measured without sample dilution as indicated by manufacturer's instructions.
Determination of MSC Phenotype
Monoclonal antibodies conjugated with either fluorescein isothiocyanate or phycoerythrin (Becton Dickinson, Le Pont de Claix, France) and directed to CD29, CD95, CD105, and CD105a to CD105e, or matched isotype control were used for immunophenotyping, according to the manufacturer's protocol. Data were acquired and analyzed on a five parameters flow cytometer (FACS Calibur; Becton Dickinson) with Cell Quest software.
Detection of MSC Apoptosis and Necrosis
MSC apoptosis and viability were determined at passage 4 in cells exposed to normoxia or to cytomix (20 ng/ml) plus hypoxia (3% O2) for 12 h using the annexin V (FITC)-7AAD kit (Beckman Coulter, Roissy Charles de Gaulle, France). Briefly, cells were collected and resuspended in binding buffer. Annexin V-FITC and propidium iodide were added, and the reaction was incubated in the dark for 15 min. Cells were analyzed by flow cytometry using FACScan flow cytometer.
Isolation and Culture of Rat AECs
The procedure of alveolar type II cell isolation from pathogen-free male Sprague-Dawley rats accorded with legislation currently in force in France and animal welfare guidelines (Ministère de la Pêche et de l'Agriculture, Agreement 5669). Alveolar type II cells were isolated from adult rats (200–250 g) by elastase digestion of lung tissue followed by sequential filtration and differential adherence on bacteriological dishes as previously described (46). Cells (purity >90%; viability >95%) were seeded either onto Transwell/Snapwell (polycarbonate membrane with a pore size of 0.4 μm; Costar, Cambridge, MA) filters or onto 6- or 12-well plastic culture dishes and cultured in a 5% CO2, 95% air atmosphere in DMEM containing 25 mM d-glucose, 10 mM HEPES, 23.8 mM NaHCO3, 2 mM l-glutamine, 10% fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, and 10 μg/ml gentamycin. Transepithelial resistance (Rte) and transepithelial potential difference were measured on day 4 using a microvoltohmeter (World Precision Instruments, Astonbury, UK). Snapwell filters with Rte <300 Ω/cm2 were discarded.
Experimental Protocols
Freshly isolated AECs were seeded on polycarbonate Transwells (Costar, Cambridge, UK) and grown for 4 days at 37°C (5% CO2) in a liquid-liquid interface. When the AECs reached confluence, one of four conditions were applied.
Protocol 1.
The media were replaced by fresh media only, and the AECs were incubated in normoxia (21% O2-5% CO2-74% N2). After 6 h, the media were replaced by fresh media and AECs were incubated for an additional 12 h in normoxia.
Protocol 2.
The medium was replaced by media containing 20 ng/ml cytomix, and the AECs were placed in a box flushed with a hypoxic gas (3% O2-5% CO2-92% N2). After 6 h, the media of the AECs were removed and replaced by DMEM plus 20 ng/ml cytomix and AECs were incubated in hypoxia for an additional 12 h.
Protocol 3.
The media were replaced by media containing 20 ng/ml cytomix, and AECs were placed in a box flushed with a hypoxic gas (3% O2-5% CO2-92% N2). After 6 h, the media of the AECs were removed and replaced by MSC-M plus 20 ng/ml cytomix and cells were incubated in hypoxia for an additional 12 h.
Protocol 4.
The media were replaced by media containing 20 ng/ml cytomix, and the AECs were placed in a box flushed with a hypoxic gas (3% O2-5% CO2-92% N2). After 6 h, the media of the AECs were removed and replaced by HCYT-MSC-M (cytomix was not added in this protocol since HCYT-MSC-M already contained cytomix) and cells were incubated in hypoxia for additional 12 h.
Measurement of epithelial permeability to protein across AECs
Measurements of protein epithelial permeability form apical to basolateral side of AECs were done 4 days after seeding, as previously described (16). AECs were exposed to normoxia or to 20 ng/ml cytomix plus hypoxia (3% O2) for 6 h and then the media were removed and replaced by 1) DMEM alone and AECs were exposed to normoxia; 2) DMEM plus cytomix (20 ng/ml) and AECs were exposed to hypoxia (3% O2); 3) media of nonexposed MSC (MSC-M) plus cytomix (20 ng/ml) and AECs were exposed to hypoxia (3% O2); and 4) medium of MSC exposed to cytomix plus hypoxia (HCYT-MSC-M) and AECs were exposed to hypoxia (3% O2). Epithelial protein permeability was then assessed by adding labeled 0.3 μCi/ml 125I-albumin only to the upper compartment, at the time of replacement of the media, and the unidirectional flux to the lower compartment was measured over 12 h. There is no hydrostatic pressure gradient between the two compartments. Aliquots of media containing 125I-albumin were retained as references for initial radioactivity (R0). Twelve hours later, apical and basolateral media were collected and medium samples were placed into assay tubes for γ counting. The radioactivity expressed as counts per minute was normalized by the weight of the samples. The epithelial permeability was then calculated as follows: (Rbaso/R0) × 100 = epithelial permeability to albumin over 12 h (%). AEC monolayers with baseline permeability >2% were not used for experiments.
Measurement of bioelectrical properties of AECs.
Spontaneous potential difference (PD) and transepithelial resistance (Rte) across AECs monolayers were measured using an epithelial voltohmeter equipped with chopstick-style electrodes (World Precision Instruments, Astonbury, UK). Equivalent short-circuit current (Ieq) was calculated by Ohm's law (Ieq = PD/Rte).
Measurement of short circuit current (Isc), transepithelial potential difference, and transepithelial resistance were performed in AECs grown 5 days after seeding, as previously described (46). Snapwell inserts were mounted in vertical diffusion chambers and were bathed with Ringer solution (pH 7.4) continuously bubbled in 5% CO2-95% air at 37°C. The apical and basolateral chambers were filled with 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, and 15 mM glucose, pH 7.4 adjusted with NaOH. The hemi-chambers were connected to a VCC MC6 voltage clamp (Physiologic Instruments, San Diego, CA) via Ag/AgCl electrodes and 3 M KCl agar bridges. Isc data were acquired by PowerLab (AD Instruments, Colorado Springs, CO) and recorded in Chart software (AD Instruments). Snapwell filters with unstable Isc or with Rte <300 Ω/cm2 were discarded (discarded filters represented <10% of filters mounted in Ussing chamber and were equally distributed in all experimental conditions). Isc was measured every 5 min by clamping the potential difference to 0 mV for 1 s, and Rte was calculated from Isc and potential difference using Ohm's law. Amiloride-sensitive Isc was determined as the difference in current with and without amiloride (10−5 M).
Experiments were also undertaken to measure sodium influx through apical amiloride-sensitive channels in basolaterally permeabilized cells as previously described (46). AECs were bathed in an apical compartment containing 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, and 15 mM glucose, pH 7.4 adjusted with NaOH, and a basolateral solution containing 10 mM NaCl, 130 mM N-methyl-d-glucamine, 4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, and 15 mM glucose, pH 7.4 adjusted with HCl for 15 min before the basolateral membrane was permeabilized by the basolateral addition of nystatin (60 mg/ml) a monovalent ionophore. This induced a rapid increase in Isc. Once the Isc reached a new steady state, amiloride (10−5M) was added in the apical bathing solution and the difference current representing the amiloride-sensitive component of the sodium current across the apical membrane (apical amiloride-sensitive current) was calculated. The voltage was clamped at 0 V for all the Isc measurements.
Other experiments were done to measure Na-K-ATPase activity in apically permeabilized AECs. Cells were bathed in an apical and basolateral compartments containing 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, and 15 mM glucose, pH 7.4 adjusted with NaOH. After the steady-state condition was reached, the apical membrane was permeabilized by apical addition of 60 mg/ml nystatin. Ouabain (1 mM) was added to the basolateral bathing solution, and the basolateral ouabain-sensitive current (representing the fall in Isc evoked by ouabain) was measured to estimate the extrusion capacity of the basolateral Na+ pump.
Western blotting of epithelial sodium channel subunits in AECs
Biotinylation and recovery of apical membrane proteins from cultured AECs were performed as previously described (46, 48). In brief, AECs grown on Transwell filters were placed on ice and washed three times with ice-cold PBS-Ca2+-Mg2+ (PBS with 0.1 mM CaCl2 and 1 mM MgCl2). Apical membrane proteins were then biotinylated by a 15-min incubation at 4°C with NHS-ss-biotin 1.25 mg/ml (Pierce, Rockford, IL) freshly diluted into biotinylation buffer (10 mM triethanolamine, 2 mM CaCl2, and 150 mM NaCl, pH 7.5) with gentle agitation. Alveolar type II cells were rinsed with PBS-Ca2+-Mg2+ glycine (100 mM) and washed in this buffer for 20 min at 4°C to quench unreacted biotin. Cells were then rinsed twice with PBS-Ca2+-Mg2+, scraped in cold PBS, and pelleted at 2,000 rpm at 4°C. Pellets were solubilized for 45 min in 20 μl of lysis buffer [1% Triton X-100, 150 mM NaCl, 5 mM EDTA, and 50 mM Tris(hydroxymethyl) aminomethane (Tris), pH 7.5] containing protease inhibitors. The lysates were clarified by centrifugation at 14,000 g for 10 min at 4°C, and supernatants were incubated overnight with packed streptavidin-agarose beads (Pierce) to recover biotinylated proteins. The beads were then pelleted by centrifugation, and aliquots of supernatants were taken to represent the unbound, intracellular pool of proteins. Biotinylated proteins were eluted from the beads by heating to 100°C for 5 min in SDS-PAGE sample buffer [containing 10% glycerol, 12.5% 0.5 M Tris·HCl (pH 6.8), 10% of 20% SDS, 5% 2-mercaptoethanol, and 2.5% of 0.05% (wt/vol) bromophenol blue]. Samples of biotinylated and nonbiotinylated proteins were resolved through 10% acrylamide gels, electroblotted, electrically transferred to nitrocellulose paper, and subsequently probed for the α- or β-rat epithelial sodium channel (ENaC) subunits. To ensure the absence of leakage of biotin into the cells, we systematically verified the absence of the intracellular protein β-actin in biotinylated extracts. Rabbit polyclonal anti-α-rat ENaC subunit and anti-β-rat ENaC subunit antibodies (14, 46) were used at the dilution 1:2,000, and mouse monoclonal anti-β-actin at the dilution 1:5,000. Quantification of rat ENaC and actin levels was obtained using National Institutes of Health Image software. The expression of γ-ENaC at the cell surface could not be investigated in this study, due to the very faint signal of biotinylated γ-ENaC in native rat AECs.
MSC media with and without KGF neutralization
To determine whether the observed effects of MSC-M were due to the presence of KGF, KGF depletion was performed using protein G/agarose beads coated with anti-KGF monoclonal antibody (R&D Systems) or with anti-IgG antibodies (as a control). MSC-conditioned media were incubated at 4°C overnight on wheel with antibodies-conjugated beads. KGF concentrations in MSC medium were measured by ELISA. AECs cultured for 96 h were exposed or not to hypoxia plus cytomix as previously described and incubated with MSC-M depleted or not from KGF. Cultures were maintained for an additional 12 h, at which point AEC monolayers were used for measurement of Ieq. In an another set of experiments, the preventive effect of recombinant human KGF (rhKGF) on hypoxia plus cytomix-induced injury was tested. AECs exposed or not to hypoxia plus cytomix were incubated for 12 h with rhKGF (200 and 400 pg/ml) before Ieq measurements were performed.
Statistical Analysis
Data are presented as means ± SD (n = 3–5 for flow cytometry experiments, ELISA, transepithelial permeability to albumin determination, and Western blot experiments; n = 4–17 for electrophysiological measurements). Statistical analyses were done by unpaired Student's t-test for comparison of concentrations of paracrine factors in conditioned media. One-way ANOVAs were performed for all other experiments and, when allowed by the F value, results were compared by the modified least significant difference (Fisher's protected least significant difference). P < 0.05 was considered significant. The following software was used: StatView (SAS Institute, Cary, NC) and Prism (GraphPad Software, La Jolla, CA).
RESULTS
Exposure to Inflammatory and Hypoxic Environment Did Not Alter MSC Viability and Specific MSC Marker Expression
MSCs were exposed to cytomix (20 ng/ml) plus hypoxia (3% O2) that is similar to the inflammatory and hypoxic environment during ALI, and they were compared with MSCs exposed to normoxia. Control MSCs as well as MSCs exposed to cytomix plus hypoxia did not express HLA-DR. The expression of the specific MSC markers CD29, CD95, CD105, CD105a, CD105b, CD105c, CD105e, and CD105f was similar in MSC exposed to cytomix plus hypoxia and in those exposed to normoxia (data not shown). Interestingly, exposure to cytomix plus hypoxia did not change the percentage of intact, apoptotic, or necrotic MSCs compared with control cells (Fig. 1). Cell morphology was also similar in both culture conditions.
Fig. 1.

Effects of cytomix and hypoxia on apoptosis and necrosis of human mesenchymal stromal cells (MSCs). The MSCs were exposed to either normoxia (21% O2) or to cytomix (20 ng/ml) plus hypoxia (3% O2) for 12 h. Apoptosis and necrosis were determined using annexin-V7AAD kit and expressed as the percentage of total cells. Experiments were repeated 3 times. SSC, side-scatter angle.
Proinflammatory and Hypoxic Environment Altered Paracrine Secretion by MSCs
Previous studies have identified several potential soluble factors that could be responsible in part for the beneficial effects of MSCs on reabsorption of alveolar edema fluid in ALI, such as IL-1ra, PGE-2, and KGF (32, 42, 43). Therefore, we focused on the release of these three mediators by MSCs. The secretion was evaluated in control condition (MSC-M) or after exposure of MSCs to cytomix plus hypoxia (HCYT-MSC-M). As shown in Fig. 2, conditioned media from MSCs exposed to cytomix plus hypoxia (HCYT-MSC-M) for 12 h altered their paracrine secretion compared with MSC-M. Cytomix plus hypoxia induced the release of IL-1ra from 0 to 134 ± 43 pg/ml and markedly increased the release of PGE2 from 77 ± 25 to 1,295 ± 184 pg/ml. By contrast, cytomix plus hypoxia (HCYT-MSC-M) induced a decrease of KGF secretion from 205 ± 4 to 109 ± 10 pg/ml compared with MSC-M. These results indicated that acute inflammation in an hypoxic environment modified the MSC secretion of soluble mediators. We were unable to detect IL-10 in the conditioned medium of MSC either in MSC-M or HCYT-MSC-M.
Fig. 2.
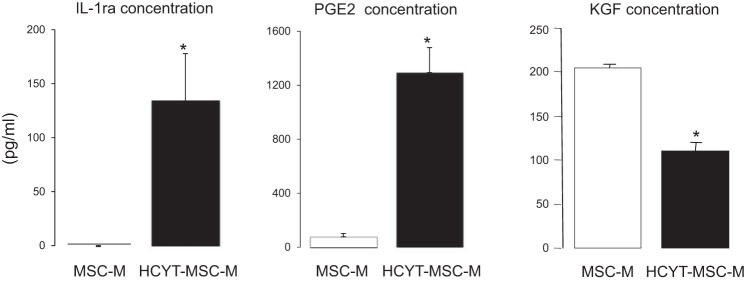
Effect of cytomix plus hypoxia on the release of soluble mediators by MSCs. The MSC were cultured in DMEM in the absence or presence of cytomix (20 ng/ml) plus hypoxia (3% O2). After 12 h, the media of nonexposed cells (MSC-M) or the media of cells exposed to cytomix (20 ng/ml) plus hypoxia (3% O2; HCYT-MSC-M) were used for the determination of IL-1 receptor antagonist (IL-1ra), PGE2, and keratinocyte growth factor (KGF) concentrations. The values represent the means ± SD of 4–6 experiments in each group. *P < 0.01 by unpaired Student's t-test.
MSC-conditioned Media Prevented the Increase in Albumin Permeability Induced by Cytomix Plus Hypoxia in AEC Monolayers
During ALI, epithelial permeability to protein is increased and favors alveolar flooding (39). To evaluate whether media of MSCs exposed or not to cytomix plus hypoxia (HCYT-MSC-M or MSC-M) could prevent the increase in epithelial permeability (16), AECs pretreated by cytomix plus hypoxia were exposed to MSC-M or HCYT-MSC-M for 12 h before measurement of epithelial permeability to radiolabeled albumin. Exposure of AECs to hypoxia plus cytomix increased by threefold epithelial permeability compared with the control condition. Treatment with MSC media for 12 h completely prevented the increase in transepithelial permeability to 125I-albumin (Fig. 3). MSC-M and HCYT-MSCM had a similar protective effect on AEC permeability to protein.
Fig. 3.
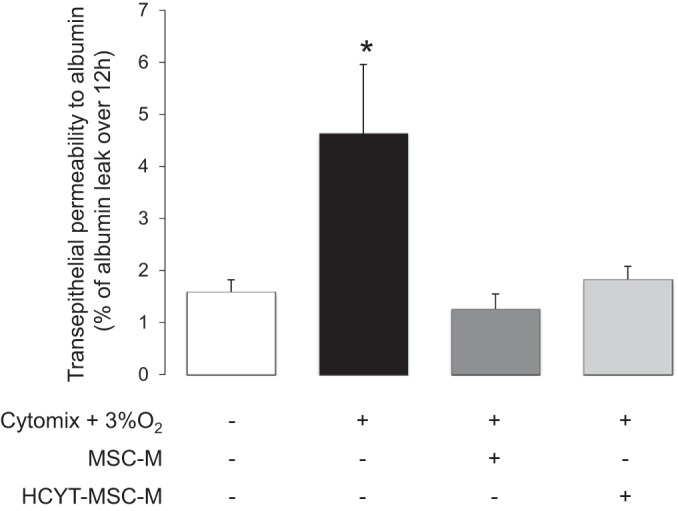
Effect of MSC medium on cytomix-hypoxia-induced decrease of epithelial permeability to albumin in cultured alveolar epithelial cells. Alveolar epithelial cells (AECs) were seeded on Transwell filters. AECs were exposed or not to cytomix (20 ng/ml) plus hypoxia (3% O2) for 6 h. Then, the media were replaced for additional 12 h by 1) DMEM alone and AECs were exposed to normoxia (21% O2); 2) DMEM plus cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2); 3) media of nonexposed MSC (MSC-M) plus cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2); and 4) medium of MSC exposed to cytomix plus hypoxia (HCYT-MSC-M) and cells were exposed to hypoxia (3% O2). Epithelial protein permeability was then assessed by adding labeled 125I-albumin only to the upper compartment, at the time of replacement of the media, and the unidirectional flux to the lower compartment was measured over 12 h. The values are the means ± SD of 3 experiments in each group. *P < 0.01, compared with control conditions [one-way ANOVA followed by Fisher's protected least significant difference (PLSD)].
MSC-Conditioned Media Prevented the Cytomix Plus Hypoxia-Induced Decrease in Total and Amiloride-Sensitive Transepithelial Na Transport in AECs
The transepithelial Na transport across AECs was evaluated by the amiloride-sensitive component of Isc (AS-Isc). In control conditions, AS-Isc represented 80% of the total Isc (Fig. 4A). Hypoxia plus cytomix decreased total Isc without change in amiloride-insensitive Isc, so that AS-Isc was reduced by 60% compared with control (Fig. 4B). Addition of MSC-M completely prevented the cytomix plus hypoxia-induced decrease in total Isc and in AS-Isc (Fig. 4, A and B) and also increased the amiloride-insensitive component of Na current. By contrast, HCYT-MSC-M had no significant protective effect on total Isc or AS-Isc.
Fig. 4.
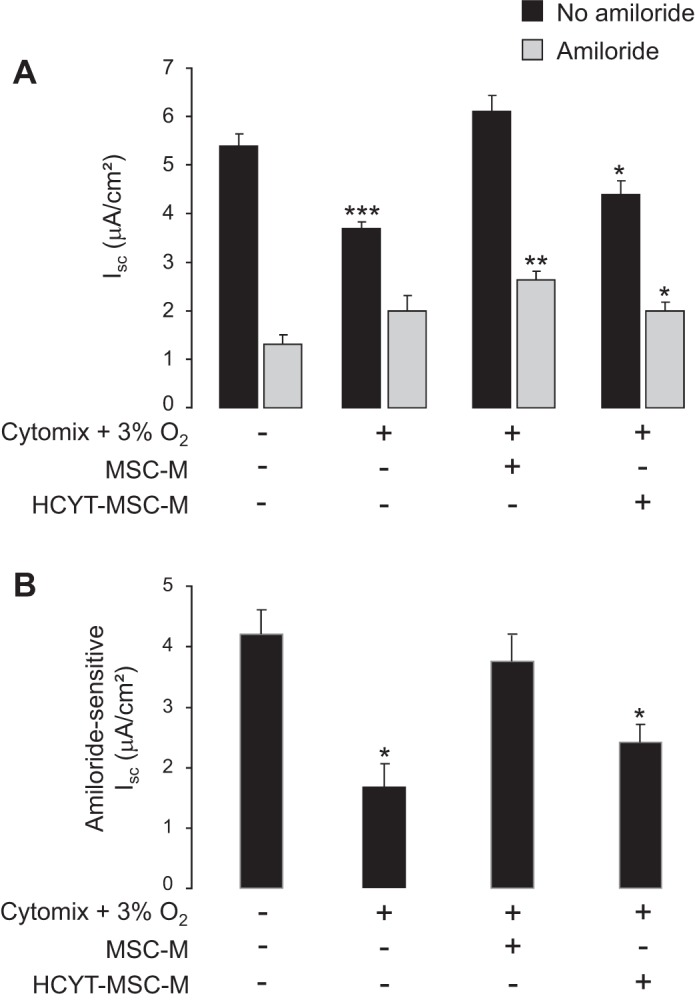
Effect of MSC-conditioned media on cytomix plus hypoxia-induced decrease of Na transport in alveolar epithelial cells. AECs grown on Snapwell filters were exposed to either normoxia or cytomix (20 ng/ml) plus hypoxia (3% O2) for 6 h. Then, the media were replaced for additional 12 h by 1) DMEM alone and AECs were exposed to normoxia (21% O2); 2) DMEM plus cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2); 3) media of nonexposed MSC (MSC-M) plus cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2); and 4) medium of MSC exposed to cytomix plus hypoxia (HCYT-MSC-M) and cells were exposed to hypoxia (3% O2). AECs were mounted into a voltage-clamp system, and short-circuit current (Isc) was measured at baseline (black bars) and after addition of amiloride (10−5 M) into the apical bath (A; gray bars). Amiloride-sensitive Isc (B) represents the difference of Isc before and after addition of amiloride. The values are the means ± SD of 4–6 filters for each condition. *P < 0.05, **P < 0.01, and ***P < 0.001, significantly different from corresponding control value (one-way ANOVA followed by Fisher's PLSD).
To further test the mechanisms, we evaluated both apical and basolateral Na+ conductance by measuring Isc on permeabilized monolayers. In the first series of experiments, permeabilization of the basolateral membrane was done in the presence of an asymmetrical Na+ gradient (140 mM apical vs. 10 mM basolateral). The results showed that hypoxia plus cytomix induced a 60% fall of apical AS-Isc and that MSC-M completely restored the apical AS-Isc current (Fig. 5A). In the second series of experiments, we tested the contribution of the Na+-K+ ATPase after permeabilization of the apical membrane. Figure 5B shows that neither hypoxia plus cytomix nor MSC-M did significantly alter the basolateral ouabain-sensitive current reflecting Na+-K+-ATPase activity.
Fig. 5.
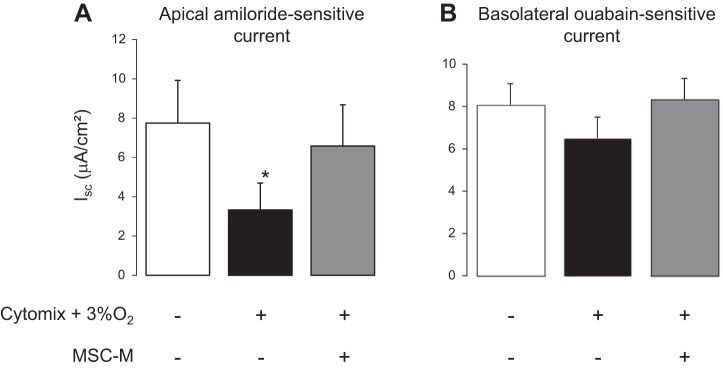
AECs grown on Snapwell filters were exposed to either normoxia or cytomix (20 ng/ml) plus hypoxia (3% O2) for 6 h. Then, the media were replaced for additional 12 h by 1) DMEM alone and AECs were exposed to normoxia (21% O2); 2) DMEM plus cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2); and 3) media of nonexposed MSC (MSC-M) plus cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2). A: AECs were mounted into a voltage-clamp system in the presence of a Na+ concentration gradient (mucosal to serosal, 140:10 mM). Addition of nystatin into the basolateral bath increased the Isc to a peak value before amiloride (10 μM) was added into the apical bath. Apical amiloride-sensitive current is the difference between Isc peak value and Isc value after amiloride addition and reflects epithelial sodium channel (ENaC) activity; B: AECs were then immediately mounted into a voltage-clamp system. After stabilization, nystatin into the apical bath evoked a slowly developing rise in Isc and ouabain (1 mM) was added to the basolateral solution once this response had reached its peak value. Basolateral ouabain-sensitive current representing the fall in Isc evoked by this application of ouabain was measured to estimate the extrusion capacity of the basolateral Na+ pump. The values are the means ± SD of 5–8 filters for each condition. *P < 0.01, compared with control conditions (one-way ANOVA followed by Fisher's PLSD).
MSC-Conditioned Media Restored the Membrane Expression of α-ENaC But Did Not Modify β-ENaC Expression After Exposure to Inflammatory and Hypoxic Conditions
The effects of hypoxia plus cytomix in the presence or absence of MSC-M on ENaC subunit expression were evaluated (Fig. 6). First, the total protein levels of α- and β-ENaC subunits were determined in AECs exposed to hypoxia plus cytomix in the presence or absence of MSC-M. Figure 6 shows that hypoxia plus cytomix induced no significant changes in the total protein expression of the α-ENaC (major band at 85 kDa and minor band at 65 kDa) and β-ENaC (95 kDa band) subunits and that MSC-M had no effect on the total protein expression of α- and β-ENaC subunits. Second, apical surface biotinylation experiments were performed to evaluate cell surface expression of α- or β-ENaC in AECs exposed to the above experimental conditions. Exposure to hypoxia plus cytomix significantly decreased α-ENaC protein expression in apical cell surface and had no effect on β-ENaC expression (Fig. 7). This result indicates that the decrease in apical AS-Isc was partly related to a decrease of α-ENaC protein in apical membrane. Incubation with MSC-M completely prevented the decrease of α-ENaC membrane expression in AECs exposed to hypoxia plus cytomix.
Fig. 6.

Effect of cytomix plus hypoxia in the presence or absence of MSC-conditioned media on total protein levels of the α- and β-subunits of ENaC in alveolar epithelial cells. AECs grown on Transwell filters were exposed to either normoxia or cytomix (20 ng/ml) plus hypoxia (3% O2). After 6 h, the media were replaced for additional 12 h by 1) DMEM alone and AECs were exposed to normoxia (21% O2); and 2) DMEM plus cytomix (20 ng/ml) and presence of cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2). Western blot experiments were performed on whole cells extracts using rabbit polyclonal antibodies raised against rat α-ENaC (A, left) and rat β-ENaC (A, right). Quantification of α-ENaC (85 and 65 kDa) and β-ENaC levels was obtained using Scion Image software, and the data were normalized for the corresponding actin signal in each lane (B). Results are expressed as the ratios of α- and β-ENaC/actin. The values are the means ± SD of 5 separate experiments (one-way ANOVA followed by Fisher's PLSD).
Fig. 7.

Effect of cytomix plus hypoxia on apical cell surface expression of α- and β-ENaC subunits in alveolar epithelial cells in the presence or absence of MSC-conditioned media. AEC grown on Transwell filters were exposed to either normoxia or cytomix (20 ng/ml) plus hypoxia (3% O2). After 6 h, the media were replaced for additional 12 h by 1) DMEM alone and AECs were exposed to normoxia (21% O2); 2) DMEM plus cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2); and 3) the medium of nonexposed MSC (MSC-M) in the presence of cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2). Then, AECs were immediately processed for apical biotinylation experiments as described in Experimental Protocols before immunoblotting. A: representative immunoblots of α-ENaC (left) and β-ENaC (right) cell surface, and of intracellular protein expression of β-actin. The validity of surface biotinylation approach in control and treated cells is attested by the cell surface expression of α- and β-ENaC but not of the intracellular protein β-actin. The quantification of biotinylated surface proteins for α-ENaC (65 kDa) (left) and β-ENaC (right) is shown in B. Quantification of biotinylated α- and β-ENaC signals was obtained using Scion Image software, and the data were normalized for the actin signal in corresponding intracellular extracts. The values are the means ± SD of 4 separate experiments. *P < 0.01, compared with control conditions (one-way ANOVA followed by Fisher's PLSD).
KGF-Depleted MSC-M Had No Protective Effect on Hypoxia Plus Cytomix-Induced Decrease in Na Transport in AECs
To define whether the beneficial effects of MSC-M were related in part to KGF, the MSC-M medium was depleted of KGF using a anti-KGF antibody (IgG were used as control). The concentration of KGF declined from 190 ± 63 to 0 pg/ml in control conditions and after depletion of KGF. KGF-depleted MSC-M had no protective effective on hypoxia plus cytomix-induced decrease in Na transport (Fig. 8A). To test whether KGF was sufficient per se to prevent the hypoxia plus cytomix-induced decrease in Na transport, we finally evaluated the potential effect of rhKGF. As shown in Fig. 8B, incubation of AECs with rhKGF (200 or 400 pg/ml) had no protective effect on hypoxia plus cytomix-induced decrease in Na transport.
Fig. 8.

Effect of MSC-conditioned media depleted of KGF and of recombinant human KGF (rhKGF) on the cytomix plus hypoxia-induced decrease in Na transport. A: AECs grown on Transwell filters were exposed to either normoxia or cytomix (20 ng/ml) plus hypoxia (3% O2). After 6 h, the media were replaced for additional 12 h by 1) DMEM alone and AECs were exposed to normoxia (21% O2); 2) DMEM plus cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2); 3) the media of nonexposed MSC (MSC-M) in the presence of cytomix (20 ng/ml) plus IgG and cells were exposed to hypoxia (3% O2); and 4) the media of nonexposed MSC (MSC-M) depleted of KGF as described in Experimental Procedures in the presence of cytomix (20 ng/ml) and cells were exposed to hypoxia (3% O2). Ieq was measured before and after addition of amiloride (10 μM) was added into the apical bath. Amiloride-sensitive Ieq represents the difference of Isc before and after addition of amiloride. The values are the means ± SD of 8–14 filters for each condition. **P < 0.01, compared with control conditions; §P < 0.05, compared with cytomix plus hypoxia; +P < 0.05, compared with cytomix plus hypoxia in presence of MSC-M plus IgG (one-way ANOVA followed by Fisher's PLSD). B: after a 6 h exposure to normoxia (white bars) or cytomix plus hypoxia (black bars), AECs were treated with rhKGF (200 or 400 pg/ml) or vehicle for additional 12 h and amiloride-sensitive Ieq was calculated. Values are means ± SD of 8–17 filters for each condition. ***P < 0.001, compared with normoxic corresponding value (one-way ANOVA followed by Fisher's PLSD).
DISCUSSION
The major findings of this study can be summarized as follows: 1) MSC exposure to an inflammatory and hypoxic environment induced substantial changes in the secretion of paracrine factors known to upregulate Na and fluid transport in ALI: IL-1ra, PGE2, and KGF. 2) The inflammatory and hypoxic insult to AECs induced a threefold increase in transepithelial permeability to albumin and a 60% decrease in the transepithelial sodium transport due to a decreased apical amiloride-sensitive Na uptake without a change in Na+-K+-ATPase activity. This decreased vectorial Na transport was associated with a decline in α-ENaC subunit expression at the apical membrane. 3) Conditioned media of MSCs exposed or not to hypoxia plus cytomix (HCYT-MSC-M and MSC-M) completely prevented the increase in transepithelial permeability to albumin. 4) MSC-M prevented cytomix-hypoxia-induced decrease of Na transport in AECs, restored amiloride-sensitive apical Na transport, and maintained α-ENaC cell surface expression. In contrast, HCYT-MSC-M had no significant effect on cytomix-hypoxia-induced decrease in Na transport. 5) secretion of KGF by MSCs was required for the protective effect of MSC-M on alveolar epithelial Na transport. This study provides the proof of concept that paracrine factors produced by MSCs in the damaged lung may have beneficial effects in vivo by preventing alveolar flooding.
In recent years, MSCs have emerged as a potential therapeutic modality for several inflammatory states, including ALI. Some studies have demonstrated benefits from intratracheal or intravenous administration of MSCs (4, 21, 41). MSCs attenuate the inflammatory response to LPS and reduce the severity of lung injury. In most ALI models, the engraftment rates of MSCs were low (26, 27, 50) and a therapeutic effect occurred within 24–48 h, suggesting that beyond cell replacement, MSCs may be releasing factors responsible for beneficial effect of cell therapy. Because the use of MSCs may carry some risks to the patient (2, 38), the administration of MSC-conditioned medium might provide an alternative therapeutic option for ALI/ARDS. In ALI models, MSCs delivered by either intratracheal or intravenous routes were primarily located in the lung microcirculation and alveoli and therefore are directly exposed to alveolar hypoxia and high levels of proinflammatory cytokines from existing and evolving inflammatory reactions. Therefore, we hypothesized that this inflammatory environment may induce changes in MSCs phenotype that could modify the paracrine secretion profile of MSCs and modulate their capacity to repair damaged epithelium. To evaluate the role of the inflammatory and hypoxic environment on MSCs and their conditioned medium, we exposed MSCs to hypoxia plus a mixture of the major biologically cytokines present in ALI pulmonary edema, IL-1β, TNF-α, and IFNγ (referred to as cytomix), as in prior studies (31). The results showed that MSCs exposed to hypoxia plus cytomix do not exhibit changes in their cell surface markers, cell survival, or the degree of apoptosis. In a mice model of endotoxin induced ALI, MSC instillation improved survival through the secretion several factors including IL-1ra (43), PGE2 (42), IL-10, and KGF (23, 34). Therefore, the present study tested the influence of inflammatory and hypoxic conditions on the secretion of these key candidates in the MSC-conditioned medium. In MSCs exposed to the hypoxic and inflammatory stimuli, the conditioned medium contained higher IL-1ra and PGE2 concentrations (10- and 4-fold, respectively) and lower KGF concentrations compared with MSCs cultured in control conditions. Hypoxia plus cytomix probably increased IL-1ra and PGE2 secretion because of the presence of IL-1β in the cytomix (43). Interestingly, IL-1β is one of the major inflammatory cytokines in pulmonary edema fluid from patients with ALI/ARDS (18, 22) and IL-1ra competes with IL-1β for IL-1 receptor binding. In the present study IL-10 was undetectable in control condition as well as after hypoxic and inflammatory insults. This result was in agreement with a prior observation that in ALI, the increase of IL-10 in the alveolar space after MSC treatment did not result from direct MSC secretion but rather as the result of PGE2 release from alveolar macrophages reprogrammed by MSCs (43). The present study is the first report showing that the combination of an hypoxic and proinflammatory insult decreases KGF secretion by MSCs. This result is unexpected since a prior study reported an increased release of growth factors such as VEGF, IGF-1, and HGF in MSCs exposed to LPS, TNF-α, or hypoxia (11).
The alteration of alveolar ion transport, lung inflammation, or/and increased endothelial permeability are well-known features of the ALI pathophysiology (17, 34, 42). Previous studies reported a beneficial effect of MSCs in a model of ALI induced by intratracheal instillation of Escherichia coli endotoxin with reduced mortality and less pulmonary edema (21, 23). Similar results were published in an ex vivo perfused human lung model, in which MSCs reversed endotoxin-induced lung injury by restoring alveolar fluid clearance and reducing pulmonary edema (34). Interestingly, in a model of ventilator-induced lung injury, a similar beneficial effect was obtained by using either MSCs or MSC-conditioned medium (23).
In this study, we tested the efficacy of MSC-conditioned media on the epithelial permeability and the vectorial sodium transport in an in vitro model of ALI (16). In this in vitro model of ALI, AECs were exposed to cytomix plus hypoxia for 6 h before treatment with MSC-conditioned media, harvested either from MSCs cultured in normal conditions (MSC-M) or from MSCs exposed to inflammatory and hypoxic insults (HCYT-MSC-M). Exposure of AECs to cytomix plus hypoxia increased paracellular permeability by threefold as previously reported (31) and a 60% decrease in apical to basolateral vectorial Na transport. To determine whether cytomix and hypoxia altered Na entry and/or extrusion, we studied Na transport after permeabilization of AEC basolateral or apical membrane. Exposure of AECs to cytomix plus hypoxia induced a decrease in Na transport that was related to a diminished apical expression of amiloride-sensitive Na channels whereas ouabain-sensitive basolateral Na transport, reflecting Na-K-ATPase activity, remained unchanged. The cytomix plus hypoxia-induced reduction of Na current was related to a reduction of the number of Na channels in the apical membrane as demonstrated by the biotinylation experiments that showed a decrease of α-ENaC apical cell surface expression while total protein content of α-ENaC was unchanged. We and other investigators have reported that short-term exposure to moderate hypoxia decrease Na transport by internalization of ENaC subunits without a change in the total protein content (10, 12, 46). Lee et al. (32) previously showed that 50 ng/ml cytomix for 24 h decreased α-ENaC protein levels in the apical membrane of human AECs in parallel with reduced α-ENaC protein content. The discrepancy concerning the effect of cytomix on ENaC protein total cell content may be explained by the fact that we used a lower concentration of cytomix than Lee et al. In the present study, the decrease in α-ENaC subunits in the apical membrane likely results from additive effects of hypoxia plus cytomix on ENaC subunit trafficking.
The most significant finding of this study is that MSC-conditioned medium restored normal epithelial permeability to protein and prevented the decrease in transepithelial Na transport. Interestingly, HCYT-MSC-M was as potent as MSC-M for preventing the decrease in transepithelial albumin permeability, suggesting that paracrine factors released by MSC exposed to the inflammatory milieu of ALI/ARDS could protect in vivo the lungs from flooding. MSC-M (but not HCYT-MSC-M) also restored cytomix plus hypoxia-induced decrease of transepithelial Na transport. From a biochemical standpoint, MSC-M restored the cell surface expression of α-ENaC, suggesting that the beneficial effect of MSC-M on vectorial Na transport was due, at least in part, to an effect on ENaC trafficking. Another explanation would be that MSC-M somehow favored the activation of ENaC channels at the cell surface by membrane-bound serine proteases (such as channel activating proteases) (47). For instance, MSC-M may prevent the inactivation of these proteases by reactive intermediates known to be present in bronchoalveolar lavage during ALI/ARDS (30). In addition, it is possible that MSC-M also stimulated apical Na transporters distinct from ENaC [such as the cyclic nucleotide gated (CNG) cationic channels (25)], inasmuch as it increased the amiloride-insensitive component of Na transport in injured AECs. However, the increase in amiloride-insensitive currents could also be due to a decrease in the sensitivity of Na channels to amiloride, as previously reported (24, 49).
Since PGE2 and IL-ra concentrations were low in MSC-M compared with HCYT-MSC-M, the higher potency of MSC-M to restore the Na transport under hypoxia plus cytomix seemed likely to be mediated by KGF. This result is remarkable because PGE2 is known to increase the intracellular cAMP, a key nucleotide for the stimulation of Na transport (13, 15, 46). In line with our results, prior studies in rats and sheep showed that PGE2 had no influence on alveolar liquid clearance (5). The very limited effect of HCYT-MSC-M despite increased levels of IL-1ra was also surprising since cytomix contained IL-1β, which has been reported to inhibit Na transport in AECs (22). Thus it seemed possible that the higher concentration of KGF in MSC-M compared with HCYT-MSC-M accounted for most of the beneficial effect of MSC-M. Indeed, several arguments favor this hypothesis. First, KGF has been reported to increase alveolar fluid transport in rat lung and to stimulate active Na ion transport in AECs by upregulating α-ENaC protein expression and Na-K-ATPase activity (6, 20, 52). Second, Lee et al. (32) reported that in an ex vivo perfused human lung, the instillation of human MSC following endotoxin-induced lung injury restored alveolar fluid clearance and increased vectorial Na transport, in part, due to KGF release, an observation recently confirmed in a live bacterial model of lung injury in ex vivo perfused human lungs (35). Third, these authors also reported that in MSC treated with siRNA KGF the medium was unable to restore cytomix-induced decreased transepithelial fluid transport over 24 h in AECs. Consistent with these observations, the current study shows that MSC-M completely depleted of KGF lost its beneficial effect on AEC amiloride-sensitive Na transport. However, incubation of AECs with rhKGF at concentrations similar to what was observed in MSC-M was unable to prevent the hypoxia plus cytomix-induced inhibition of Na transport. All together, our data indicate that secreted KGF probably plays a role in the therapeutic effect of the MSC media, although it is not sufficient per se to induce this effect.
One limitation of the present study is that our in vitro model combined cells originating from two different species, i.e., rat AECs and human MSCs. Unfortunately, human AEC lines are not really suitable for electrophysiological experiments in Ussing chamber, namely because of low endogenous expression of ENaC subunits and low transepithelial resistance. Primary rat AECs are usually considered as good surrogates for human AECs, which are very difficult to isolate. It is relevant to emphasize that previous studies have shown that human MSCs were effective in vivo in several rodent models of injury (7, 36, 37). One reason for that may be that most paracrine factors secreted by MSCs (such as KGF, IL-1-ra, or PGE2) show considerable homology across species. Indeed, it is well recognized that recombinant human proteins (rhKGF and rhIL-1-ra) have a biological effect on rodent AECs (17, 45). The fact that we observed a clear beneficial effect of human MSC media on rat AECs indicates that our model, despite the species difference, still represents a valuable tool for studying in vitro the crosstalk between AECs and MSCs.
In conclusion, the conditioned media of MSCs restored epithelial permeability to normal and completely prevented the decrease in amiloride-sensitive apical Na transport and α-ENaC cell surface expression induced by inflammatory and hypoxic insults, suggesting that the use of MSC-conditioned media may be relevant as a therapeutic in ALI. Secretion of the paracrine factor KGF by MSCs was required for the protective effect on alveolar epithelial Na transport.
GRANTS
This work was supported by Legs Poix from Chancellerie des Universités de Paris. A. Goolaerts is the recipient of a European Respiratory Society/Marie Curie Joint Research Fellowship-Number MC 1219-2009. The research leading to these results has received funding from the European Respiratory Society and the European Community's Seventh Framework Program FP7/2007–2013–Marie Curie Actions under Grant Agreement RESPIRE, PCOFUND-GA-2008-229571. The work was also supported by National Heart, Lung, and Blood Institute Grant HL-51854 (to M. A. Matthay).
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.G., N.P.-R., V.V., Y.U., T.G., and N.D. performed experiments; A.G., N.P.-R., V.V., Y.U., T.G., N.D., C.I.P., and C.C. analyzed data; A.G., J.L., T.G., and C.I.P. prepared figures; A.G., N.P.-R., J.L., V.V., Y.U., N.D., C.I.P., and M.A.M. approved final version of manuscript; J.L., C.I.P., M.A.M., and C.C. conception and design of research; J.L., C.I.P., and C.C. drafted manuscript; T.G., C.I.P., M.A.M., and C.C. interpreted results of experiments; C.I.P., M.A.M., and C.C. edited and revised manuscript.
REFERENCES
- 1.Abreu SC, Antunes MA, Pelosi P, Morales MM, Rocco PR. Mechanisms of cellular therapy in respiratory diseases. Intensive Care Med 37: 1421–1431, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Amariglio N, Hirshberg A, Scheithauer BW, Cohen Y, Loewenthal R, Trakhtenbrot L, Paz N, Koren-Michowitz M, Waldman D, Leider-Trejo L, Toren A, Constantini S, Rechavi G. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med 6: e1000029, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnulf B, Lecourt S, Soulier J, Ternaux B, Lacassagne MN, Crinquette A, Dessoly J, Sciaini AK, Benbunan M, Chomienne C, Fermand JP, Marolleau JP, Larghero J. Phenotypic and functional characterization of bone marrow mesenchymal stem cells derived from patients with multiple myeloma. Leukemia 21: 158–163, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, Mitsialis SA, Kourembanas S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med 180: 1122–1130, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berthiaume Y, Folkesson HG, Matthay MA. Indomethacin does not influence alveolar liquid clearance in anesthetized sheep or rats. Exp Lung Res 25: 517–530, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Borok Z, Danto SI, Dimen LL, Zhang XL, Lubman RL. Na+-K+-ATPase expression in alveolar epithelial cells: upregulation of active ion transport by KGF. Am J Physiol Lung Cell Mol Physiol 274: L149–L158, 1998 [DOI] [PubMed] [Google Scholar]
- 7.Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, Morando L, Busca A, Falda M, Bussolati B, Tetta C, Camussi G. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol 20: 1053–1067, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cepkova M, Brady S, Sapru A, Matthay MA, Church G. Biological markers of lung injury before and after the institution of positive pressure ventilation in patients with acute lung injury. Crit Care 10: R126, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Tredget EE, Liu C, Wu Y. Analysis of allogenicity of mesenchymal stem cells in engraftment and wound healing in mice. PLoS One 4: e7119, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clerici C, Matthay MA. Hypoxia regulates gene expression of alveolar epithelial transport proteins. J Appl Physiol 88: 1890–1896, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Crisostomo PR, Wang Y, Markel TA, Wang M, Lahm T, Meldrum DR. Human mesenchymal stem cells stimulated by TNF-α, LPS, or hypoxia produce growth factors by an NF-κB- but not JNK-dependent mechanism. Am J Physiol Cell Physiol 294: C675–C682, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Dada LA, Sznajder JI. Hypoxic inhibition of alveolar fluid reabsorption. Adv Exp Med Biol 618: 159–168, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Downs CA, Kriener LH, Yu L, Eaton DC, Jain L, Helms MN. β-Adrenergic agonists differentially regulate highly selective and nonselective epithelial sodium channels to promote alveolar fluid clearance in vivo. Am J Physiol Lung Cell Mol Physiol 302: L1167–L1178, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duc C, Farman N, Canessa CM, Bonvalet JP, Rossier BC. Cell-specific expression of epithelial sodium channel α, β and γ subunits in aldosterone-responsive epithelia from the rat: localization by in situ hybridization and immunocytochemistry. J Cell Biol 127: 1907–1921, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Factor P, Adir Y, Mutlu GM, Burhop J, Dumasius V. Effects of beta2-adrenergic receptor overexpression on alveolar epithelial active transport. J Allergy Clin Immunol 110: S242–246, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Fang X, Neyrinck AP, Matthay MA, Lee JW. Allogeneic human mesenchymal stem cells restore epithelial protein permeability in cultured human alveolar type II cells by secretion of angiopoietin-1. J Biol Chem 285: 26211–26222, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frank JA, Pittet JF, Wray C, Matthay MA. Protection from experimental ventilator-induced acute lung injury by IL-1 receptor blockade. Thorax 63: 147–153, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Geiser T, Atabai K, Jarreau PH, Ware LB, Pugin J, Matthay MA. Pulmonary edema fluid from patients with acute lung injury augments in vitro alveolar epithelial repair by an IL-1beta-dependent mechanism. Am J Respir Crit Care Med 163: 1384–1388, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Gotts JE, Matthay MA. Mesenchymal stem cells and acute lung injury. Crit Care Clin 27: 719–733, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guery BP, Mason CM, Dobard EP, Beaucaire G, Summer WR, Nelson S. Keratinocyte growth factor increases transalveolar sodium reabsorption in normal and injured rat lungs. Am J Respir Crit Care Med 155: 1777–1784, 1997 [DOI] [PubMed] [Google Scholar]
- 21.Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol 179: 1855–1863, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Howard M, Roux J, Iles KE, Miyazawa B, Christiaans S, Anjum N, Dickinson DA, Goolaerts A, Matthay MA, Pittet JF. Activation of the heat shock response attenuates the interleukin 1beta-mediated inhibition of the amiloride-sensitive alveolar epithelial ion transport. Shock 39: 189–196, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ionescu L, Byrne RN, van Haaften T, Vadivel A, Alphonse RS, Rey-Parra GJ, Weissmann G, Hall A, Eaton F, Thebaud B. Stem cell conditioned medium improves acute lung injury in mice: in vivo evidence for stem cell paracrine action. Am J Physiol Lung Cell Mol Physiol 303: L967–L977, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji HL, Zhao RZ, Chen ZX, Shetty S, Idell S, Matalon S. δENaC: a novel divergent amiloride-inhibitable sodium channel. Am J Physiol Lung Cell Mol Physiol 303: L1013–L1026, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kemp PJ, Kim KJ, Borok Z, Crandall ED. Re-evaluating the Na+ conductance of adult rat alveolar type II pneumocytes: evidence for the involvement of cGMP-activated cation channels. J Physiol 536: 693–701, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kotton DN, Fabian AJ, Mulligan RC. Failure of bone marrow to reconstitute lung epithelium. Am J Respir Cell Mol Biol 33: 328–334, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotton DN, Ma BY, Cardoso WV, Sanderson EA, Summer RS, Williams MC, Fine A. Bone marrow-derived cells as progenitors of lung alveolar epithelium. Development 128: 5181–5188, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis SJ. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 105: 369–377, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, Sdrimas K, Fernandez-Gonzalez A, Kourembanas S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 126: 2601–2611, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazrak A, Nita I, Subramaniyam D, Wei S, Song W, Ji HL, Janciauskiene S, Matalon S. Alpha(1)-antitrypsin inhibits epithelial Na+ transport in vitro and in vivo. Am J Respir Cell Mol Biol 41: 261–270, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JW, Fang X, Dolganov G, Fremont RD, Bastarache JA, Ware LB, Matthay MA. Acute lung injury edema fluid decreases net fluid transport across human alveolar epithelial type II cells. J Biol Chem 282: 24109–24119, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JW, Fang X, Gupta N, Serikov V, Matthay MA. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci USA 106: 16357–16362, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JW, Fang X, Krasnodembskaya A, Howard JP, Matthay MA. Concise review: mesenchymal stem cells for acute lung injury: role of paracrine soluble factors. Stem Cells 29: 913–919, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JW, Gupta N, Serikov V, Matthay MA. Potential application of mesenchymal stem cells in acute lung injury. Expert Opin Biol Ther 9: 1259–1270, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbott J, Matthay MA. Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria. Am J Respir Crit Care Med 187: 751–760, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, Semprun-Prieto L, Delafontaine P, Prockop DJ. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 5: 54–63, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee RH, Seo MJ, Reger RL, Spees JL, Pulin AA, Olson SD, Prockop DJ. Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proc Natl Acad Sci USA 103: 17438–17443, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lepperdinger G, Brunauer R, Jamnig A, Laschober G, Kassem M. Controversial issue: is it safe to employ mesenchymal stem cells in cell-based therapies? Exp Gerontol 43: 1018–1023, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 122: 2731–2740, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol 33: 319–327, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC, Stewart DJ. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med 4: e269, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med 15: 42–49, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ortiz LA, Dutreil M, Fattman C, Pandey AC, Torres G, Go K, Phinney DG. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc Natl Acad Sci USA 104: 11002–11007, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ortiz LA, Gambelli F, McBride C, Gaupp D, Baddoo M, Kaminski N, Phinney DG. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc Natl Acad Sci USA 100: 8407–8411, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panos RJ, Rubin JS, Csaky KG, Aaronson SA, Mason RJ. Keratinocyte growth factor and hepatocyte growth factor/scatter factor are heparin-binding growth factors for alveolar type II cells in fibroblast-conditioned medium. J Clin Invest 92: 969–977, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Planès C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. Hypoxia and beta 2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem 277: 47318–47324, 2002 [DOI] [PubMed] [Google Scholar]
- 47.Planès C, Leyvraz C, Uchida T, Angelova MA, Vuagniaux G, Hummler E, Matthay MA, Clerici C, Rossier B. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am J Physiol Lung Cell Mol Physiol 288: L1099–L1109, 2005 [DOI] [PubMed] [Google Scholar]
- 48.Prulière-Escabasse V, Planès C, Escudier E, Fanen P, Coste A, Clerici C. Modulation of epithelial sodium channel trafficking and function by sodium 4-phenylbutyrate in human nasal epithelial cells. J Biol Chem 282: 34048–34057, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Randrianarison N, Clerici C, Ferreira C, Fontayne A, Pradervand S, Fowler-Jaeger N, Hummler E, Rossier BC, Planès C. Low expression of the β-ENaC subunit impairs lung fluid clearance in the mouse. Am J Physiol Lung Cell Mol Physiol 294: L409–L416, 2008 [DOI] [PubMed] [Google Scholar]
- 50.Rojas M, Xu J, Woods CR, Mora AL, Spears W, Roman J, Brigham KL. Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am J Respir Cell Mol Biol 33: 145–152, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005 [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Folkesson HG, Jayr C, Ware LB, Matthay MA. Alveolar epithelial fluid transport can be simultaneously upregulated by both KGF and beta-agonist therapy. J Appl Physiol 87: 1852–1860, 1999 [DOI] [PubMed] [Google Scholar]
- 53.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 163: 1376–1383, 2001 [DOI] [PubMed] [Google Scholar]