

Abstract
Defective colonic Na+ and Cl− absorption is a feature of active ulcerative colitis (UC), but little is known about changes in colonic K+ transport. We therefore investigated colonic K+ transport in a rat model of dextran sulfate-induced colitis. Colitis was induced in rat distal colon using 5% dextran sulfate sodium (DSS). Short-circuit current (Isc, indicating electrogenic ion transport) and 86Rb (K+ surrogate) fluxes were measured in colonic mucosa mounted in Ussing chambers under voltage-clamp conditions in the presence of mucosal orthovanadate (a P-type ATPase inhibitor). Serum aldosterone was measured by immunoassay. Control animals exhibited zero net K+ flux. By contrast, DSS-treated animals exhibited active K+ secretion, which was inhibited by 98, 76, and 22% by Ba2+ (nonspecific K+ channel blocker), iberiotoxin (IbTX; BK channel blocker), and TRAM-34 (IK channel blocker), respectively. Apical BK channel α-subunit mRNA abundance and protein expression, and serum aldosterone levels in DSS-treated animals, were enhanced 6-, 3-, and 6-fold respectively, compared with controls. Increasing intracellular Ca2+ with carbachol (CCH), or intracellular cAMP with forskolin (FSK), stimulated both active Cl− secretion and active K+ secretion in controls but had no or little effect in DSS-treated animals. In DSS-induced colitis, active K+ secretion involves upregulation of apical BK channel expression, which may be aldosterone-dependent, whereas Cl− secretion is diminished. Since similar ion transport abnormalities occur in patients with UC, diarrhea in this disease may reflect increased colonic K+ secretion (rather than increased Cl− secretion), as well as defective Na+ and Cl− absorption.
Keywords: aldosterone, chloride secretion, colitis, potassium channels, potassium secretion
dysregulation of ion transport across the inflamed colonic epithelium is a major contributor to the pathogenesis of diarrhea in ulcerative colitis (UC; Refs. 13, 27). The main ion transport abnormalities are decreases in Na+ and Cl− absorption (rather than increased Cl− secretion), which results in a decrease in net water absorption (27). The dominant transport defects are the virtual disappearance of electrogenic Na+ absorption (reflecting decreased apical Na+ entry via amiloride-sensitive Na+ channels and decreased basolateral Na+ extrusion mediated by Na+-K+-ATPase) and impaired electroneutral NaCl absorption (reflecting downregulation of both apical Na+:H+ exchange isoform-3 [NHE3] and apical Cl−:HCO3− exchange; Refs. 2, 10, 34, 35, 39).
In addition, some patients with severe UC develop hypokalemia, which may reflect increased fecal K+ losses due, at least in part, to enhanced K+ secretion across the inflamed colonic epithelium (28). However, the mechanism(s) responsible for increased colonic K+ secretion in UC are unclear. Apical BK channel expression is increased in both acute and quiescent UC (28), but the precise role that enhanced BK channel expression plays in increased K+ secretion is unknown, even though BMS (a BK channel opener) and DC-EBIO [an opener of intermediate conductance K+ (IK) channels] induce BK and IK channel-mediated K+ secretion respectively, in control rat distal colon (21, 29). In addition, in control rat distal colon, cAMP activates BK channel-mediated K+ secretion, while aldosterone increases BK channel expression and induces BK channel-mediated K+ secretion (29, 31). The aim of this study was to determine whether the expression of BK and/or IK channels was increased in DSS-induced colitis in rat distal colon and, if so, whether this resulted in enhanced colonic K+ secretion. Our results indicate that in this experimental model of chronic colitis, increased colonic K+ secretion reflects upregulation of apical BK channel expression, possibly as a result of increased serum aldosterone levels.
METHODS AND MATERIALS
Preparation of animals.
Chronic active UC was induced in male Sprague-Dawley rats (Charles River, Raleigh, NC) weighing 126–150 g by feeding 5% DSS in water ad libitum for 5 alternate wk. Both control and DSS-treated rats were given standard rat chow. Other rats were fed a Na+-free diet for 7 days to induce secondary hyperaldosteronism (31). Control and dietary Na+-depleted (“aldo-treated”) rats were given water ad libitum. The experimental protocols were approved by the West Virginia University Institutional Animal Care and Use Committee.
Histology.
Distal colons removed from anaesthetized control and DSS-treated rats were washed with ice-cold saline and fixed overnight in 10% neutral formalin. Sections (5 μm) cut from paraffin-embedded tissues were mounted on glass slides and stained with hematoxylin and eosin for histological evaluation. Coded slides were analyzed in blinded fashion by two investigators. The features considered were reactive epithelial changes, acute inflammatory changes within the lamina propria, acute cryptitis, crypt abscesses, crypt distortion, acute ulcer, healing ulcer, lymphoid aggregates, and transmural inflammation.
Cytokine assay.
Proinflammatory cytokine levels in distal colonic epithelial cells from control and DSS-treated rats were quantified using a rat cytokine/chemokine panel (Milliplex MAP kit; Millipore). Mucosal sheets were freed of underlying muscle, manually homogenized using tight-fit Teflon-homogenizer andcentrifuged (15 min at 2,000 g), and the resultant supernatants were used for cytokine assays. Filter plates dampened with assay buffer, were vacuum filtered before addition of either standard, control, or samples to appropriate wells. Premixed beads were added to each well, which were incubated overnight while shaking at 4°C. Following three washes, detection antibodies (25 μl) were added to each well and incubated for 2 h at room temperature. The wells were then treated with streptavidin-phycoerythrin (25 μl/well) and incubated for 30 min at room temperature. After three washes, beads resuspended in Milliplex sheath fluid (150 μl) were measured using the BioPlex Suspension System. Mucosal scrapings from four different control and DSS-treated rat distal colons were individually homogenized to prepare supernatants for cytokine assays. Cytokine levels were expressed as picograms per milligrams of protein. Protein was measured as previously described (18).
Aldosterone assays.
Serum aldosterone levels were measured using the aldosterone enzyme immunoassay kit, according to the manufacturer's protocol (Enzo Life Sciences). Blood was collected directly from heart of six different anesthetized normal and UC rats and allowed to coagulate. Serum was collected by brief centrifugation. Triplicate assays were performed on all serum samples, and aldosterone levels were expressed as picomoles per milliliters of serum.
Nitric oxide assays.
Distal colons obtained from four different control and DSS-treated rats were used for total nitric oxided (NO) measurements. Each distal colon was divided into 10 segments, which were homogenized in Ringer solution and centrifuged (15 min at 2,000 g), and NO was measured in the resultant supernatants using a total NO detection kit, according to the manufacturer's instruction (Enzo Life Sciences). In brief, NO that interacts covalently with target molecules (proteins) were measured as nitrate (NO3). Thus the total NO detection kit measured protein NO3 as an indicator of NO. This assay included conversion of NO3 to nitrite (NO2) by nitrate reductase, followed by the colorimetric detection of NO2 as a colored azo dye product at 540 nM. The total NO levels were expressed as picomoles per milligrams of protein.
Luminal stool and water content measurement.
The luminal stool and water contents were measured in nonfasted control and DSS-treated rat distal colon, which was that segment between the stripped proximal colon and the rectum. Before the distal colon was removed from anesthetized animals, both ends of the segment were tied with thread to prevent loss of the luminal contents. By gently squeezing along the colonic segment, the luminal contents were emptied into a weighing boat and weighed immediately (i.e., wet stool weight). The lumen of control colon contained hard stool pellets, whereas the lumen of DSS-treated colon contained soft stools with mucus and traces of blood. Dry stool weight was measured after drying for 48 h at 60°C. The luminal water content was calculated as the difference between the wet stool weight and the dry stool weight.
Ussing chamber studies.
Distal colons excised from euthanized rats were flushed with ice-cold saline and opened along their mesenteric borders, and mucosal sheets were gently separated from the serosal muscular layers. Two distal (1 cm proximal to rectum) segments obtained from each animal were mounted in snap wells with an opening of 1.12 cm2. Snap wells placed in sliders were inserted into EasyMount Ussing chambers (Physiological Instruments, San Diego, CA) and bathed both sides with equal volumes (5.0 ml) of Ringer solution containing the following (in mM): 115 NaCl, 25 NaHCO3, 2.4 K2HPO4, 0.4 KH2PO4, 1.2 CaCl2, 1.2 MgCl2 and 10 glucose (pH 7.4), maintained at 37°C, and gassed with 5% CO2-95% O2. 86Rb (K+ surrogate; PerkinElmer, Billerica, MA) fluxes, short-circuit current (Isc; a measure of net electrogenic ion transport) and transepithelial conductance (G) were measured across the mucosal sheets under voltage-clamp conditions at 0 mV as previously described (21). Thus, by removing the transepithelial electrical potential difference present under open-circuit (mimicking in vivo) conditions, the net flux of the ionic species under study reflected its active (i.e., potential-independent) transport. The Isc and G were measured every 20 s using an automated multichannel voltage/current-clamp instrument (Physiological Instruments). Positive Isc represented either electrogenic Na+ absorption or electrogenic Cl− secretion, whereas negative Isc represented electrogenic K+ secretion.
For K+ flux studies, a trace of 86RbCl (1 μCi/chamber) was added to either the mucosal (m) or serosal (s) bath solutions. After a 45-min equilibration period, mucosa-to-serosa (m-s) and serosa-to-mucosa (s-m) K+ fluxes were measured under voltage-clamp conditions. Net fluxes were calculated from the difference between the m-s and s-m fluxes in tissue pairs that were matched based on differences in basal G values of <10%. Positive and negative values represented active absorption and active secretion, respectively. Epithelial sheets from control and DSS-treated rat distal colon exhibited basal G values of 5.5 ± 0.7 and 7.8 ± 0.8 mS, respectively, while the value in distal colon from aldo-treated rats was 29.3 ± 3.1 mS. Forskolin (FSK) significantly increased G in control distal colon from 5.5 ± 0.7 to 18.2 ± 1.6 mS but had no effect on G in either DSS-treated or aldo-treated distal colon (data not shown). Distal colonic epithelial sheets from control, DSS-treated, and aldo-treated animals exhibited stable values of G throughout the flux period (up to 60 min), indicating sustained tissue viability.
K+ fluxes were measured over 15-min periods. At the end of each period, 0.5-ml samples were withdrawn from the bath opposite to the isotope-containing side. Following sample removal, 0.5 ml of regular Ringer were added to maintain bath volume. Basal fluxes were measured immediately following an equilibrium period. Following the basal flux period, either carbachol (CCH; 100 μM), a cholinergic agonist that transiently increases cellular free Ca2+, or FSK (10 μM), an adenylate cyclase inhibitor that increases cellular cAMP levels, was added to the serosal bath. In additional experiments, the effects of mucosal Ba2+ (a nonspecific K+ channel blocker), TRAM-34 (a specific IK channel blocker; 10 μM), and iberiotoxin (IbTX; a BK channel blocker; 100 nM) were examined on the changes in Isc, G, and K+ fluxes induced by CCH and FSK. The K+ fluxes are presented as microequivalents per hour per centimeter squared, while Isc are presented as microamperes per centimeter squared. G values are presented as mSiemens (mS).
Western blots.
Western blot analyses were performed on epithelial cell homogenates of distal colon using anti-Kcnn4-abc (4), BK α-subunit (MaxiKα; Santa Cruz Biotechnology, Hercules, CA), and actin (Santa Cruz Biotechnology) antibodies, as described previously (31). Epithelial cells suspended (1:20) in ice-cold lysis buffer [50 mM Tris, pH 8.0, 0.5% SDS, 1 mM PMSF, 4 μg/ml pepstatin A, and one tablet of complete protease inhibitor/50-ml solution (Roche Applied Science, Indianapolis, IN)] were homogenized with a tight fit Teflon homogenizer. The homogenate was centrifuged (15 min at 2,000 g), and 16-μl aliquots of supernatant were mixed with equal volumes of Laemmli buffer and heated at 95°C for 5 min. Heated aliquots were immediately placed in liquid nitrogen and stored at −80°C. Frozen samples were heated at 40°C for 1–2 min, and 10-μl samples (20 μg protein) resolved on 14% polyacrylamide gels were transferred onto nitrocellulose membranes. Blots were incubated with primary antibody [Kcnma1α (1:400); anti-Kcnn4-abc (1:3,000); and β-actin (1:2,500)] and then with horseradish peroxidase-conjugated goat anti-rabbit IgG, and immune complexes were detected using enhanced chemiluminescence (GE Healthcare, Buckinghamshire, UK). The stripped blots were processed with anti-actin antibody and horseradish peroxidase-conjugated donkey anti-mouse IgG. Arbitrary units of BKα and IK (Kcnn4b and Kcnn4c) proteins normalized to actin were quantitated using Personal Densitometer SI (Molecular Dynamics).
Real-time-PCR.
Total RNA for real-time-PCR (RT-PCR) analysis was prepared from colonocytes from control and DSS-treated rat distal colon using TRIzol (Invitrogen, Carlsbad, CA). RT-PCR was performed by a two-step method, as described previously (4). In brief, first-strand cDNA was synthesized from total RNA using SuperScript III and random hexamers (Invitrogen). The first-strand cDNA template (5 ng) and TaqMan universal PCR master mix, together with the TaqMan gene expression assay, was used to amplify rat BK α-subunit according to the manufacturer's instructions (Applied Biosystems, Foster City, CA). Custom designed primers used to amplify rat IK (Kcnn4b or Kcnn4c)-specific fragments were as follows: Kcnn4b (sense 5′-GGCCACATAGCTGCCTGTTA; antisense 5′-TCCTTGAGCTCAGTCCTTCG-3′) primers (500 nM each) and 100 nM TaqMan probes (5′ FAM-TCAGGACCCACAGAAGAATCAGGCT-TAMRA 3′); and Kcnn4c (sense 5′-CTGGGTTGCAAGGAGGTC-3′; antisense 5′-CATACCAGCAGCTCCAGCA-3′) primers (500 nM each) and 100 nM TaqMan probes (5′ FAM-CTGTTCATGACTGACAACGGGCTCC-TAMRA 3′). Rat β-actin amplified using TaqMan gene expression assay served as an internal control. Threshold cycle (Ct) values of the BKα-subunit, Kcnn4b, and Kcnn4c transcripts were normalized to the internal control. Differential expression of BKα and Kcnn4b/c was calculated according to the 2−ΔΔCt method, as previously described (17).
Statistics.
Results represent means ± SE of six tissue pairs from six rats. Statistical analyses were performed using unpaired or paired Students t-test or Bonferroni's one-way ANOVA post hoc test using Originpro 8.0 (OriginLab, Northampton, MA). P < 0.05 was considered to be statistically significant.
Stock solutions.
Stock solutions were DSS (5% in water; MP Biomedicals), amiloride (20 mM in methanol, Sigma-Aldrich), TRAM-34 (50 mM in DMSO; Tocris Bioscience, Ellisville, MO), IbTX (100 μM in Ringer solution; Sigma-Aldrich), and charybdotoxin (20 μM in Ringer solution; Tocris Bioscience, Ellisville, MO). All other molecular grade chemicals used were purchased from Sigma-Aldrich.
RESULTS
Colonic histology and proinflammatory cytokines.
Control distal colon and the inflammatory response in DSS-treated distal colon were evaluated histologically and by assay of proinflammatory cytokines (Fig. 1 and Table 1). Control distal colon exhibited straight crypts and an intact surface epithelium, without any inflammatory cells in the lamina propria (Fig. 1A). By contrast, in DSS-treated distal colon, there was loss and distortion of the crypts (Fig. 1B), crypt abscess (Fig. 1, C and D), and an excess of inflammatory cells within the lamina propria (Fig. 1D), these being the typical histological features of human UC (9). Levels of the proinflammatory cytokines TNF-α, IL-1β, IL-6, and INF-γ were significantly increased in epithelial cells from DSS-treated distal colon (Table 1). These findings are similar to those previously reported in the mouse model of chronic UC induced by DSS treatment (1) and establish our experimental model of rat colitis as one suitable for ion transport studies in this disease.
Fig. 1.

Hematoxylin and eosin staining of distal colon from control and dextran sulfate sodium (DSS)-treated rats. A: in controls, crypts were straight and the surface epithelium was intact (×20). In DSS-treated rats, there was marked inflammation, ulceration, and loss of crypts (arrow; B), together with distorted crypts (CD) and crypt abscesses (CA; ×20; C), and CA and acute inflammatory cells in the lamina propria (×40; D).
Table 1.
Proinflammatory cytokine levels in control and DSS-treated rat distal colon
| Cytokine, pg/mg protein |
|||
|---|---|---|---|
| Cytokines | Control | DSS treated | P |
| TNF-α | 10.5 ± 0.01 | 17.7 ± 0.6 | <0.02 |
| IL-1β | 48.2 ± 2.3 | 106.8 ± 0.9 | <0.001 |
| IL-6 (GRO/KC) | 76.2 ± 1.4 | 178.9 ± 0.9 | <0.001 |
| INF-γ | 51.8 ± 2.3 | 175.8 ± 2.2 | <0.001 |
DSS, dextran sulfate sodium.
Luminal stool and water contents.
Bloody and often profuse diarrhea is the commonest symptom in patients with active UC (13, 27). To establish whether the luminal water content was increased in DSS-treated rat colon, the luminal stool and water contents in distal colon were measured. The wet weight of the luminal contents from DSS-treated distal colon was 2.2-fold greater than that from control distal colon (control vs DSS-treated colon: 0.88 ± 0.10 vs. 1.95 ± 0.18 g/colon; P < 0.001). By contrast, the dry weights of the luminal contents were identical in DSS-treated and control distal colon (Fig. 2B). The greater wet weight in DSS-treated distal colon reflected a 3.3-fold increase in the weight of luminal water in DSS-treated colon (1.47 ± 0.17 g/colon) compared with that in controls (0.45 ± 0.07 g/colon; P < 0.001; Fig. 2C). The increased luminal water content in DSS-treated rat distal colon is perhaps not surprising given that the animals develop diarrhea, and most likely reflects enhanced water secretion and/or inhibition of water absorption.
Fig. 2.

Luminal stool and water contents of control and DSS-treated rat distal colon. A: wet stool weight was measured immediately after luminal contents emptied from colon. B: dry stool weight was measured after 48 h of drying the wet stool in 60°C oven. C: weight of luminal water in each colon was calculated by subtracting dry stool weight from the corresponding wet stool weight. Data represent means ± SE values from 6 different animals in both groups. *P < 0.001, compared with control.
BK channel expression.
It has been suggested that enhanced apical BK channel expression in human UC may underlie increased colonic K+ secretion, resulting in excessive fecal K+ losses in some patients with severe disease (28). We therefore evaluated apical BK channel expression in DSS-treated distal colon by Western blot analyses. In keeping with previous studies in human UC (28), BK α-subunit protein expression was enhanced approximately threefold in plasma membranes from DSS-treated distal colon (Fig. 3, A and C). Since IK channels are also present in rat distal colon (4), Western blot analyses were performed using Kcnn4a-b antibody, which detects apical (37 kDa) and basolateral (40 kDa) IK channel proteins (4). As shown in Fig. 3, B and D, neither apical nor basolateral IK channel expression was altered in DSS-treated distal colon. In additional studies, acute colitis induced by just 1 wk of 5% DSS treatment had no effect on either BK α-subunit or IK protein expression in the distal colon (data not shown).
Fig. 3.
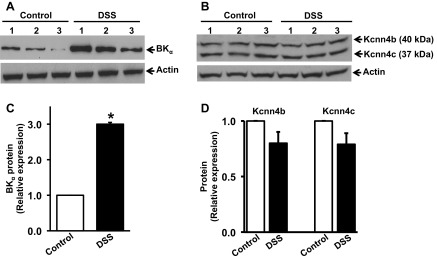
Large conductance K+ (BKα) and intermediate conductance K+ (IK; Kcnn4b and Kcnn4c) channel-specific protein expression in control and DSS-treated rat distal colon. A and B: Western blots of BKα and IK (Kcnn4b/c) channel proteins respectively, following which the blots were stripped and stained with β-actin antibody (see methods and materials). Each lane represents epithelial cell homogenate prepared from a single animal. C and D: relative expression of BKα and IK (Kcnn4b/c) channel proteins, respectively, from control (open bars) and DSS-treated rats (closed bars) were calculated from densitometry analyses of data in A and B, respectively. *P < 0.001, compared with control.
We next determined whether the increased BK α-subunit protein expression in DSS-treated distal colon occurred at a transcriptional or posttranscriptional level by measuring BK α-subunit mRNA abundance in colonic epithelial cells. RT-PCR analyses indicated that BK α-subunit mRNA abundance was enhanced approximately sixfold in DSS-treated distal colon (Fig. 4A), which suggests that increased BK channel expression in DSS-treated distal colon reflects regulation at the level of transcription. By contrast, in the case of IK channels, the abundances of both apical and basolateral IK channel-specific mRNAs were significantly increased in DSS-treated distal colon (Fig. 4, B and C), despite there being no changes in IK channel protein expression (Fig. 3, B and D).
Fig. 4.

Large conductance K+ (BKα) and intermediate conductance K+ (IK; Kcnn4b and Kcnn4c) channel-specific mRNA abundance in control and DSS-treated rat distal colon. BKα (A)-, Kcnn4b (B)-, and Kcnn4c (C)-specific mRNA from control (open bars) and DSS-treated (closed bars) rats quantified by RT-PCR. Data represent means ± SE values from 3 different animals in both groups. *P < 0.001, compared with control; £P < 0.001, compared with control.
Ion transport studies.
Ion flux studies were performed to identify whether Ca2+- or cAMP-induced Cl− secretion, which drive fluid secretion and/or K+ secretion, is altered in DSS-treated distal colon. We avoided epithelial sodium channel (ENaC)-related Isc and interference by apical H-K-ATPase-mediated active K+ absorption by measuring Isc and K+ fluxes in the mucosal presence of 0.1 mM amiloride (to inhibit ENaC) and 1 mM VO4 (to inhibit apical H-K-ATPase; Ref. 6). Control distal colon exhibited a positive Isc, reflecting a mucosa-negative transepithelial membrane potential under basal condition (Fig. 5A). However, in DSS-treated distal colon, there was a negative Isc, reflecting a mucosa-positive transepithelial membrane potential, and possibly K+ secretion (Fig. 5B). This observation differs from other studies, which showed a marked decrease in the mucosa-negative transepithelial membrane potential (but not a mucosa-positive transepithelial membrane potential) in a DSS-induced model of acute colitis in mouse colon (24). This disparity may reflect a species difference, a technical difference (stripped mucosa vs. whole intestine), or the different chronicities of the colitis.
Fig. 5.

Effect of carbachol (CCH) on Isc and net K+ fluxes in control and DSS-treated rat distal colon. Time course of short-circuit current (Isc; A and B) and net K+ fluxes (C and D) before and after serosal addition of CCH (50 μM) under voltage-clamp conditions in control and DSS-treated rat distal colon. Net K+ fluxes were measured in the absence (open bars) and presence (closed bars) of CCH. *P < 0.001, compared with basal value in controls; £P < 0.001 compared with basal value in controls.
Electrogenic Cl− secretion.
In control distal colon, serosal CCH (100 μM) that elicits a transient increase in intracellular free Ca2+ enhanced the Isc, reflecting stimulation of electrogenic Cl− secretion (Fig. 5A). In DSS-treated distal colon, CCH had no effect on Isc (Fig. 5B), and a similar failure of CCH to stimulate Cl− secretion has been reported in the mouse model of DSS-induced colitis (14, 24). The absence of CCH-induced Cl− secretion suggests that either the Ca2+-dependent Cl− channel (CaCC) and/or the Ca2+-dependent K+ channels that provide the driving force for electrogenic Cl− secretion, or the protein kinase C (PKC) signaling pathway, may be impaired in DSS-treated distal colon.
cAMP-dependent Cl− secretion was also examined in control and DSS-treated distal colon. In control distal colon, serosal FSK stimulated electrogenic Cl− secretion, as previously described (29), and subsequent addition of CCH further enhanced the Cl− secretory response (Fig. 6A). In DSS-treated distal colon, cAMP-induced Cl− secretion was reduced by 80%, while the addition of CCH had no further effect (Fig. 6B).
Fig. 6.

Effect of forskolin (FSK) on Isc and net K+ fluxes in control and DSS-treated rat distal colon. Time course of Isc (A and B) and net K+ fluxes (C and D) before and after serosal addition of FSK (10 μM), with subsequent serosal addition of CCH (50 μM), under voltage-clamp conditions, in control and DSS-treated rat distal colon. Net K+ fluxes were measured in the absence (open bars) and presence (closed bars) of FSK. *P < 0.001, compared with basal values in controls; £P < 0.001, compared with basal value in controls.
Electrogenic K+ secretion.
In parallel experiments, net K+ fluxes were measured in control and DSS-treated distal colon (Fig. 5, C and D). In controls, net K+ flux was zero under basal condition (in presence of mucosal VO4), while CCH stimulated active K+ secretion (basal 0.02 ± 0.04 μeq·h−1·cm−2 vs. CCH −0.12 ± 0.05 μeq·h−1·cm−2; P < 0.001; Fig. 5C). By contrast, in DSS-treated colon, there was significant active K+ secretion under basal conditions (−0.42 ± 0.06 μeq·h−1·cm−2) compared with controls (0.02 ± 0. μeq·h−1·cm−2; P < 0.001), although CCH did not elicit any additional K+ secretory response. As shown in Fig. 6C, FSK induced active K+ secretion in control distal colon, but subsequent addition of CCH did not alter FSK-induced K+ secretion (Fig. 6C), despite enhancing FSK-enhanced Cl− secretion (Fig. 6A). In DSS-treated distal colon, active K+ secretion was present under basal conditions, but neither cAMP nor CCH had any additional effect (Fig. 6D). These observations indicate that upregulation of BK channel expression in DSS-treated distal colon (Fig. 3, A and C) is associated with a substantial amount of active K+ secretion, which is insensitive to FSK and CCH.
A variety of K+ channel blockers were used to identify the different types of K+ channel that might be involved in the active K+ secretory process in DSS-treated distal colon. Active K+ secretion was inhibited completely by Ba2+ (a nonspecific K+ channel blocker), inhibited 25% by TRAM-34 (an IK channel blocker), and inhibited 80% by IbTX (a BK channel blocker; Fig. 7), which suggests that active K+ secretion in DSS-treated distal colon is mediated primarily via apical BK channels.
Fig. 7.

Effect of K+ channel blockers on net K+ secretion in DSS-treated rat distal colon. Net K+ secretion was measured in the absence (basal) and in the presence of either TRAM-34 (50 μM), iberiotoxin (IbTX; 100 nM), or Ba2+ (3 mM). *P < 0.05, compared with basal; £P < 0.001, compared with basal; $P < 0.05, compared with IbTX.
Roles of aldosterone and NO.
Aldosterone enhances apical BK channel expression and stimulates BK channel-mediated K+ secretion, while NO nitrosylates and activates BK channels expressed (15, 26, 31, 32). Based on previous studies, we envisaged that in DSS-treated rats impaired colonic Na+ absorption might result in raised serum aldosterone levels and that mucosal inflammation might increase NO concentrations in colonic epithelial cells (36, 38). We found that serum aldosterone and epithelial cell NO concentrations were enhanced by 5.8- and 2.7-fold, respectively, in DSS-treated rats compared with controls (Table 2). Increased serum aldosterone levels in DSS-treated rats were similar to those in rats fed a high-K+ diet but markedly lower than those in rats fed a Na+-free diet. These observations suggest that in DSS-treated rats aldosterone may have a role in the upregulation of BK channel expression and aldosterone and/or NO may be involved in the active K+ secretory process.
Table 2.
Serum aldosterone and epithelial cell nitric oxide concentrations in control and DSS-treated rat distal colon
| Control | DSS Treated | P | |
|---|---|---|---|
| Aldosterone, pg/ml serum | 236.2 ± 27.8 | 1,386.6 ± 208.2 | <0.001 |
| Total NO, pmol/mg protein | 239.8 ± 26.9 | 651.5 ± 85.2 | <0.001 |
NO, nitric oxide.
To further explore aldosterone-enhanced BK channel-mediated K+ secretion, the effects of FSK on Isc and K+ transport were studies in aldo-treated distal colon. As shown in Fig. 8A, before the addition of FSK, the negative Isc (−50.8 ± 5.6 μA/cm2) was consistent with K+ secretion and the addition of FSK reversed the Isc to a positive value (148 ± 11.8 μA/cm2), reflecting stimulation of electrogenic Cl− secretion. Although net K+ secretion was present under basal conditions in aldo-treated distal colon, as previously described (31), FSK did not further enhance K+ secretion (Fig. 8B), a situation similar to that seen in DSS-treated distal colon (Fig. 5D). Taken together, these observations suggest that aldosterone may have a regulatory role in BK channel-mediated K+ secretion in DSS-treated distal colon.
Fig. 8.

Effect of forskolin (FSK) on Isc and K+ fluxes in aldo-treated rat distal colon Time course of Isc (A) and K+ (B) fluxes before and after serosal addition of FSK (10 μM), under voltage-clamp conditions, in aldo-treated rat distal colon. Mucosal-to-serosa (m-s) and serosa-to-mucosa (s-m), and net K+ fluxes, were measured during 3 consecutive 15-min periods in the absence (white bars) and in the presence (grey and black bars) of FSK.
Given that previous studies have indicated that NO activates BK channels (26) and mucosal NO levels were significantly raised in DSS-treated distal colon (Table 2), we examined the effect of NO donor (DPTA-NONOate; a slow NO donor) on Isc and K+ fluxes in control distal colon. The addition of DPTA-NONOate initially stimulated Isc, which then declined to less than the basal value (Fig. 9A). The initial rise in Isc suggests that NO activated Cl− secretion, as reported previously (33), although in that study the effect of NO on Isc was followed for only 5 min. The decrease in Isc we observed at later time points after the addition of DPTA-NONOate is consistent with NO-stimulated active K+ secretion (Fig. 9B). We also studied the effect of cAMP on NO-stimulated Isc and K+ secretion, using 8-bromoadenosine 3′,5′-cyclic monophosphate (8BrcAMP) instead of FSK, since NO inhibits adenylate cyclase. 8BrcAMP in the continued presence of DPTA-NONOate did not affect Isc but further enhanced net K+ secretion. The observation that 8BrcAMP further enhanced K+ secretion already activated by NO suggests that NO and cAMP stimulate active K+ secretion by nitrosylation and phosphorylation, respectively.
Fig. 9.

Effect of nitric oxide (NO) on Isc and net K+ fluxes in control rat distal colon. Time course of Isc (A) and net K+ fluxes (B) under voltage-clamp conditions before and after the addition of NONOate (10 μM), with subsequent addition of 8-bromoadenosine 3′,5′-cyclic monophosphate (8BrcAMP; 100 μM), in control rat distal colon. Following basal Isc and K+ flux measurements, Isc and K+ fluxes were measured during 2 consecutive 15-min periods, first in presence of NONOate and then in the presence of NONOate/8BrcAMP. NONOate was added to both the mucosal and the serosal sides of the tissues, while 8BrcAMP was added only to serosal side. *P < 0.001, compared with basal; £P < 0.001, compared with NONOate.
DISCUSSION
In the present study, administration of 5% DSS to rats for 5 alternate wk resulted in chronic active inflammation and an increase in the luminal water content in the distal colon, which demonstrated similar clinical and histological features to those seen in human UC. Recent studies have revealed enhanced colonic BK channel expression in human UC (28), which could conceivably result in increased colonic K+ secretion (3, 12) and excessive fecal K+ losses in this disease. We found that DSS-treated rat distal colon exhibited a significant level of active K+ secretion, which was associated with a marked enhancement of BK channel expression. This conclusion is based on the following observations: 1) under basal conditions, a serosa-positive transepithelial potential difference was present in control distal colon, whereas a serosa-negative potential difference was present in DSS-treated distal colon, consistent with the induction of active K+ secretion in this experimental model of colitis (Figs. 5 and 6); 2) mucosal IbTX (a BK channel blocker) inhibited active K+ secretion in DSS-treated distal colon (Fig. 7); and 3) BK-specific mRNA abundance and protein expression in DSS-treated distal colon were increased six- and threefold, respectively, compared with the levels in control distal colon. Furthermore, BK channel activity stimulates the transcription factor NF-κB, which upregulates proinflammatory cytokine production (23). It has been suggested that immune dysregulation has a pathogenetic role in UC (30, 37), and in keeping with this notion, antibiotics inhibit the colonic inflammatory response induced by DSS, which is similar to that in patients with UC (22, 25).
Although the precise mechanism of BK channel upregulation in DSS-induced colitis is unclear, we found in DSS-treated animals that serum aldosterone and mucosal NO concentrations were increased 6.9- and 2.7-fold, respectively, compared with controls (Table 2). It is possible that hyperaldosteronism occurred secondary to excessive fecal Na+ losses in DSS-treated distal colon, since NHE3-mediated Na+ absorption is inhibited in this experimental model of colitis (Nanda Kumar NS, Binder HJ, Rajendran VM; unpublished observations), and NHE3-mediated electroneutral Na+ absorption and ENaC-mediated electrogenic Na+ absorption are both markedly impaired or absent in patients with active UC (2, 34). Hyperaldosteronism has been shown to enhance BK channel expression and induces BK channel-mediated active K+ secretion in mammalian colon (31, 32), and it is possible that the increases in BK channel expression and net K+ secretion we observed in DSS-treated distal colon reflected, at least in part, the increase in serum aldosterone concentration. Hyperaldosteronism secondary to feeding either a high-K+ diet or a Na+-free diet increase BK channel expression and induce net K+ secretion in rat distal colon (7, 20). However, it should be noted that dietary K+ enrichment results in a modest increase in serum aldosterone concentration, which is associated with net K+ secretion alone, whereas dietary Na+ depletion results in a much higher serum aldosterone concentration, which is associated with stimulation of ENaC-mediated electrogenic Na+ absorption, in addition to net K+ secretion (7, 11, 20, 31). Interestingly, serum aldosterone concentrations in the DSS-treated animals were similar to those reported in rats fed a high-K+ diet (20), and both cases exhibit an increase in K+ secretion without any change in ENaC-mediated Na+ absorption.
The total NO concentration in cells from DSS-treated distal colon was substantially higher than in control distal colon (Table 2), raising the possibility that NO may have a role in K+ secretion in this model of colitis. In addition to our observation that the NO donor NONOate stimulated K+ secretion in control distal colon (Fig. 9), NO has been shown to activate BK channels directly by nitrosylation, and indirectly by cGMP-dependent phosphorylation (8, 15, 19). NO also increases the number of active BK channels in colonic myocyte membranes (16). However, it should be noted that net K+ secretion in distal colon from aldo-treated animals (Fig. 8B) was 12-fold greater than net K+ secretion stimulated by NO in distal colon from control animals (Fig. 9B), which suggests that aldosterone and NO stimulate colonic K+ secretion by different mechanisms.
BK channels consist of a pore-forming α-subunit and a regulatory β-subunit. Two characteristically distinct splice variants of the BKα-subunit are present in mammalian colon, a 58 amino acid peptide that is present in the COOH terminus of the STREX variant being absent from the ZERO variant. cAMP inhibits the STREX variant but activates the ZERO variant. RT-PCR analyses and IbTX-inhibition of cAMP-stimulated K+ secretion have confirmed that ZERO transcripts encode apical BK channels in rat and human colon (unpublished observations) (5), and the BK α-subunit protein has been localized to the apical membrane of colonic epithelial cells using immunofluorescence (28, 32, 40). The addition of cAMP had no further stimulatory effect on aldosterone-induced K+ secretion (Fig. 8B), possibly because the BK channels were already maximally activated. However, cAMP elicited a significant increase in NO-induced K+ secretion (Fig. 9B), which most likely reflected the additive effect of phosphorylation and nitrosylation on BK channel activity. This leads us to speculate that in both human UC and DSS-induced colitis a combination of raised intracellular concentrations of NO and cAMP (secondary to raised mucosal prostaglandin E2 concentrations) may result in sufficient BK channel activity to sustain K+ secretion across the inflamed colonic mucosa.
In summary, we have demonstrated that a significant level of active K+ secretion occurs in DSS-induced colitis and this is mediated by upregulated apical BK channels. Furthermore, increases in serum aldosterone and/or mucosal NO concentrations may enhance BK channel expression/activity as part of the K+ secretory process. Based on these findings, we speculate that similar changes in human UC may account for the excessive fecal K+ losses and hypokalemia that sometimes occur in patients with this disease.
GRANTS
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases Grant RO1-DK-018777 (to V. M. Rajendran).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: B.M.K. and V.M.R. performed experiments; B.M.K., G.I.S., and V.M.R. approved final version of manuscript; G.I.S. and V.M.R. conception and design of research; G.I.S. and V.M.R. analyzed data; G.I.S. and V.M.R. interpreted results of experiments; G.I.S. and V.M.R. edited and revised manuscript; V.M.R. prepared figures; V.M.R. drafted manuscript.
REFERENCES
- 1.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis 15: 341–352, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amasheh S, Barmeyer C, Koch CS, Tavalali S, Mankertz J, Epple HJ, Gehring MM, Florian P, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Cytokine-dependent transcriptional down-regulation of epithelial sodium channel in ulcerative colitis. Gastroenterology 126: 1711–1720, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Archampong EQ, Harris J, Clark CG. The absorption and secretion of water and electrolytes across the healthy and the diseased human colonic mucosa measured in vitro. Gut 13: 880–886, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barmeyer C, Rahner C, Yang Y, Sigworth FJ, Binder HJ, Rajendran VM. Cloning and identification of tissue-specific expression of KCNN4 splice variants in rat colon. Am J Physiol Cell Physiol 299: C251–C263, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Tian L, MacDonald SH, McClafferty H, Hammond MS, Huibant JM, Ruth P, Knaus HG, Shipston MJ. Functionally diverse complement of large conductance calcium- and voltage-activated potassium channel (BK) alpha-subunits generated from a single site of splicing. J Biol Chem 280: 33599–33609, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Del Castillo JR, Rajendran VM, Binder HJ. Apical membrane localization of ouabain-sensitive K+-activated ATPase activities in rat distal colon. Am J Physiol Gastrointest Liver Physiol 261: G1005–G1011, 1991 [DOI] [PubMed] [Google Scholar]
- 7.Foster ES, Jones WJ, Hayslett JP, Binder HJ. Role of aldosterone and dietary potassium in potassium adaptation in the distal colon of the rat. Gastroenterology 88: 41–46, 1985 [DOI] [PubMed] [Google Scholar]
- 8.Fukao M, Mason HS, Britton FC, Kenyon JL, Horowitz B, Keef KD. Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J Biol Chem 274: 10927–10935, 1999 [DOI] [PubMed] [Google Scholar]
- 9.Geboes K. Histopathology of Crohn's diseas and ulcertive colitis. In: Inflammatory Bowel Disease, edited by Satsangi JSL, Colombel JF, Fiocci C, Lofberg R, Pemberton J, Rutgeerts P. London, UK: Churchill Livingstone, 2003, p. 255–276 [Google Scholar]
- 10.Greig ER, Boot-Handford RP, Mani V, Sandle GI. Decreased expression of apical Na+ channels and basolateral Na+,K+-ATPase in ulcerative colitis. J Pathol 204: 84–92, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Halevy J, Budinger ME, Hayslett JP, Binder HJ. Role of aldosterone in the regulation of sodium and chloride transport in the distal colon of sodium-depleted rats. Gastroenterology 91: 1227–1233, 1986 [DOI] [PubMed] [Google Scholar]
- 12.Harris J, Shields R. Absorption and secretion of water and electrolytes by the intact human colon in diffuse untreated proctocolitis. Gut 11: 27–33, 1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hawker PC, McKay JS, Turnberg LA. Electrolyte transport across colonic mucosa from patients with inflammatory bowel disease. Gastroenterology 79: 508–511, 1980 [PubMed] [Google Scholar]
- 14.Hirota CL, McKay DM. Loss of Ca-mediated ion transport during colitis correlates with reduced ion transport responses to a Ca-activated K channel opener. Br J Pharmacol 156: 1085–1097, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lang RJ, Harvey JR, McPhee GJ, Klemm MF. Nitric oxide and thiol reagent modulation of Ca2+-activated K+ (BKCa) channels in myocytes of the guinea-pig taenia caeci. J Physiol 525: 363–376, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lang RJ, Watson MJ. Effects of nitric oxide donors, S-nitroso-l-cysteine and sodium nitroprusside, on the whole-cell and single channel currents in single myocytes of the guinea-pig proximal colon. Br J Pharmacol 123: 505–517, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-delta delta C(T)] method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275, 1951 [PubMed] [Google Scholar]
- 19.Mandala M, Heppner TJ, Bonev AD, Nelson MT. Effect of endogenous and exogenous nitric oxide on calcium sparks as targets for vasodilation in rat cerebral artery. Nitric Oxide 16: 104–109, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Martin RS, Jones WJ, Hayslett JP. Animal model to study the effect of adrenal hormones on epithelial function. Kidney Int 24: 386–391, 1983 [DOI] [PubMed] [Google Scholar]
- 21.Nanda Kumar NS, Singh SK, Rajendran VM. Mucosal potassium efflux mediated via Kcnn4 channels provides the driving force for electrogenic anion secretion in colon. Am J Physiol Gastrointest Liver Physiol 299: G707–G714, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98: 694–702, 1990 [DOI] [PubMed] [Google Scholar]
- 23.Papavlassopoulos M, Stamme C, Thon L, Adam D, Hillemann D, Seydel U, Schromm AB. MaxiK blockade selectively inhibits the lipopolysaccharide-induced I kappa B-alpha/NF-kappa B signaling pathway in macrophages. J Immunol 177: 4086–4093, 2006 [DOI] [PubMed] [Google Scholar]
- 24.Perez-Navarro R, Ballester I, Zarzuelo A, Sanchez de Medina F. Disturbances in epithelial ionic secretion in different experimental models of colitis. Life Sci 76: 1489–1501, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Perse M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol 2012: 718617, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rechkemmer G, Halm DR. Aldosterone stimulates K secretion across mammalian colon independent of Na absorption. Proc Natl Acad Sci USA 86: 397–401, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandle GI, Hayslett JP, Binder HJ. Effect of glucocorticoids on rectal transport in normal subjects and patients with ulcerative colitis. Gut 27: 309–316, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandle GI, Perry MD, Mathialahan T, Linley JE, Robinson P, Hunter M, MacLennan KA. Altered cryptal expression of luminal potassium (BK) channels in ulcerative colitis. J Pathol 212: 66–73, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Sandle GI, Rajendran VM. Cyclic AMP-induced K+ secretion occurs independently of Cl− secretion in rat distal colon. Am J Physiol Cell Physiol 303: C328–C333, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology 134: 577–594, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Singh SK, O'Hara B, Talukder JR, Rajendran VM. Aldosterone induces active K+ secretion by enhancing mucosal expression of Kcnn4c and Kcnma1 channels in rat distal colon. Am J Physiol Cell Physiol 302: C1353–C1360, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sorensen MV, Matos JE, Sausbier M, Sausbier U, Ruth P, Praetorius HA, Leipziger J. Aldosterone increases KCa1.1 (BK) channel-mediated colonic K+ secretion. J Physiol 586: 4251–4264, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stoner MC, Kellum JM. Both serotonin and a nitric-oxide donor cause chloride secretion in rat colonocytes by stimulating cGMP. Surgery 130: 236–241, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Sullivan S, Alex P, Dassopoulos T, Zachos NC, Iacobuzio-Donahue C, Donowitz M, Brant SR, Cuffari C, Harris ML, Datta LW, Conklin L, Chen Y, Li X. Downregulation of sodium transporters and NHERF proteins in IBD patients and mouse colitis models: potential contributors to IBD-associated diarrhea. Inflamm Bowel Dis 15: 261–274, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker NM, Simpson JE, Yen PF, Gill RK, Rigsby EV, Brazill JM, Dudeja PK, Schweinfest CW, Clarke LL. Down-regulated in adenoma Cl/HCO3 exchanger couples with Na/H exchanger 3 for NaCl absorption in murine small intestine. Gastroenterology 135: 1645–1653.e3, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whittle BJ. Nitric oxide–a mediator of inflammation or mucosal defence. Eur J Gastroenterol Hepatol 9: 1026–1032, 1997 [DOI] [PubMed] [Google Scholar]
- 37.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448: 427–434, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci USA 93: 6770–6774, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao F, Juric M, Li J, Riederer B, Yeruva S, Singh AK, Zheng L, Glage S, Kollias G, Dudeja P, Tian DA, Xu G, Zhu J, Bachmann O, Seidler U. Loss of downregulated in adenoma (DRA) impairs mucosal HCO3(−) secretion in murine ileocolonic inflammation. Inflamm Bowel Dis 18: 101–111, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Halm ST, Halm DR. Role of the BK channel (KCa1.1) during activation of electrogenic K+ secretion in guinea pig distal colon. Am J Physiol Gastrointest Liver Physiol 303: G1322–G1334, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]