

Abstract
The effects of 1H-[1,2,4]-oxadizaolo[4,3-]quinoxaline-1-one (ODQ), an inhibitor of the activation of soluble guanylate cyclase (sGC) on responses to NO donors acetylcholine (ACh) and bradykinin (BK) were investigated in the pulmonary and systemic vascular beds of the rat. In these studies the administration of ODQ in a dose of 5 mg/kg iv attenuated vasodilator responses to five different NO donors without inhibiting responses to ACh and BK in the systemic and pulmonary vascular beds of the rat. Vasodilator responses to ACh were not inhibited by l-NAME or the transient receptor vanilloid type 4 (TRPV4) antagonist GSK-2193874, which attenuated vasodilator responses to the TRPV4 agonist GSK-1016790A. ODQ did not inhibit vasodilator responses to agents reported to act in an NO-independent manner or to vasoconstrictor agents, and ODQ did not increase blood methemoglobin levels, suggesting that off target effects were minimal. These results show that ODQ in a dose that inhibited NO donor-mediated responses did not alter vasodilator responses to ACh in the pulmonary and systemic vascular beds and did not alter systemic vasodilator responses to BK. The present results indicate that decreases in pulmonary and systemic arterial pressures in response to ACh are not mediated by the activation of sGC or TRPV4 channels and that ODQ can be used to study the role of the activation of sGC in mediating vasodilator responses in the rat.
Keywords: pulmonary vascular bed, transient receptor vanilloid type, vasodilation
cyclic guanosine monophosphate (cGMP) has an important role in the regulation of vascular smooth muscle tone and is synthesized from guanosine triphosphate (GTP) by soluble guanylate cyclase (sGC), the receptor for nitric oxide (NO) (2, 29, 32, 41). sGC is a heterodimeric enzyme, and binding of NO to the heme-containing subunit activates sGC (2). In contrast, oxidation of the heme-containing subunit reduces the sensitivity of the enzyme to NO (7, 25, 45, 48, 49). Pharmacological agents and genetic alterations that selectively modulate sGC activity are useful in the study of the physiological function of the enzyme in regulating vascular tone and mediating vasodilator responses (19, 35, 43).
Although the role of NO in the regulation of baseline tone is well established, the role of NO in mediating vasodilator responses to acetylcholine (ACh) and bradykinin (BK) is controversial (4, 12, 34). A large number of studies show that vasorelaxant responses to ACh are mediated by the NO-cGMP pathway; however, other studies indicate that NO is not involved (13, 18, 20). It has been hypothesized that vessel size, vascular segment studied, method used, and species may account for differences in the role of NO in mediating responses to ACh and BK reported in the literature (17, 34). Studies in isolated first-order gracilis muscle arterioles from endothelial NOS (eNOS) knockout mice show that responses to ACh were only slightly attenuated, and studies in eNOS and cGMP-dependent protein kinase type-1 (cGKI) knockout mice show that vasodepressor responses to ACh are preserved, suggesting that NO and G kinase do not play a major role in mediating these responses (18, 28). Inasmuch as eNOS- and cGKI-deficient rats have not yet been developed, current studies are limited to the use of pharmacological inhibitors in this species.
1H-[1,2,4]-oxadizaolo[4,3-]quinoxaline-1-one (ODQ) is an agent that has been shown to inhibit NO activation of sGC by oxidizing the heme moiety on the β-subunit of the enzyme (9, 11, 39, 52). ODQ has been used to study activation of sGC by NO in biochemical and isolated vessel experiments and has been used in a small number of in vivo cardiovascular studies (9, 11, 39, 52). Studies with ODQ in the rat show that vasodilator responses to the NO donor sodium nitroprusside (SNP) are inhibited, whereas vasodilator responses to chemical activators of sGC are enhanced (35, 43). The present study was undertaken to investigate the effects of ODQ on cardiovascular responses in the pulmonary and systemic vascular beds of the intact rat. In these experiments the effect of ODQ on responses on NO-dependent and NO-independent vasodilator agents and responses to vasoconstrictor agents were investigated. These results show that ODQ did not attenuate decreases in pulmonary and systemic arterial pressures in response to ACh in a dose that inhibits vasodilator responses to a variety of NO donors without inhibiting responses to NO-independent vasodilator agents or responses to vasoconstrictor agents. In these studies, ODQ did not alter blood methemoglobin levels.
It has been reported that vasodilator responses to ACh are mediated by hyperpolarization, and it has been hypothesized that transient receptor vanilloid type 4 (TRPV4) channels are involved in mediating vasodilator responses to ACh (10, 40, 51). In the present study the effect of TRPV4 blockade on vasodilator responses to ACh was investigated. The present results suggest that ODQ can be used to study the role of NO and sGC in mediating vasodilator responses to NO donors and that vasodilator responses to ACh are not mediated by NO and the activation of sGC or the activation of TRPV4 receptors in the rat.
METHODS
The Institutional Animal Care and Use Committee of the Tulane University School of Medicine approved the experimental protocol employed in these studies, and all procedures were conducted in accordance with institutional guidelines. In these experiments, adult male Sprague-Dawley rats (Charles Rivers) weighing 325–450 g were anesthetized with 100 mg/kg ip Inactin (Sigma-Aldrich) and placed in the supine position on an operating table. Supplemental doses of Inactin were administered intraperitoneally to maintain a uniform level of anesthesia. Body temperature was maintained with a heating lamp. The trachea was cannulated with a short segment of PE-240 tubing to maintain a patent airway. The animals spontaneously breathed room air. A femoral artery was catheterized with PE-50 tubing for measurement of systemic arterial pressure. The left jugular and femoral veins were catheterized with PE-50 tubing for intravenous injections and infusions of agents. For pulmonary arterial pressure measurement, a specially designed 3-Fr single-lumen catheter with a curved tip and radio-opaque marker was passed from the right jugular vein into the main pulmonary artery (PA) under fluoroscopic guidance (Picker-Surveyor Fluoroscope) as previously described (22, 36). Pulmonary and systemic arterial pressures were measured with Namic Perceptor DT transducers (Boston Scientific), digitized by a Biopac MP100 data acquisition system (Biopac Systems), and stored on a Dell personal computer (PC). Cardiac output was measured by the thermodilution technique with a Cardiomax II computer (Columbus Instruments). A known volume (0.2 ml) of room temperature 0.9% NaCl solution was injected in the jugular vein catheter with the tip near the right atrium, and changes in blood temperature were detected by a 1.5-Fr thermistor microprobe catheter (Columbus Instruments) positioned in the aortic arch from the left carotid artery. The indicator dilution curve data were stored on the PC.
Each experimental series was carried out in a separate group of rats, and, in the first set of experiments, the effect of ODQ in a dose of 5 mg/kg iv on responses to the NO donors SNP, sodium nitrite (NaNO2), glyceryl trinitrate (GTN), diethylamine NONOate (DEA/NO), and S-nitrosoglutathione (SNGT) was investigated.
In the second set of experiments, the effect of ODQ on responses to vasodilator agents that act by NO-independent mechanisms was investigated. In the third set of experiments, the effect of ODQ on responses to vasoconstrictor agents was evaluated. In the fourth set of experiments, the effect of ODQ on responses to ACh and BK was investigated, and, in the fifth set of experiments, the effects of l-NAME, and a combination of l-NAME and ODQ on responses to ACh were investigated. The effect of ODQ on the vasodilator response to a subthreshold dose of the sGC activator BAY 60-2770 was also investigated.
In another group of experiments, the effect of the TRPV4 antagonist GSK-2193874 on responses to ACh was investigated.
In ex vivo experiments, PA were isolated and dissected from the lung and cut into rings (∼2 mm in length) from large, medium, and small pulmonary arterial branches (∼300, 200, and 100 μm in diameter) and bathed in PSS solution (containing in mM): 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 KH2PO4, 1.2 MgSO4·7H2O, 25 NaHCO3, and 11 glucose at pH = 7.4. The PA rings were mounted in a small-vessel dual-chamber myograph, bathed in PSS solution at 37°C, and oxygenated with a gas mixture of 95% O2 and 5% CO2. Isometric tension was measured as previously described (18, 27, 33, 44). The rings were initially stretched with a resting tension equivalent to a luminal pressure of 30 mmHg (3.9 kPa). The normalization was performed by distending the vessel and measuring micrometer and force readings. The wall tension is the force divided by the wall length, and the LaPlace relationship can be used to determine the effective pressure, which was set to 30 mmHg or 3.9 kPa. After precontraction to ∼80% of maximum tension induced by the thromboxane A2 mimic U-46619 (10−6 M) and a steady contraction was achieved, cumulative dose-response curves were then obtained for ACh (10−8 to 10−5 M) and SNP (10−8 to 10−5 M) in the presence or absence of ODQ (10−5 M).
For determination of methemoglobin levels, a 200-μl arterial blood sample was withdrawn from a femoral artery and analyzed with a Radiometer NPT7 (Copenhagen, Denmark) blood gas analyzer.
For determination of cGMP levels, PA segments from the rats were dissected and cut into rings and incubated with ODQ (10−5 M) or SNP (10−4 M) or the combination of ODQ and SNP. After 1 h incubation, the PA rings were frozen in liquid nitrogen. The cGMP measurements were performed using an enzyme-linked immunosorbent assay kit from Cayman Chemical (Ann Arbor, MI) according to the manufacturer's protocol and as previously described (1, 6).
Drugs.
ODQ (Cayman Chemical), ACh, BK, fasudil, PGE1, angiotensin II, phenylephrine, histamine, albuterol, sodium meclofenamate, SNP, NaNO2, GTN, DEA/NO, SNGT, l-NAME, Nω-nitro-l-arginine methyl ester hydrochloride, and HOE-140 (Sigma-Aldrich) were dissolved in 0.9% NaCl, and apocynin was dissolved in dimethyl sulfoxide (DMSO). BAY 60-2770 was obtained from Dr. Johannes-Peter Stasch of the Institute of Cardiovascular Research (Pharma Research Centre, Bayer, Wuppertal, Germany) and was dissolved in Transcutol-Cremophor EL-0.9% NaCl solution (10:10:80) (42). U-46619 (Cayman Chemical) was dissolved in 95% ethyl alcohol and diluted in 0.9% NaCl solution. The TRPV4 agonist GSK-1016790A (Smith Klein Glaxo) and the TRPV4 antagonist GSK-2193874 (Smith Klein Glaxo) were prepared in 1% DMSO/20% Captisol (sulfobutyl ether-β-cyclodextrin; CyDex, Lenexa, KS) and saline. The doses of GSK-1016790A and GSK-2193874 were determined from previous studies and pilot data (37, 47).
The hemodynamic data are expressed as means ± SE and were analyzed using paired and group t-tests and ANOVA for repeated measures. The criteria used for statistical significance was P < 0.05.
RESULTS
Effect of ODQ on NO donor responses.
The effect of ODQ on responses to SNP, NaNO2, GTN, DEA/NO, and SNGT was investigated in the intact chest rat, and the data are summarized in Fig. 1. The intravenous injections of the five NO donors produced dose-related decreases in pulmonary and systemic arterial pressure and no change or small increases in cardiac output (Fig. 1). After administration of ODQ in a dose of 5 mg/kg iv, the decreases in pulmonary and systemic arterial pressures in response to the NO donors were significantly attenuated, whereas changes in cardiac output were not altered (Fig. 1).
Fig. 1.

Bar graphs showing the effects of iv injection of sodium nitroprusside (SNP), diethylamine NONOate (DEA/NO), S-nitrosoglutathione, sodium nitrite, and glyceryl trinitrate on pulmonary and systemic arterial pressure and cardiac output before and after treatment with 1H-[1,2,4]-oxadizaolo[4,3-]quinoxaline-1-one (ODQ, 5 mg/kg iv). n, No. of experiments. *P < 0.05 compared with control, paired analysis.
Effect of ODQ on responses to NO-independent vasodilator agents.
The effects of ODQ on responses to vasodilator agents that act by NO-independent mechanisms were investigated, and the intravenous injections of albuterol, isoproterenol, histamine, fasudil, and PGE1 produced decreases in systemic and pulmonary arterial pressures with small effects on cardiac output, and responses to these agents were not altered after administration of 5 mg/kg iv ODQ (Fig. 2A).
Fig. 2.

A: bar graphs showing responses to the iv injections of the vasodilators albuterol, isoproterenol, fasudil, histamine, and PGE1. B: iv injections of the vasoconstrictors U-46619, phenylephrine, and angiotensin II and acute ventilatory hypoxia (10% O2-90% N2 gas) on pulmonary and systemic arterial pressure and cardiac output before and after treatment with ODQ (5 mg/kg iv). n, No. of experiments. *P < 0.05 compared with control, paired analysis.
Effect of ODQ on responses to vasoconstrictor agents.
The intravenous injections of angiotensin II and phenylephrine produced dose-related increases in pulmonary and systemic arterial pressures, and exposure to acute ventilatory hypoxia (10% O2) produced an increase in pulmonary arterial pressure and a decrease in systemic arterial pressure. The hemodynamic responses to phenylephrine and ventilatory hypoxia were not altered after administration of 5 mg/kg iv ODQ, whereas increases in pulmonary arterial pressure in response to angiotensin II were significantly increased (Fig. 2B). The enhanced response to angiotensin II in the pulmonary vascular bed after ODQ treatment was not reduced by treatment with the NADPH oxidase inhibitor apocyanin (300 μmol/kg iv) (Fig. 3). The intravenous injections of U-46619 increased pulmonary arterial pressure in a dose-related manner, had no consistent effect on systemic arterial pressure, and decreased cardiac output (Fig. 2B). The increases in pulmonary arterial pressure in response to intravenous injections of U-46619 were not changed after administration of ODQ (Fig. 2B).
Fig. 3.
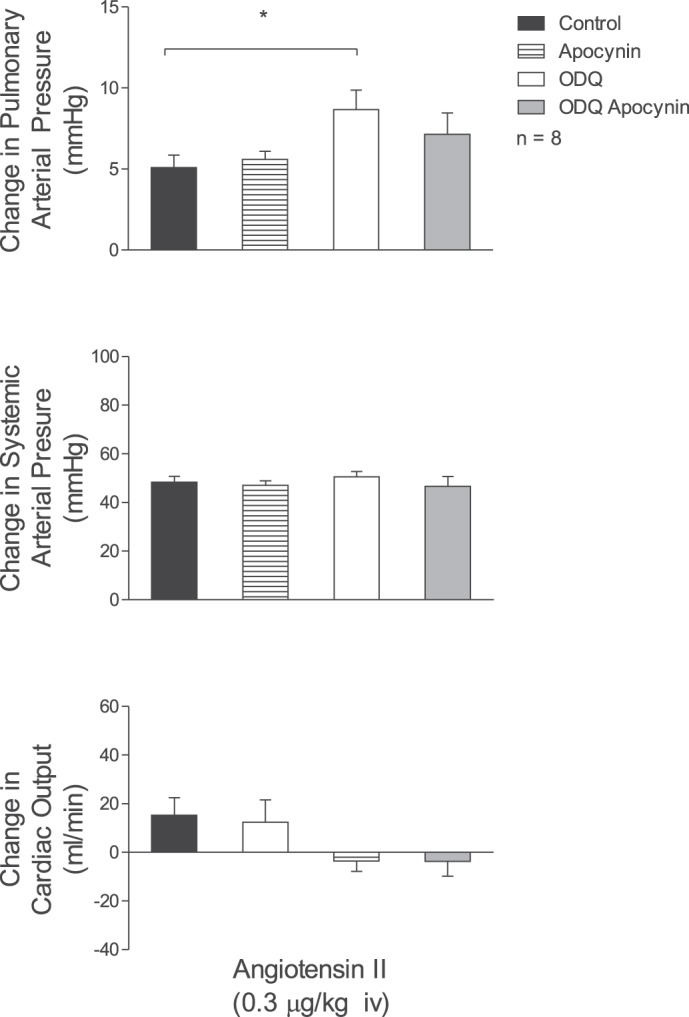
Bar graphs showing the effects of iv injection of angiotensin II (0.3 μg/kg) on pulmonary and systemic arterial pressure and cardiac output in control settings, after treatment with apocynin (300 μmol/kg), after ODQ (5 mg/kg) treatment, and after combined treatment with ODQ (5 mg/kg) and apocynin. n, No. of experiments. *P < 0.05.
Effect of ODQ on responses to BAY 60-2770.
The effect of ODQ on the response to a subthreshold or threshold dose of the sGC activator BAY 60-2770 was investigated, and the intravenous injection of BAY 60-2770 in a dose of 1 μg/kg had little or no effect on pulmonary or systemic arterial pressure (0 ± 0 and −4 ± 2 mmHg, respectively) in the control period but produced significant decreases in these pressures after treatment with 5 mg/kg iv ODQ (Fig. 4).
Fig. 4.
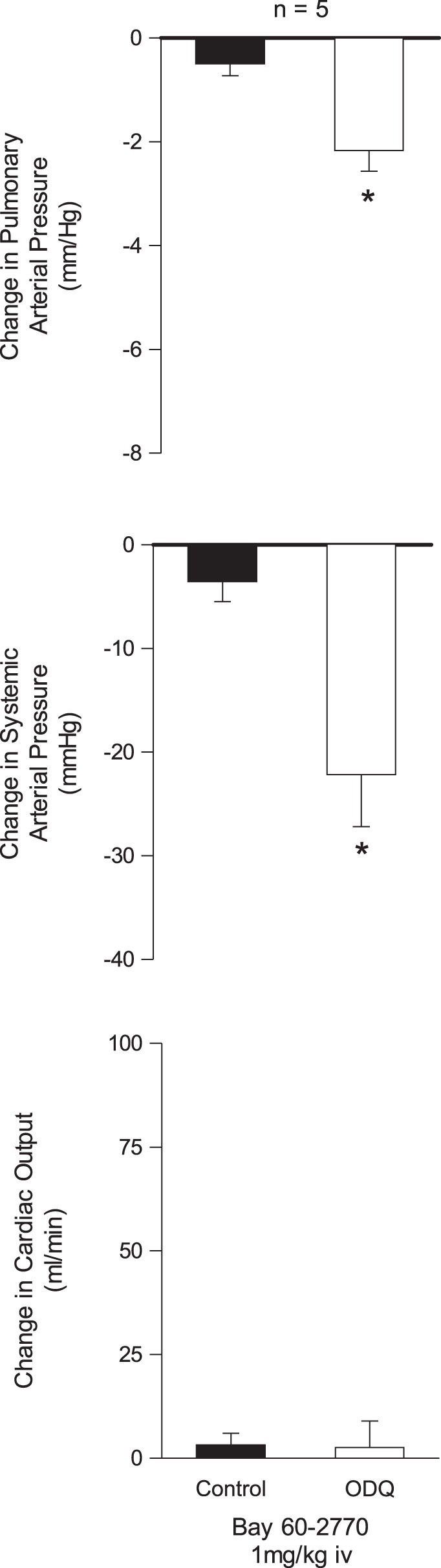
Bar graphs showing the effects of iv injection of a subthreshold dose (1 mg/kg) of the soluble guanylate cyclase (sGC) activator BAY 60-2770 on pulmonary and systemic arterial pressure and cardiac output before and after treatment with ODQ (5 mg/kg iv). *P < 0.05 compared with control.
Effect of ODQ on responses to ACh and BK.
The effect of ODQ on responses to ACh and BK was investigated in the rat, and these data are summarized in Figs. 5 and 6. The intravenous injections of ACh in doses of 0.3–3 μg/kg produced dose-related decreases in systemic arterial pressure and at higher doses studied decreases in pulmonary arterial pressure with no change or small increases in cardiac output that were probably baroreceptor reflex mediated (Fig. 5). The increase in cardiac output is most likely caused by the decrease in afterload and a reflex increase in heart rate. The decreases in pulmonary and systemic arterial pressures in response to intravenous injections of ACh were not altered after administration of 5 mg/kg iv ODQ (Fig. 5).
Fig. 5.
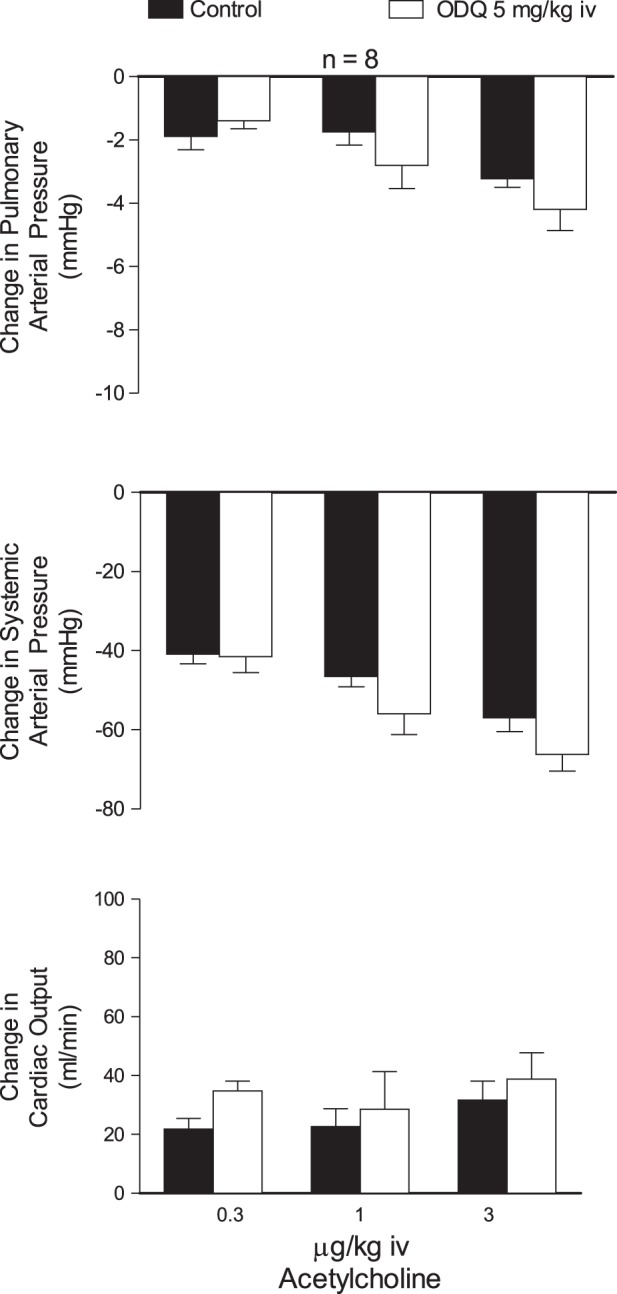
Bar graphs showing the effects of iv injections of ACh (0.3 to 3 μg/kg) on pulmonary and systemic arterial pressure and cardiac output before and after treatment with ODQ (5 mg/kg iv). n, No. of experiments.
Fig. 6.

Bar graphs showing the effects of iv injections of bradykinin on pulmonary and systemic arterial pressure and cardiac output before and after treatment with ODQ (5 mg/kg iv) (A), HOE-140 (0.1 mg/kg iv) (B), sodium meclofenomate (5 mg/kg iv) (C), l-NAME (50 mg/kg iv) (D), and phentolamine (0.5 mg/kg iv) (E). n, No. of experiments. *P < 0.05.
The intravenous injections of BK produced dose-related decreases in systemic arterial pressure and dose-related increases in pulmonary arterial pressure with small changes in cardiac output (Fig. 6A). The decreases in systemic arterial pressure and increases in pulmonary arterial pressure in response to intravenous injections of BK were not altered after administration of 5 mg/kg iv ODQ (Fig. 6A). The decreases in systemic arterial pressure and increases in pulmonary arterial pressure in response to BK were significantly reduced after administration of the BK B2-receptor antagonist HOE-140 in a dose of 0.1 mg/kg iv (Fig. 6B) but were not altered after administration of the cyclooxygenase inhibitor sodium meclofenamate in a dose of 5 mg/kg iv or l-NAME in a dose of 50 mg/kg iv (Fig. 6, C and D). The increases in pulmonary arterial pressure in response to BK were significantly reduced after administration of the α-receptor antagonist phentolamine in a dose of 0.5 mg/kg iv that significantly reduced the pressor response intravenous injection of the α1-receptor agonist phenylephrine (Fig. 6E).
Effect of ODQ on SNP and ACh responses in isolated PA.
The effects of ODQ on vasorelaxant responses to ACh and SNP were investigated in large (∼300-μm)-, medium (∼200-μm)-, and small (∼100-μm)-diameter PA rings precontracted with U-46619. The endothelium-dependent relaxation response to ACh was greater in large arteries (87%) compared with medium (51%) and small (27%) arteries. The incubation with ODQ (10−5 M) had no significant effect on endothelium-dependent relaxation responses to ACh in large, medium, and small PA segments (Fig. 7, A, B, and C). Interestingly, endothelium-independent relaxation responses to SNP were independent of vessel size and were attenuated by ODQ. The incubation with 10−5 M ODQ shifted the dose-response curve for SNP to the right in large artery rings, indicating a reduction in sensitivity to the NO donor. In medium- and small-sized arteries, ODQ also reduced the maximal response to SNP (Fig. 7, A, B, and C).
Fig. 7.

Graphs showing SNP- and ACh-induced vessel relaxation after precontraction with U-46619 on large (∼300 μm diameter) (A), medium (∼200 μm diameter) (B), and small (∼100 μm diameter) (C) pulmonary artery rings before and after treatment with 10 μM ODQ. n, No. of experiments.
The incubation with ODQ had no significant effect on baseline tension in the isolated PA ring segments.
Effect of l-NAME and ODQ on responses to ACh in the rat.
The effects of l-NAME alone and in combination with ODQ on responses to ACh in the rat were investigated, and these data are summarized in Fig. 7. The administration of l-NAME in a dose of 50 mg/kg iv significantly increased pulmonary and systemic arterial pressures and decreased cardiac output (Table 1). However, l-NAME treatment did not attenuate decreases in pulmonary and systemic arterial pressures in response to intravenous injections of ACh, and deceases in pulmonary and systemic arterial pressures were enhanced, an effect that was probably related to the increase in tone (Fig. 8A). Previous studies have shown that decreases in pulmonary arterial pressures in response to vasodilator agents are dependent on the level of baseline vasoconstrictor tone (23, 24). The effect of l-NAME in combination with ODQ on the response to ACh was investigated, and, after intravenous administration of 50 mg/kg l-NAME and 5 mg/kg iv ODQ, decreases in pulmonary and systemic arterial pressures in response to intravenous injections of ACh were not reduced (Fig. 8B).
Table 1.
Effect of l-NAME on systemic and pulmonary arterial pressure and on cardiac output
| Systemic Arterial Pressure, mmHg | Pulmonary Arterial Pressure, mmHg | Cardiac Output, ml/min | |
|---|---|---|---|
| Control | 105 ± 4 | 19 ± 1 | 113 ± 7 |
| l-NAME (50 mg/kg iv) (n = 10) | 146 ± 4* | 31 ± 2* | 73 ± 7* |
Values are means ± SE; n, no. of eperiments. l-NAME, NG-nitro-l-arginine methyl ester.
P < 0.05 compared with corresponding control.
Fig. 8.

Bar graphs showing the effect of ACh (0.3–3 μg/kg iv) on pulmonary and systemic arterial pressure before and after treatment with l-NAME (50 mg/kg iv) (A) and after treatment with l-NAME (50 mg/kg iv) and a combination of l-NAME (50 mg/kg iv) and ODQ (5 mg/kg iv) (B). n, No. of experiments. *P < 0.05.
Effect of ODQ on baseline hemodynamic values and methemoglobin levels.
The intravenous injection of ODQ in a dose of 5 mg/kg iv had no significant effect on baseline systemic and pulmonary arterial pressures or cardiac output and on blood methemoglobin levels (Table 2).
Table 2.
Effect of ODQ on hemodynamic parameters and methemeoglobin formation
| Systemic Arterial Pressure, mmHg | Pulmonary Arterial Pressure, mmHg | Cardiac Output, ml/min | %MetHb, g/dl | |
|---|---|---|---|---|
| Control | 102 ± 4 | 19 ± 1 | 115 ± 3 | 0.94 ± 0.3 |
| ODQ (5 mg/kg iv) (n = 10) | 112 ± 5 | 19 ± 1 | 108 ± 4 | 2.18 ± 0.5 |
Values are means ± SE; n, no. of eperiments.
MetHb, methemoglobin.
Effect of ODQ and SNP on cGMP levels.
Colorimetric determination of cGMP content was performed in the lysate from PA. The cGMP level in arteries incubated with SNP (10−4 M) was significantly higher compared with values in control arteries or arteries incubated with ODQ (10−5 M) or arteries incubated with ODQ and SNP (Fig. 9).
Fig. 9.
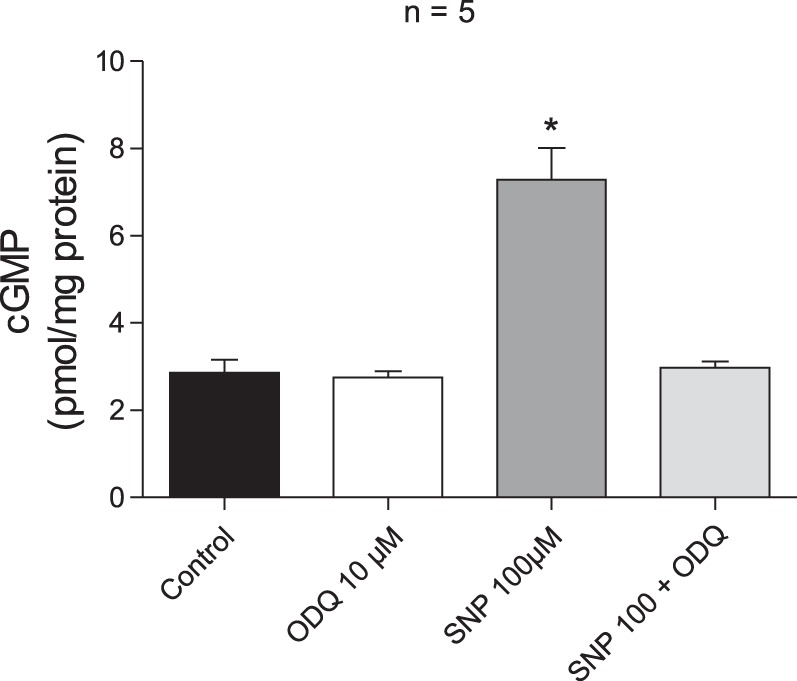
Bar graph showing cGMP levels in pulmonary arterial segments under control conditions, following treatment with 10 μM ODQ, 100 μM SNP, and combined treatment with 100 μM SNP and 10 μM ODQ. n, No. of experiments. *P < 0.05 compared with control.
Role of TRPV4 channels.
It has been suggested that vasodilator responses to ACh are mediated in part by hyperpolarization, and it has been hypothesized that TRPV4 channels are involved in mediating the response to ACh in resistance arteries from the mouse mesentery (40, 46, 51). To investigate the role of TRPV4 channels in mediating decreases in pulmonary and systemic arterial pressures in response to ACh in the rat, the effect of the TRPV4 antagonist GSK-2193874 was evaluated. The intravenous injection of the TRPV4 agonist GSK-1016790A (6 μg/kg), SNP (1 μg/kg), and ACh (1 μg/kg) produced decreases in pulmonary and systemic arterial pressures (Fig. 10). The decreases in pulmonary and systemic arterial pressures in response to intravenous injections of the TRPV4 agonist GSK-1016790A were significantly attenuated by the intravenous administration of the TRPV4 antagonist GSK-2193874 (300 μg/kg), whereas decreases in pulmonary and systemic arterial pressures in response to intravenous injections of ACh were not changed (Fig. 10).
Fig. 10.
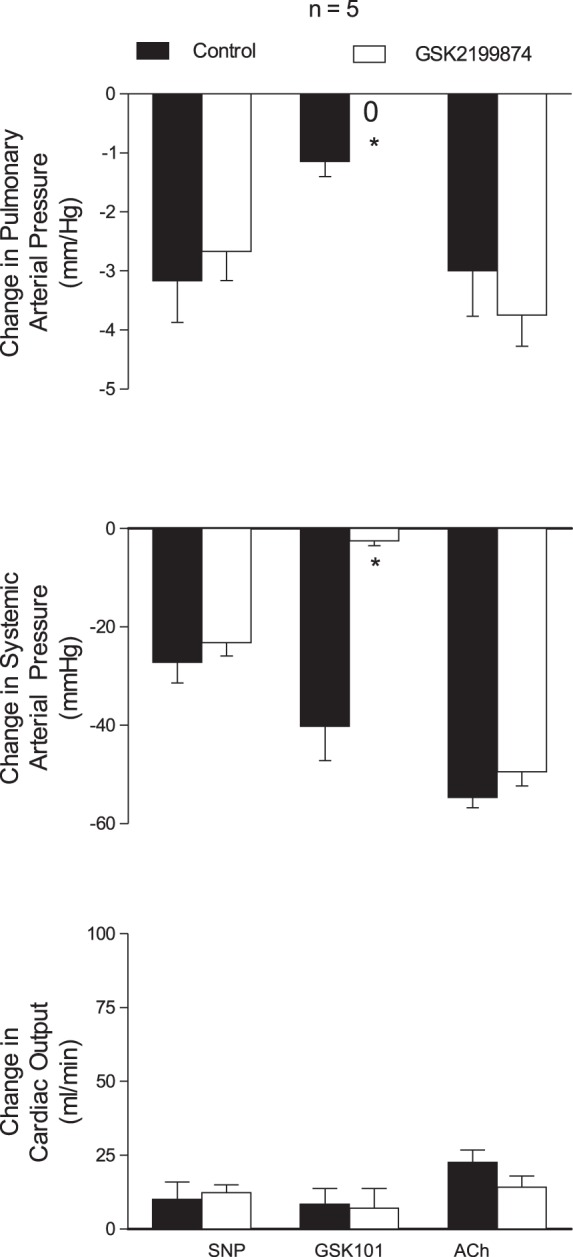
Bar graphs showing the effects of the transient receptor vanilloid type 4 (TRPV4) antagonist GSK-2192874 (300 μg/kg iv) on changes in pulmonary and systemic arterial pressure and cardiac output in response to iv injections of SNP (1 μg/kg), the TRPV4 agonist GSK-1016790A (6 μg/kg), and ACh (1 μg/kg). n, No. of experiments. *P < 0.05 compared with control.
DISCUSSION
ODQ is an agent that inhibits the activation of sGC by NO and provides a useful probe for studying the role of sGC activation in mediating vasorelaxant responses to NO (9, 11, 39, 52). ODQ has been used in many biochemical and isolated blood vessel studies but has only been used in few cardiovascular studies in the intact animal (9, 11, 14, 35, 39, 52). ODQ has recently been shown to inhibit vasodilator responses to the NO donor SNP in the rat (35). In the present study, ODQ was used to investigate responses to five different NO donors, endothelium-dependent and -independent vasodilator agents, and vasoconstrictor agents in the intact chest rat. The results of these studies show that decreases in pulmonary and systemic arterial pressures in response to intravenous injections of the NO donors DEA/NO, NaNO2, GTN, SNGT, and SNP are attenuated by ODQ in a dose that had no significant effect on responses to vasodilator agents that act by NO-independent mechanisms. These data provide support for the concept that decreases in pulmonary and systemic pressures in response to the NO donors are mediated by the activation of sGC in resistance vessel elements in the pulmonary and systemic vascular beds in the intact chest rat and that ODQ is useful for investigating the role of sGC activation in mediating NO-dependent responses in vivo in the rat. In addition to not altering responses to vasodilator agents that act by NO-independent mechanisms, ODQ did not inhibit responses to vasoconstrictor agents, providing support for the hypothesis that the inhibitory effect of ODQ is selective for NO-sGC-cGMP pathway-mediated responses. Although ODQ attenuated responses to the NO donors in the rat in a selective manner, decreases in pulmonary and systemic arterial pressures in response to intravenous injections of ACh were not decreased by ODQ. These data are consistent with results of studies with the NOS inhibitor l-NAME, indicating that decreases in systemic arterial pressure and regional vascular resistance in response to ACh in the rat are not attenuated, suggesting that responses are not mediated by NO (8, 12–14, 17). The results with ODQ in the pulmonary vascular bed of the intact rat are consistent with results in isolated pulmonary vessels where vasorelaxant responses to the NO donor SNP were attenuated, whereas vasorelaxant responses to ACh were not altered by ODQ in various-sized arterial rings precontracted with U-46619. These results are in agreement with studies in cGKI and eNOS knockout mice in which vasodepressor responses to SNP were attenuated in cGKI knockout mice, whereas vasodepressor responses to ACh are not altered in cGKI or eNOS knockout mice (19, 28). The results of pharmacological studies in the rat and studies in knockout mice provide support for the hypothesis that the sGC-cGMP-cGK pathway has an important role in mediating the vasodilator response to the NO donors but is not involved in mediating decreases in pulmonary and systemic arterial pressure in response to ACh in the intact mouse or rat (8, 12–14, 17, 28).
The present results are not in agreement with a number of studies in the literature, including previous studies from our own laboratory in several laboratory species (3, 8, 31). The reason for the different results with ACh is uncertain and is complicated by the large increases in baseline pulmonary and systemic arterial pressures induced by the NOS inhibitor l-NAME and by species (21, 23, 24). In this regard, when systemic arterial pressure was restored to baseline values after l-NAME treatment with an SNP infusion, the decreases in systemic arterial pressure in response to ACh were not altered by l-NAME in the rat (8). In previous studies in the rat hindlimb vascular bed, vasodilator responses to ACh were attenuated by apamin and charybdotoxin, providing support for the hypothesis that they were mediated by the opening calcium-activated K+ channels (8).
The intravenous injection of BK produced an increase in pulmonary arterial pressure and a decrease in systemic arterial pressure, and these responses were not altered by ODQ. The increase in pulmonary arterial pressure and decrease in systemic arterial pressure were attenuated by HOE-140, indicating that these opposite responses were mediated by BK B2 receptors (26). Responses to BK were not altered by sodium meclofenamate or l-NAME, and the increase in pulmonary arterial pressure was attenuated by phentolamine, suggesting a role for α-adrenergic receptors in mediating the pulmonary pressor response. The observation that responses to BK were not inhibited by sodium meclofenamate, l-NAME, or ODQ suggests that the release of cyclooxygenase products or the activation of sGC by NO was not involved in mediating or modulating responses to the peptide.
Treatment with ODQ reduced responses to NO donors without altering vasodilator responses to NO-independent agents. The inhibition of sGC by ODQ is induced by oxidation of the heme iron on the enzyme reducing the sensitivity to NO and increasing responses to sGC activator agents (35, 43, 52). The present data are consistent with previous results and show that responses to a low or subthreshold dose of the sGC activator BAY 60-2770 are markedly enhanced by ODQ, suggesting that the inactivated or oxidized enzyme is stimulated by a subthreshold dose sGC activator (30, 35). The enhanced pressor response to angiotensin II in the pulmonary vascular bed observed after treatment with ODQ was not antagonized by apocyanin, suggesting that acute responses were not dependent on NADPH oxidase activation and may involve an effect of the peptide on NO formation, which is counterregulatory and can be observed when NO-sGC-cGMP signaling is inhibited. These data suggest that the acute pressor response to angiotensin II is not modulated by the activation of NADPH oxidase (15). Although it has been reported that ODQ can oxidize other heme-containing enzymes, the observation that methemoglobin levels were not significantly increased suggests that off-target effects were minimal in the present experiments.
Treatment with ODQ had no significant effect on baseline pulmonary and systemic arterial pressures; are consistent with previous studies in the fetal lamb, ovine fetus, and rat knockout models; and are consistent with the normal systemic arterial pressure in cGKI knockout mice (5, 35, 42). A transient elevation in pulmonary and systemic arterial pressure occurred during the infusion; however, these effects were short-lived and were not present after 10–15 min. In contrast, the intravenous administration of 50 mg/kg iv l-NAME produced elevations of systemic and pulmonary arterial pressure and decreases in cardiac output that persist for several hours.
The observation that systemic arterial pressure was not different in wild-type and cGKI knockout mice is supported by the present data with ODQ in the rat (28). The lack of effect of ODQ on baseline pressure may be explained in part by the observation that ODQ had no effect on basal sGC activity but inhibited the activation of the enzyme by NO (52).
It has been reported that ACh-induced vasodilation is mediated by hyperpolarization, and it has been suggested that TRPV4 channels are involved in mediating the endothelium-dependent vasodilator response to the muscarinic agonist (10, 40, 51).
Recent studies support the hypothesis that TRPV4 activation plays an important role in mediating agonist-induced vasodilation (50). Studies in TRPV4−/− mice show that cholinergic agonist-induced vasodilator responses in isolated resistance arteries and decreases in systemic arterial pressure in response to intravenous injections of ACh were attenuated (10, 51). In contrast, it has been reported that vasodilation in response to ACh in isolated carotid arteries from TRPV4−/− mice was not attenuated compared with responses in wild-type mice (16). In addition vasorelaxant responses to ACh were not attenuated by TRPV4 antagonists in isolated PA from the rat (46). In the present study, the hypothesis that vasodilator responses to ACh are mediated by activation of TRPV4 channels was investigated in experiments with the TRPV4 antagonist GSK-2193874. The TRPV4 antagonist reduced vasodilator response to the TRPV4 agonist GSK-1016790A but had no significant effect on vasodilator responses to ACh. These data indicate that decreases in pulmonary and systemic arterial pressures in response to intravenous ACh are not mediated by the activation of TRPV4 channels in the rat and are consistent with results in isolated vessel studies (16, 46). The reason for the difference in results in regard to the role of TRPV4 in mediating cholinergic vasodilator responses is uncertain.
Recent studies have shown a cGMP-independent mechanism for activating PKG1α that involves enzyme dimerization (38). It is possible that cGMP-independent activation of PKG may be involved in mediating decreases in pulmonary and systemic arterial pressure in response to ACh in the present study in the rat.
In summary, results of the present study show that ODQ inhibits the vasodilator response to NO donors without altering responses to vasodilator agents that act by NO-independent mechanisms in the rat. ODQ did not inhibit vasoconstrictor responses or increase methemoglobin blood levels, suggesting that off target effects were minimal. ODQ did not inhibit responses to ACh and BK, suggesting the vasodilator responses are not mediated by the activation of sGC and are similar to results with ACh in cGKi knockout mice (28). Although it has been hypothesized that TRPV4 channels mediate vasodilator responses to ACh, responses to the muscarinic agonist in isolated resistance vessel were not altered by the TRPV4 antagonist (40, 46).
The present results are consistent with the hypothesis that vasodilator responses to ACh are mediated by membrane hyperpolarization. However, the activation of sGC or TRPV4 channels is not involved in mediating vasodilator responses to ACh in the pulmonary and systemic vascular beds in the rat.
Limitations
In regard to limitations of the present in vivo study in the rat, the observation that decreases in pulmonary and systemic arterial pressures in response to intravenous injection of ACh are not inhibited by ODQ or the TRPV4 antagonist GSK-2192874 do not exclude a role for sGC activation or TRPV4 channels in mediating vasodilator responses to ACh in some vascular segments or beds in the rat or in vessel segments isolated from large and small arteries. The present results do indicate, however, that the overall physiological response to ACh, which is a decrease in pulmonary and systemic arterial pressure in the rat in vivo in response to systemic (iv) injection of ACh, is not mediated in large part by activation of sGC or TRPV4 channels. It is quite possible that vasodilator responses in some vascular segments or domains in the intact rat are inhibited. However, this contribution is difficult to evaluate when overall decreases in systemic or pulmonary arterial pressures are measured.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-62000 and HL-77421.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: E.A.P. and P.J.K. conception and design of research; E.A.P., M.K., and S.-K.C. performed experiments; E.A.P., M.K., S.-K.C., K.M., and P.J.K. analyzed data; E.A.P., S.-K.C., K.M., B.D.N., and P.J.K. interpreted results of experiments; E.A.P., M.K., and B.D.N. prepared figures; E.A.P., B.D.N., and P.J.K. drafted manuscript; E.A.P., K.M., B.D.N., A.L.H., and P.J.K. edited and revised manuscript; E.A.P., M.K., S.-K.C., B.D.N., A.L.H., and P.J.K. approved final version of manuscript.
REFERENCES
- 1.Amin AH, Abd Elmageed ZY, Nair D, Partyka MI, Kadowitz PJ, Belmadani S, Matrougui K. Modified multipotent stromal cells with epidermal growth factor restore vasculogenesis and blood flow in ischemic hind-limb of type II diabetic mice. Lab Invest 90: 985–996, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA 74: 3203–3207, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellan JA, Minkes RK, McNamara DB, Kadowitz PJ. N omega-nitro-l-arginine selectively inhibits vasodilator responses to acetylcholine and bradykinin in cats. Am J Physiol Heart Circ Physiol 260: H1025–H1029, 1991 [DOI] [PubMed] [Google Scholar]
- 4.Brandes RP, Schmitz-Winnenthal FH, Feletou M, Godecke A, Huang PL, Vanhoutte PM, Fleming I, Busse R. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc Natl Acad Sci USA 97: 9747–9752, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chester M, Tourneux P, Seedorf G, Grover TR, Gien J, Abman SH. Cinaciguat, a soluble guanylate cyclase activator, causes potent and sustained pulmonary vasodilation in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 297: L318–L325, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi SK, Galan M, Partyka M, Trebak M, Belmadani S, Matrougui K. Chronic inhibition of epidermal growth factor receptor tyrosine kinase and extracellular signal-regulated kinases 1 and 2 (ERK1/2) augments vascular response to limb ischemia in type 2 diabetic mice. Am J Pathol 180: 410–418, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craven PA, DeRubertis FR. Requirement for heme in the activation of purified guanylate cyclase by nitric oxide. Biochim Biophys Acta 745: 310–321, 1983 [DOI] [PubMed] [Google Scholar]
- 8.Dabisch PA, Liles JT, Baber SR, Golwala NH, Murthy SN, Kadowitz PJ. Analysis of l-NAME-dependent and -resistant responses to acetylcholine in the rat. Am J Physiol Heart Circ Physiol 294: H688–H698, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Dierks EA, Burstyn JN. The deactivation of soluble guanylyl cyclase by redox-active agents. Arch Biochem Biophys 351: 1–7, 1998 [DOI] [PubMed] [Google Scholar]
- 10.Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, Brayden JE. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol 297: H1096–H1102, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galle J, Mulsch A, Busse R, Bassenge E. Effects of native and oxidized low density lipoproteins on formation and inactivation of endothelium-derived relaxing factor. Arterioscler Thromb 11: 198–203, 1991 [DOI] [PubMed] [Google Scholar]
- 12.Garland JG, McPherson GA. Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. Br J Pharmacol 105: 429–435, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerova M. Acetylcholine and bradykinin induce paradoxically amplified hypotensive response in hypertensive NO-deficient rats. Physiol Res 48: 249–257, 1999 [PubMed] [Google Scholar]
- 14.Gerova M, Kristek F. Efficiency of NO donors in substituting impaired endogenous NO production: a functional and morphological study. Physiol Res 50: 165–173, 2001 [PubMed] [Google Scholar]
- 15.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74: 1141–1148, 1994 [DOI] [PubMed] [Google Scholar]
- 16.Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, Liedtke W, Hoyer J, Kohler R. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PloS one 2: e827, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoepfl B, Rodenwaldt B, Pohl U, De Wit C. EDHF, but not NO or prostaglandins, is critical to evoke a conducted dilation upon ACh in hamster arterioles. Am J Physiol Heart Circ Physiol 283: H996–H1004, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta JA, Falck JR, Shesely EG, Koller A, Kaley G. EDHF mediates flow-induced dilation in skeletal muscle arterioles of female eNOS-KO mice. Am J Physiol Heart Circ Physiol 280: H2462–H2469, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Huang A, Sun D, Smith CJ, Connetta JA, Shesely EG, Koller A, Kaley G. In eNOS knockout mice skeletal muscle arteriolar dilation to acetylcholine is mediated by EDHF. Am J Physiol Heart Circ Physiol 278: H762–H768, 2000 [DOI] [PubMed] [Google Scholar]
- 20.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377: 239–242, 1995 [DOI] [PubMed] [Google Scholar]
- 21.Hyman AL, De Witt BJ, Gumusel B, Hao Q, Kadowitz PJ, Lippton HL. l-NAME enhances responses to atrial natriuretic peptide in the pulmonary vascular bed of the cat. J Appl Physiol 90: 2101–2108, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Hyman AL, Hao Q, Tower A, Kadowitz PJ, Champion HC, Gumusel B, Lippton H. Novel catheterization technique for the in vivo measurement of pulmonary vascular responses in rats. Am J Physiol Heart Circ Physiol 274: H1218–H1229, 1998 [DOI] [PubMed] [Google Scholar]
- 23.Hyman AL, Kadowitz PJ. Influence of tone on responses to acetylcholine in the rabbit pulmonary vascular bed. J Appl Physiol 67: 1388–1394, 1989 [DOI] [PubMed] [Google Scholar]
- 24.Hyman AL, Kadowitz PJ. Tone-dependent responses to acetylcholine in the feline pulmonary vascular bed. J Appl Physiol 64: 2002–2009, 1988 [DOI] [PubMed] [Google Scholar]
- 25.Ignarro LJ, Degnan JN, Baricos WH, Kadowitz PJ, Wolin MS. Activation of purified guanylate cyclase by nitric oxide requires heme. Comparison of heme-deficient, heme-reconstituted and heme-containing forms of soluble enzyme from bovine lung. Biochim Biophys Acta 718: 49–59, 1982 [DOI] [PubMed] [Google Scholar]
- 26.Jansakul C, Tachanaparuksa K, Mulvany MJ, Sukpondma Y. Relaxant mechanisms of 3,5,7,3′,4′-pentamethoxyflavone on isolated human cavernosum. Eur J Pharmacol 691: 235–244, 2012 [DOI] [PubMed] [Google Scholar]
- 27.Kassan M, Galan M, Partyka M, Saifudeen Z, Henrion D, Trebak M, Matrougui K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arteriosclerosis Throm Vasc Biol 32: 1652–1661, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koeppen M, Feil R, Siegl D, Feil S, Hofmann F, Pohl U, de Wit C. cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension 44: 952–955, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Kots AY, Martin E, Sharina IG, Murad F. A short history of cGMP, guanylyl cyclases, and cGMP-dependent protein kinases. Handb Exp Pharmacol 1–14, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lasker GF, Pankey EA, Frink TJ, Zeitzer JR, Walter KA, Kadowitz PJ. The sGC activator BAY 60-2770 has potent erectile activity in the rat. Am J Physiol Heart Circ Physiol 304: H1670–H1679, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McMahon TJ, Hood JS, Bellan JA, Kadowitz PJ. N-omega-nitro-l-arginine methyl ester selectively inhibits pulmonary vasodilator responses to acetylcholine and bradykinin. J Appl Physiol 71: 2026–2031, 1991 [DOI] [PubMed] [Google Scholar]
- 32.McNamara DB, Kadowitz PJ, Hyman AL, Ignarro LJ. Adenosine 3′,5′-monophosphate formation by preparations of rat liver soluble guanylate cyclase activated with nitric oxide, nitrosyl ferroheme, S-nitrosothiols, and other nitroso compounds. Can J Physiol Pharmacol 58: 1446–1456, 1980 [DOI] [PubMed] [Google Scholar]
- 33.Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res 41: 19–26, 1977 [DOI] [PubMed] [Google Scholar]
- 34.Ott IM, Alter ML, von Websky K, Kretschmer A, Tsuprykov O, Sharkovska Y, Krause-Relle K, Raila J, Henze A, Stasch JP, Hocher B. Effects of stimulation of soluble guanylate cyclase on diabetic nephropathy in diabetic eNOS knockout mice on top of angiotensin II receptor blockade. PloS one 7: e42623, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pankey EA, Bhartiya M, Badejo AM, Jr, Haider U, Stasch JP, Murthy SN, Nossaman BD, Kadowitz PJ. Pulmonary and systemic vasodilator responses to the soluble guanylyl cyclase activator, BAY 60-2770, are not dependent on endogenous nitric oxide or reduced heme. Am J Physiol Heart Circ Physiol 300: H792–H802, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pankey EA, Thammasiboon S, Lasker GF, Baber S, Lasky JA, Kadowitz PJ. Imatinib attenuates monocrotaline pulmonary hypertension and has potent vasodilator activity in pulmonary and systemic vascular beds in the rat. Am J Physiol Heart Circ Physiol 305: H1288–H1296, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pankey EA, Zsombok A, Lasker GF, Kadowitz PJ. Analysis of the responses to the Trpv4 agonist Gsk1016790a in the pulmonary vascular bed of the intact chest rat. Am J Physiol Heart Circ Physiol 306: H33–H40, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel D, Kandhi S, Kelly M, Neo BH, Wolin MS. Dehydroepiandrosterone promotes pulmonary artery relaxation by NADPH oxidation-elicited subunit dimerization of protein kinase G-1α. Am J Physiol Lung Cell Mol Physiol 306: L383–L391, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt K, Graier WF, Kostner GM, Mayer B, Bohme E, Kukovetz WR. Oxidized low-density lipoprotein antagonizes the activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor but does not interfere with its biosynthesis. Cell Signal 3: 361–367, 1991 [DOI] [PubMed] [Google Scholar]
- 40.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336: 597–601, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 123: 2263–2273, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stasch JP, Schmidt P, Alonso-Alija C, Apeler H, Dembowsky K, Haerter M, Heil M, Minuth T, Perzborn E, Pleiss U, Schramm M, Schroeder W, Schroder H, Stahl E, Steinke W, Wunder F. NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. Br J Pharmacol 136: 773–783, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, HSA, Meurer S, Deile M, Taye A, Knorr A, Lapp H, Muller H, Turgay Y, Rothkegel C, Tersteegen A, Kemp-Harper B, Muller-Esterl W, Schmidt HH. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest 116: 2552–2561, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steeds RP, Thompson JS, Channer KS, Morice AH. Response of normoxic pulmonary arteries of the rat in the resting and contracted state to NO synthase blockade. Br J Pharmacol 122: 99–102, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stone JR, Marletta MA. Spectral and kinetic studies on the activation of soluble guanylate cyclase by nitric oxide. Biochemistry 35: 1093–1099, 1996 [DOI] [PubMed] [Google Scholar]
- 46.Sukumaran SV, Singh TU, Parida S, Narasimha Reddy CE, Thangamalai R, Kandasamy K, Singh V, Mishra SK. TRPV4 channel activation leads to endothelium-dependent relaxation mediated by nitric oxide and endothelium-derived hyperpolarizing factor in rat pulmonary artery. Pharmacol Res 78C: 18–27, 2013 [DOI] [PubMed] [Google Scholar]
- 47.Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, Gordon E, Lozinskaya I, Elefante L, Qin P, Matasic DS, James C, Tunstead J, Donovan B, Kallal L, Waszkiewicz A, Vaidya K, Davenport EA, Larkin J, Burgert M, Casillas LN, Marquis RW, Ye G, Eidam HS, Goodman KB, Toomey JR, Roethke TJ, Jucker BM, Schnackenberg CG, Townsley MI, Lepore JJ, Willette RN. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 4: 148–159, 2012 [DOI] [PubMed] [Google Scholar]
- 48.Wedel B, Harteneck C, Foerster J, Friebe A, Schultz G, Koesling D. Functional domains of soluble guanylyl cyclase. J Biol Chem 270: 24871–24875, 1995 [DOI] [PubMed] [Google Scholar]
- 49.Wedel B, Humbert P, Harteneck C, Foerster J, Malkewitz J, Bohme E, Schultz G, Koesling D. Mutation of His-105 in the beta 1 subunit yields a nitric oxide-insensitive form of soluble guanylyl cyclase. Proc Natl Acad Sci USA 91: 2592–2596, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang DX, Gutterman DD. Transient receptor potential channel activation and endothelium-dependent dilation in the systemic circulation. J Cardiovasc Pharmacol 57: 133–139, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M, Gutterman DD. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53: 532–538, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao Y, Brandish PE, DiValentin M, Schelvis JP, Babcock GT, Marletta MA. Inhibition of soluble guanylate cyclase by ODQ. Biochemistry 39: 10848–10854, 2000 [DOI] [PubMed] [Google Scholar]