

Abstract
Highly sensitive and multiplexed detection of clinically relevant proteins in biologically complex samples is crucial for the advancement of clinical proteomics. In recent years, aptamers have emerged as useful tools for protein analysis due to their specificity and affinity for protein targets as well as their compatibility with particle-based detection systems. In this study, we demonstrate the highly sensitive detection of human alpha-thrombin on encoded hydrogel microparticles functionalized with an aptamer capture sequence. We use static imaging and microfluidic flow-through analysis techniques to evaluate the detection capabilities of the microgels in sandwich assay formats that utilize both aptamers and antibodies for the reporting of target-binding events. Buffers and reagent concentrations were optimized to provide maximum reaction efficiency while still maintaining an assay with a simple workflow that can be easily adapted to the multiplexed detection of other clinically relevant proteins. The three-dimensional, non-fouling hydrogel immobilization scaffold used in this work provides three logs of dynamic range, with a limit of detection of 4 pM using a single aptamer capture species and without the need for spacers or signal amplification.
Introduction
The development of sensitive and high-throughput protein detection assays capable of multiplexed analysis is crucial for advancing both medical diagnostics and biological discovery. The majority of existing protein detection schemes rely on the use of antibodies for both target capture and reporting.1 Despite high affinities for their targets and well-characterized epitope interactions, antibodies have several major disadvantages, including expensive and time consuming selection processes, in vivo production requirements, and storage instability.2–3
An emerging class of molecules called aptamers has shown promise as an alternative to antibody-based protein detection. Aptamers are short nucleic acid sequences that bind to their targets with high specificity and with dissociation constants in the low nM range.2, 4 They are selected through an iterative process known as SELEX (Systematic Evolution of Ligands by Exponential Enrichment) in which large random libraries of nucleic acid fragments are tested against the target of interest for specific and effective binding.5–7 Several aptamers can be selected against a single target, allowing for the use of multiple aptamers to either enrich target affinity through avidity effects or to develop sandwich-based target capture and reporting.4, 7–8 Protein capture is facilitated by the folding of the nucleic acid probe sequence into a tertiary structure upon interaction with a particular epitope of the target.4–5
Aptamers’ stability in solution and the relatively simple in vitro selection and production processes used for their generation provide an attractive alternative to the antibody-based methods currently used in the field of proteomics. Due to the stringency imposed by the iterative in vitro selection process, aptamers bind to their protein targets with high specificity and thus generally exhibit less cross-reactivity with other non-target proteins than antibodies.3, 7 These qualities make the aptamer a desirable bioassay reagent for applications in complex biological samples such as plasma, serum, and urine. The primary challenge in integrating aptamers into biosensing platforms is their sensitivity to the ionic environment. Specific ionic species are required for aptamers to fold into the complex structures necessary to maintain target binding, and aptamers which are selected under different temperature, pH and ionic conditions may not always be compatible with each other.4, 9 The need for unique buffers and experimental conditions for each aptamer can lead to complications during multiplexing or when using poorly characterized biological samples. However, since aptamers may be a better option than antibodies in certain applications, there is a strong need to further characterize their performance when integrated with next-generation sensing platforms.
A number of protein detection technologies utilize microarrays or encoded microparticles to enable multiplexing, a crucial component in clinical proteomic profiling. Although the spatial encoding provided by microarrays allows analysis of thousands of targets simultaneously, these systems suffer from low throughput, long incubation times, and inability to accommodate rapid probe-set modification.10 An alternative system introduced by Luminex uses polystyrene beads doped with fluorescent dyes to generate spectral codes. The use of such a particle array leads to faster assay kinetics, high throughput analysis, and flexibility in assay design.10 However, the Luminex encoding system offers a maximum of only 500 codes and relies on a complex array of lasers and photomultiplier tubes to decode particles and quantify target. Additionally, coefficients of variation from this system are often high, especially in complex samples.11 Other next generation technologies have been developed using barcoded metallic rods, doped rare earth materials, or photoresist materials for encoding and detection, but these technologies lack high-throughput analysis schemes, often demonstrate poor sensitivity, and have complicated protocols for particle fabrication.10, 12–13
In this study, we interface aptamer-based protein sensing with a graphically-encoded particle array previously developed and validated for DNA, microRNA and antibody-based protein quantification.14–17 We use microfluidic stop-flow lithography (SFL) to generate microgel particles bearing spatially-segregated regions for encoding and target capture.14, 18 Particles are simultaneously polymerized, encoded, and functionalized with probe (Figure 1a). Our encoding scheme allows for the production of millions of unique codes using permutations of unpolymerized holes of varying dimensions in the fluorescently-doped “code” region of our particles. This expansive coding library is important for multiplexed analysis and for the pooling of particles from different incubation reactions for rapid post-assay analysis. Acrydite-modified probes can be readily incorporated into the three-dimensional gel network of a separate “probe” region, providing detection capabilities and biomolecule incorporation efficiencies that are superior to surface-based sensing platforms.15, 19–20 Furthermore, biological interactions tend to be more thermodynamically favorable inside the hydrated gel matrix, which offers probes more freedom for movement and provides an environment conducive for folding into appropriate tertiary structures with less steric hindrance.20 Here we investigate these gel-specific detection advantages in the context of aptamer-based sandwich assays.
Figure 1.
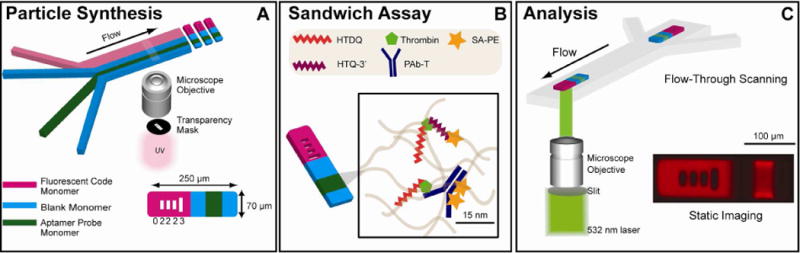
Assay workflow. (A) Particles bearing aptamer probe are synthesized using SFL. The code stream contains 0.6% rhodamine acrylate. The schematic shows a particle bearing the graphical code “02223”. Particles with additional chemistries can be synthesized by altering the number of streams in the channel. (B) Particles are used to detect thrombin using a sandwich assay in which the reporting ligand is biotinylated to faciliate labeling with SA-PE. The two reporting ligands shown are a DNA aptamer (HTQ-3′) and a polyclonal antibody against thrombin (PAb-T). (C) Fluorescently labeled complexes are analyzed using either flow-through scanning or static imaging. Custom MATLAB scripts are used to extract individual particle profiles from scan data and images. Static particle image shown is false-colored.
Our study is performed using well-characterized aptamers against alpha-thrombin, a serine protease which plays a key role in the blood clotting cascade.21 Since alpha-thrombin catalyzes the formation of fibrin from fibrinogen, its concentration in the bloodstream is of considerable clinical importance. The measurement of alpha-thrombin in the blood is important for the diagnosis and monitoring of several cardiovascular disorders.22 The protein has even been suggested as a potential biomarker for Alzheimer’s disease and other neurodegenerative disorders.23–24 The DNA aptamers used in this study, designated HTQ and HTDQ, interact with the fibrinogen and heparin exosites of the protein, respectively. In previous surface-based studies, HTQ has exhibited a Kd of 2.6 nM when binding with alpha-thrombin, while HTDQ has exhibited a Kd of 0.5 nM.21, 25 The availability of aptamers that bind at different sites of the protein allows us to develop a sandwich assay, with distinct “capture” and “reporter” aptamers (Figure 1b). Although several studies have examined detection performance of these aptamers for alpha-thrombin sensing in microarrays and other surface based particle arrays, these molecular probes have not been previously characterized in the context of a hydrogel environment.25–35 Here, we assess the detection performance of the HTQ and HTDQ aptamers within our hydrogel particles with respect to concentration of probe in the particles, type of reporting moiety, and composition of buffer at various stages of the assay. We show that by using an optimized buffer system, it is possible to extend our system to multiplexing alongside more traditional antibody-based detection schemes, and we characterize the sensitivity, specificity, and dynamic range of alpha-thrombin detection using different reporting ligands and our high-throughput analysis system (Figure 1c). We also examine the effect of incorporating thymine spacers into the aptamer probe. Finally, we examine the ability of the particles to discriminate and bind probe in the presence of interfering proteins commonly found in complex biological matrices.
Experimental Section
Particle Synthesis
Fabrication methods for particles bearing DNA and antibody probes have been described in detail elsewhere.15–17 Briefly, monomer solutions were mixed using different ratios of poly(ethylene glycol) diacrylate (PEG-DA 700, MW 700 g/mol, Sigma Aldrich), poly(ethylene glycol) (PEG 200, MW 200 g/mol, Sigma Aldrich), Darocur 1173 (Sigma Aldrich), and 3X Tris-EDTA (TE, USB Corporation) buffer. Monomer solutions were loaded into a 38 μm tall polydimethylsiloxane (PDMS) microfluidic device for synthesis. Particles were rinsed and stored in 1X TE with 0.05% (v/v) Tween-20 (T-20, TET) at 4°C after synthesis. Further details about synthesis can be found in the Supporting Information.
Reagents
DNA aptamers were purchased from Integrated DNA Technologies (IDT) with appropriate modifications (see Supporting Information). Capture probe aptamers were purchased with an acrydite modification (Acryd) on the 5′ end to facilitate integration into the PEG-DA gel matrix of the particles upon polymerization. Reporter probe aptamers were purchased with biotin modifications with a “GGG” spacer on either the 3′ or 5′ end to enable fluorophore labeling. Aptamers were reconstituted and aliquoted in 1X TE and stored at −20°C. Phosphate Buffered Saline (PBS, 1X) was purchased from Cell Gro (21-040-CV) with the following formulation: 1 mM KH2PO4, 155 mM NaCl, 6 mM Na2HPO4. Human alpha-thrombin, human serum albumin and IgGs from human serum were purchased from Sigma Aldrich (St. Louis, MO) and reconstituted into phosphate buffered saline (PBS) containing 0.1% (w/v) BSA and stored at −20°C. Polyclonal biotinylated antibodies against alpha-thrombin and IL-2 and monoclonal capture antibodies against IL-2 were purchased from R&D Systems (BAF1148, BAF202, MAB202) and stored in PBS with 0.1% BSA at −20°C. Human prothrombin, beta-thrombin, and gamma-thrombin were purchased from Haematologic Technologies (Essex Junction, VT) and stored at −20°C.
Protein Assay
All protein assays were conducted in 0.65 mL Eppendorf tubes in a total reaction volume of 50 μL at 25°C. Protein serial dilutions were performed in PBS with 0.1% T-20 (PBST). Addition of reagents to tubes was done in the following order: reaction buffer, exogenous protein, particles. Reaction volumes were agitated at 1800 RPM using a thermoshaker (Rio). After the initial incubation, particles were rinsed three times using the buffer for the subsequent incubation step, and after each rinse, the reaction volume was brought back to 50 uL.
Reporter ligands and streptavidin-phycoerythrin (SA-PE, Invitrogen) were diluted in PBST. Target incubations lasted 2.5 hours, reporter incubations lasted 1.5 hours, and SA-PE incubations lasted 30 minutes. The latter two reactions were performed in the same buffer. Approximately 25 particles of each type were used in each assay. HTQ-3′ was provided at 406.4 nM, and PAb-T was provided at either 16.7 nM or at 10 nM, depending on the assay. PAb-IL-2 and SA-PE were provided at concentrations of 6.67 nM and 13.3 nM, respectively.
Data Analysis
Prior to imaging, particles were re-suspended in 1X TE with 0.05% T-20 and 25% PEG 400 (PTET) to gain maximum image contrast. Eppendorfs were vortexed gently for 45 seconds and 13 μL of solution was deposited onto a 24 mm × 60 mm slide and sandwiched with an 18 mm × 18 mm cover slip. Particles were imaged under fluorescence using a CCD camera (Andor Clara) at an exposure time of 0.05 s. Unless otherwise noted, HTQ-3′ labeled particles were imaged within 10 minutes of their last rinse, and PAb-T labeled particles were imaged within 30 minutes of their last rinse. After acquisition, images were aligned and cropped in ImageJ (NIH), and width-averaged fluorescent profiles were extracted using a custom MATLAB (Mathworks) script.
Prior to microfluidic scanning, particles were re-suspended in 50 μl PTET, and the scanning system was set up using methods detailed elsewhere.17 Briefly, a 532 nm green laser was used to excite samples through a 4 μm × 90 μm chrome-coated glass slit. Particles were flowed past this excitation window at speeds up to 50 cm/s in a microfluidic flow-focusing device with a height of 37.5 μm. Laser-induced fluorescence was collected by a photomultiplier tube (PMT, Hamamatsu H7422-40), amplified, and reported as a temporal voltage signal which was then analyzed using a custom MATLAB script. Additional details regarding data analysis are provided in the Supporting Information.
Results & Discussion
Detection Scheme & Workflow
Since there are two unique aptamers against alpha-thrombin, we developed a sandwich-assay approach to capture and label the bound proteins (Figure 1b). Our selection of the capture aptamer was informed primarily by affinity characteristics and folding structure of the two available sequences; we sought to minimize potential dissociation of complexes during assay rinse steps. HTDQ has a greater affinity for alpha-thrombin and a more stable binding structure than HTQ.21, 25 Although both aptamers rely on the formation of a G-quadruplex structure upon target binding, HTDQ forms an additional duplex, giving it greater stability.25, 36 Previous detection platforms utilizing these aptamer sequences have found that HTDQ performs well as a capture aptamer when used in a sandwich format.26, 28 HTDQ was therefore used as the gel-embedded capture sequence.
Rather than including extensive spacers between the conjugation point and the aptamer sequences, necessary for several surface-based systems to achieve effective target capture and reporting, we employed only short “GGG” spacers to distance biotin modifications from the HTQ reporter sequence to enable flurophore labeling.25–27, 37 Due to the solution-like nature of target-probe interactions inside hydrogels, we expected that long spacers were largely unnecessary, and in past assays with our gel particles, we have attained high levels of sensitivity without them.15, 20 We hypothesize that our flexible and porous hydrogel scaffold gives us a key advantage in terms of aptamer folding and protein capture inside the particles. The three-dimensional nature of the gel microenvironment distances probe molecules further apart from each other than a typical microspot assay is able to, minimizing steric hindrance and providing the necessary flexibility for aptamers to fold into target-binding configurations.20 Before moving forward, we incubated HTDQ-embedded gel particles with fluorescently labeled alpha-thrombin to assess preliminary performance. Previous surface-based assays utilizing labeled thrombin have reported limits of detection in the low nM range9, 38 but in this model assay, the gel particles exceeded these systems in sensitivity with a limit of detection of 65.9 pM (Figure S-2) Unless otherwise noted, all further experimentation and optimization were conducted with only “GGG” spacers in the reporter sequences.
Three different reporting ligands were evaluated in our study: the HTQ aptamer with a 3′ biotin modification (HTQ-3′), the HTQ aptamer with a 5′ biotin modification (HTQ-5′), and a biotinylated polyclonal antibody directed against alpha-thrombin (PAb-T). All three reporters were tested in an initial screening assay with particles that had been incubated with 100 pM alpha-thrombin. HTQ-5′ labeled complexes with less than 10% the efficiency of HTQ-3′ or PAb-T, presumably due to the biotin modification on the 5′ end of the aptamer interfering with the ability to fold into the G-quadruplex structure necessary for prolonged interaction with alpha-thrombin, an observation that is corroborated by previous studies.36 Since HTQ-3′ and PAb-T clearly outperformed HTQ-5′, all further assays were conducted using only these two reporters.
Effect of Probe Concentration on Assay Sensitivity
Assuming a target-probe reaction that follows first order Langmuir kinetics, a higher concentration of probe molecules in solution will result in a greater number of target-probe complexes formed at the time of equilibrium and better assay sensitivity. As previously noted, surface-based systems are challenged by steric limitations, allowing for a maximum of only ~104 molecules/μm2 to be immobilized onto the construct; increasing the amount of probe in the spotting solution does not necessarily achieve a greater probe density in these systems.20, 39 In contrast, gel substrates can demonstrate at least one order of magnitude greater effective surface density than surface microspots. For example, a typical gel particle that contains probe conjugated at a concentration of 10 μM relative to probe region volume and is 35 μm tall has an effective surface probe density of ~105 molecules/μm2 when the particle surface is vertically projected onto its base. Additionally, when compared to a microspot loaded with probe to maximum capacity (which provides approximate separation of 10 nm between molecules), a typical gel particle provides an approximate separation of 55 nm between molecules and can accommodate more probe.19
For our microgel particle system, the maximum feasible probe concentration was dictated by the chemical nature of the monomer and the experimental requirements of SFL. In order to maintain miscibility with the monomer and to provide sufficient monomer loading volumes for particle synthesis procedures, the maximum monomer probe concentration investigated was 300 μM. The lowest evaluated monomer probe concentration, 100 μM, was chosen based on starting concentrations that have been successful in past nucleic acid assays with the gel particles.15 For completeness, we also decided to investigate an intermediate monomer probe concentration of 200 μM in our analysis.
Particles with a coded region and three spatially-segregated probe-bearing regions were synthesized to investigate the effects of probe-loading. The three monomers loaded for the three distinct probe streams contained the aforementioned concentrations of the capture probe (300 μM, 200 μM, 100 μM). Incorporation of probe into the particles at the time of synthesis depends primarily on the composition of the monomer. Monomer mixtures containing higher volume fractions of PEG-DA 700 and lower volume fractions of PEG 200 will produce gels with a higher cross-linking density and thus more biomolecule probe incorporation. However, composition is also tuned to produce a gel network with pores that are large enough to allow for the penetration of large biomolecules and labeling reagents. Based on previous studies examining probe incorporation as a function of monomer composition, we assumed a 10% probe incorporation efficiency during UV polymerization and estimated the probe concentrations within the polymerized particles (the number of probe molecules per unit volume of the gel probe region) to be 30 μM, 20 μM, and 10 μM for the three probe regions (Figure 2a).40 In order to avoid fluorescent bleed-over from the code region and to ensure the same geometry for all probe strips, blank regions containing neither dyes nor probes were included after the code region and at the end of the particle.
Figure 2.
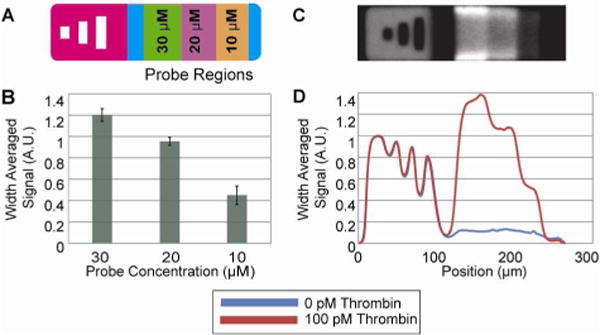
Evaluation of probe concentration. (A) Schematic of particle bearing the barcode “123” and three distinct probe regions with different concentrations of HTDQ probe. The blue regions before and after the probes are blank regions containing only monomer. (B) Bar graph showing the fluorescent signal produced from the three different probe regions from a sandwich assay. Data represent the average of 5 particles. (C) Static image of a particle incubated with 100 pM thrombin and labeled with PAb-T. Regions bearing higher concentrations of probe in the monomer clearly show higher fluorescent signal. (D) Static scan showing the average of 5 particles incubated with 100 pM thrombin (red) and 0 pM thrombin (blue).
Particles were incubated with 100 pM alpha-thrombin and labeled with 16.7 nM of PAb-T, which did not require buffer optimization prior to use. The resulting fluorescent signature clearly indicated a proportional relationship between monomer probe concentration and number of labeled complexes at equilibrium (Figure 2b–d). There was a 2.11X increase in signal between the 10 μM and the 20 μM regions, and a 2.67X increase in signal between the 10 μM and the 30 μM regions of the gradient particles. In order to achieve maximum sensitivity, a 300 μM monomer probe concentration was used for all other particles in this study. Although our synthesis methodology provides a simple workflow in which particles are encoded and functionalized in a single step, this leads to a slightly higher consumption of probe per assay. We find that our probe consumption at the aforementioned monomer concentration of 300 μM (~7 pmol/assay) is greater than for a traditional microarray utilizing 15–30 nL of a 25 μM spotting solution per microspot (~1–2.5 pmol/assay)25, 35, but is less than for competing particle array systems which have utilized up to 50 pmol/assay29. It is possible to use alternate functionalization schemes if necessary in future applications to consume aptamer probe more efficiently.
Optimization of Aptamer Buffers
Aptamers have been criticized due to their frequent incompatibility with universal buffers such as PBS.3–4 Different aptamers require unique ions to effectively fold target-binding structures, creating a need to carefully design incubation buffers.9 In the general approach for assembling these buffers, aptamer selection conditions are mimicked.3 However, such buffer-matching often leads to highly specialized buffer systems that may preclude multiplexing with other targets or probes. Additionally, care needs to be taken in addressing buffer compatibility of the environment-sensitive hydrogel particles.
In order to address both of these concerns, we built a simpler buffer system which included only the key ions necessary to optimize target binding and was pH adjusted for compatibility in a clinical setting. These criteria provided the maximum versatility and flexibility for future applications. HTQ-3′ buffer conditions were engineered first to ensure stability of the HTDQ-bound alpha-thrombin and to accommodate HTQ′s sensitivity to incubation conditions.25, 36
There is conflicting literature regarding the role of the K+ and Na+ ions for the successful folding of HTQ.30–31, 41 To parametrically study the effect of these ions on aptamer performance, we designed a series of buffers employing a phosphate base to make pH adjustments simple and to provide a suitable environment in which biomolecules could retain integrity. Four buffers were tested: 1) PBS with 0.1% T-20 (PBST), 2) 10 mM phosphate with 0.1% T-20, 3) 10 mM phosphate with 10 mM KCl and 0.1% T-20, 4) 10 mM phosphate with 50 mM KCl and 0.1% T-20. All buffers were pH adjusted to 7–7.4 prior to use.
In all assays, particles were incubated with 100 pM alpha-thrombin in PBST prior to the HTQ-3′ incubation. PBST had the worst performance, indicating that the Na+ ion was detrimental towards HTQ-3′ performance. Optimal performance was observed with the buffer containing 10 mM phosphate with10 mM KCl and 0.1% T-20 (Figure 3a–d). Too much potassium salt disrupted HTQ-3′ performance, but its complete elimination was also suboptimal. All further HTQ-3′ incubations were carried out in 10 mM phosphate with 10 mM KCl and 0.1% T-20.
Figure 3.
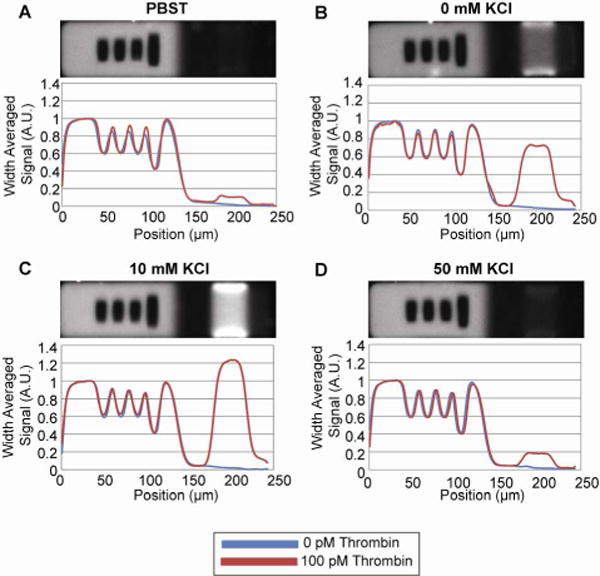
HTQ-3′ buffer optimization. In all cases, static scans depict incubations with 100 pM thrombin (red) and with 0 pM thrombin (blue). Reporter and SA-PE incubations were carried out in (A) PBST, (B) 10 mM phosphate with 0.1% T-20, (C) 10 mM phosphate with 10 mM KCl and 0.1% T-20, and (D) 10 mM phosphate with 50 mM KCl and 0.1% T-20. Maximum signal was achieved with the conditions from (C)
Modification of the target capture buffer was not investigated based on preliminary results from the 100 pM alpha-thrombin incubations carried out in both PBST and 10 mM phosphate with 10 mM KCl and 0.1% T-20 and followed by HTQ-3′ incubation in 10 mM phosphate with 10 mM KCl and 0.1% T-20. It appeared that although Na+ seemed to disrupt HTQ-3′ binding, it was crucial for HTDQ-alpha-thrombin binding. Fluorescent signal was 75X higher when the target incubation was conducted in PBST. These favorable results indicated no need to additionally modify this physiologically matched target incubation buffer.
Buffer System Compatibility with Multiplexed Assays
A proof of concept multiplexing assay was designed to assess the universality of the chosen buffer systems. Alpha-thrombin (100 pM) and interleukin-2 (IL-2, 0.667 pM), a cytokine relevant in the immune response to inflammation,42 were simultaneously detected using different graphically encoded particles. The chosen target concentrations facilitated comparisons to thrombin particles from previous assays and tested the detection capability of IL-2 particles close to their previously measured detection limit of 0.073 pM.17 IL-2 particles carried monoclonal antibody capture probes and were labeled using a polyclonal biotinylated antibody (PAb-IL-2). PAb-IL-2/HTQ-3′ and PAb-IL-2/PAb-T (16.7 nM) incubations were carried out in the previously optimized thrombin reporter buffer. The multiplexed environment did not impact the robust detection of either target, and the observed fluorescent signals were comparable to results from performing the assays in a single-plex manner (Figure 4). The one minor modification necessary for the PAb-IL-2/HTQ-3′multiplexing reaction was the addition of 200 μg/mL Bovine Serum Albumin (BSA) in the reporter step. In the absence of BSA, particles seemed to accumulate particulate matter throughout the course of the assay. Despite the need for this small adjustment, the relative compatibility with other clinically relevant targets makes the HTQ-3′ buffer system suitable for multiplexing applications. It should be noted that the reporter reaction did not require further optimization when using PAb-T. Most importantly, aptamer folding was not hindered by the presence of other proteins in solution.
Figure 4.
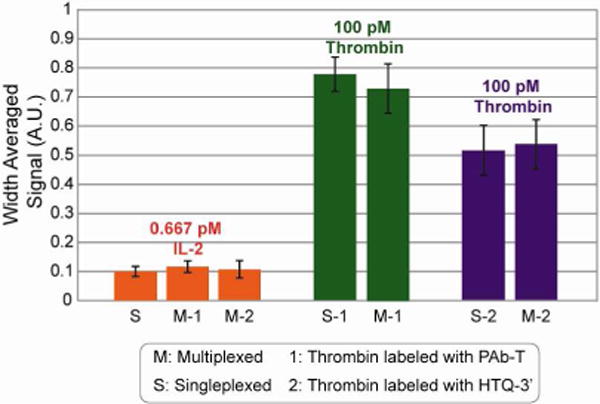
Multiplexing demonstration. 100 pM thrombin and 0.667 pM IL-2 were detected simultaneously (M) using different particles. Assays were performed using both thrombin detection ligands (1:PAb-T, 2: HTQ-3′) and an IL-2 detection antibody (PAb-IL2). Reporter steps were carried out in optimal buffers (PBST for PAb-T and 10 mM phosphate with 10 mM KCl and 0.1% T-20 for HTQ-3′). Signals are compared side by side with target signal attained using a singleplex detection for both targets (S). Target signal is unaltered in multiplexed format and the HTQ-3′ buffer does not have an inhibitory effect on IL-2 reporter binding.
Evaluation of Detection Capability
Particle performance was assessed using a high-throughput microfluidic scanner capable of decoding and quantifying signal from the soft gel particles at rates up to 25 particles per second.17 The ability to analyze several particles in a short time frame is an important assay feature for targets such as alpha-thrombin which are examined primarily in a diagnostic context. The tendency of HTQ-3′ to dissociate from the target soon after the final assay rinse additionally made such high speed analysis an essential feature. When HTQ-3′-labeled particles were monitored over time, there was a 40% drop in fluorescent signal in just 10 minutes, and a 75% drop in fluorescent signal over the course of 60 minutes (Figure S-3). Using the microfluidic scanner, it was possible to analyze the particles before any significant dissociation could occur. All HTQ-3′ particles were accordingly scanned within 2 minutes of their last rinse. Although the same loss in signal was not seen with PAb-T labeled particles over the course of 60 minutes (Figure S-3), analysis occurred within 10 minutes of their last rinse. PAb-T assay concentration was additionally optimized from 16.7 nM to 10 nM before calibration points were attained to minimize nonspecific adherence of the antibody to the particles and simultaneously maintain dynamic range and sensitivity.
Calibration curves for assays with both reporting ligands are plotted and sample scan data is provided in Figure 5. Extrapolation of the data points (Figure S-1) within the linear regime of the curves leads to an LOD of 21.7 pM with detection over a dynamic range of 2 logs using HTQ-3′ (mean intra-trial coefficient of variation (CV) = 7%) and an LOD of 4.09 pM with detection over a dynamic range of 3 logs using PAb-T (mean CV = 9%). It should be noted that the upper end of our dynamic range is determined by saturation limit of our detector and not by saturation of available probe sites at the time of analysis. If necessary, a different PMT could be used to extend the dynamic range of the assay. Because it was possible to use HTQ-3′ in 40X excess of PAb-T in reporting steps without any significant non-specific accumulation on the particle, HTQ-3′ labeled particles demonstrate greater signal than PAb-T labeled particles for alpha-thrombin concentrations greater than 100 pM. The low CVs observed in our assays make it necessary to analyze only 8 – 10 particles from each incubation, in contrast to other particle-based arrays that require the analysis of over 1000 beads for reliable quantification.43–44
Figure 5.
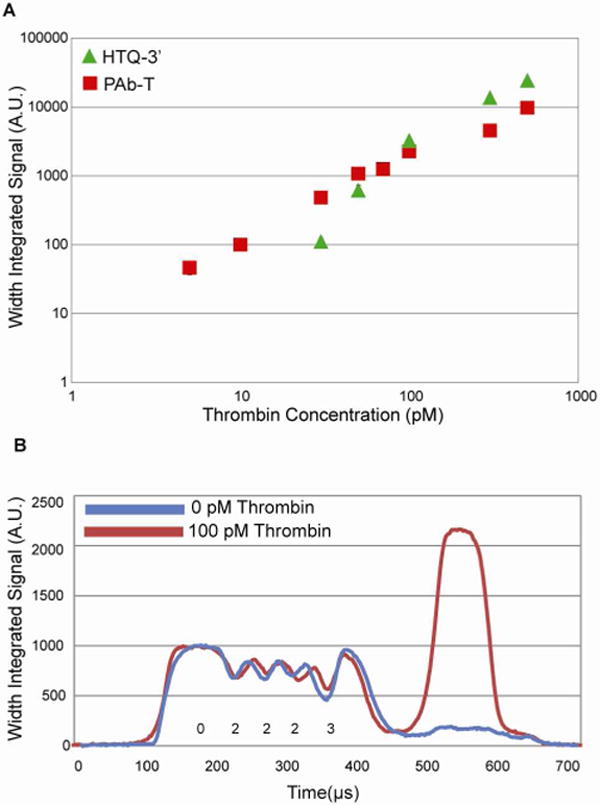
Detection capability of HTQ-3′ and PAb-T. (A) Calibration curves for HTQ-3′ (green triangle) and PAb-T (red square). Data was generated using flow-through scanning of particles after sandwich assay. Signal is linear over a range of 2 logs for HTQ-3′ and a range of 3 logs for PAb-T. CVs are provided in Table 1. (B) Particle profiles derived from scanner data. Sample scans are shown for target incubations with 100 pM thrombin (red) and 0 pM thrombin (blue). In both cases, particles were labeled with 10 nM PAb-T.
Ultimately, labeling with PAb-T provided maximum sensitivity, but it is important to recognize that labeling with HTQ-3′ led to only ~5X higher LOD, despite a lower binding affinity for alpha-thrombin. The only caveat is the need for analysis immediately after the last rinse. Typical surface based thrombin assays using these aptamers achieve sensitivities ranging from 500 pM – 5 nM3, 25, 29. Some systems have been able to boost performance by exploiting avidity effects or by using enzyme assisted or substrate based signal amplification schemes to lower limits of detection (5 fM – 1 nM)25–26, 28, 34–35. In comparison, the results attained using the gel particles surpass results presented in several of these competing platforms that use fluorescence based or electrochemical detection and that incorporate signal amplification.25–28, 30–31, 36 Based on our study, we believe that the ability of our platform to outperform many surface based detection systems is primarily due to the porous and non-fouling nature of our substrate and our ability to increase effective assay probe concentration as mentioned previously. A full table of CVs for calibration points is available in the Supporting Information (Table S-5).
Effect of Thymine Spacers on Detection Capability
A brief study was performed to examine the effect of spacers on the detection capability of the gel particles. Thymine spacers have been found to be beneficial in microarray systems, with signal enhancement typically being proportional to the length of the spacer in the regime where immobilization efficiency is not compromised.25, 27, 37 Accordingly, the HTDQ aptamer probe was modified at the 5′ end with a 6-thymine spacer (HTDQ-6T) and a 12-thymine spacer (HTDQ-12T) to assess spacers of varying length.
Particles with three distinct probe regions (HTDQ, HTDQ-6T and HTDQ-12T) were synthesized using a monomer probe concentration of 300 μM. After incubation with alpha-thrombin, particles were labeled using both HTQ-3′ and PAb-T. Using the spacers seemed to provide moderate signal enhancement when particles were labeled with PAb-T (Figure 6) for four different alpha-thrombin concentrations (10 pM, 50 pM, 100 pM and 500 pM), but there was no statistically significant enhancement when particles were labeled with HTQ-3′ (Table S-6). In PAb-T incubations, HTDQ-6T regions consistently showed less signal enhancement than HTDQ-12 regions. The highest signal increase demonstrated by the PAb-T labeled particles was a 2.26X increase in fluorescent signal between the HTDQ and the HTDQ-12T regions of the particles incubated with 100 pM alpha-thrombin. Since the enhancement was even less (1.65X increase in signal) on the lower end of the PAb-T dynamic range (10 pM alpha-thrombin), the LOD would not be substantially altered as a result of using the spacers. These results are in stark contrast to the 4X – 10X enhancement experienced by surface-based systems as a result of such spacers, and serve to further emphasize the importance of a using a gel substrate which offers flexibility and space for biomolecules to fold and bind effectively.25 Based on the additional cost of adding thymine spacers into the aptamer sequences and the minimal gain in sensitivity, it is unnecessary to use long spacers when immobilizing probes into a hydrogel.
Figure 6.
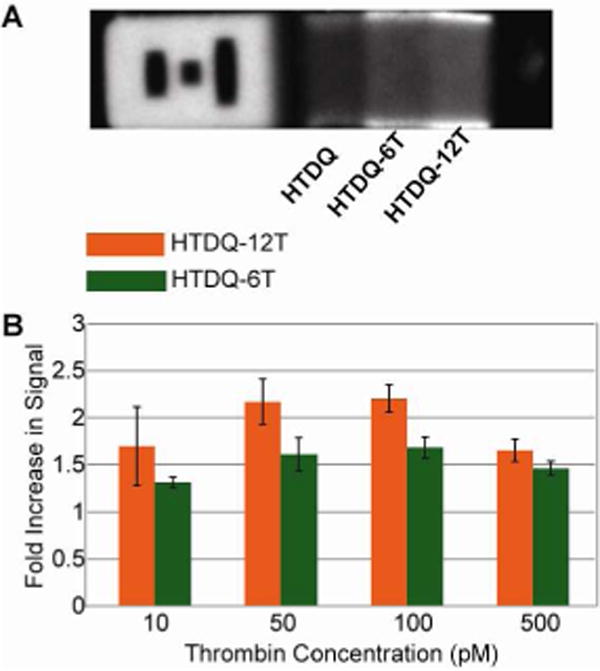
Evaluation of the effect of spacers with the HTDQ probe. Gradient particles were synthesized using HTDQ, HTDQ with a 6-thymine spacer at the 5′ end (HTDQ-6T), and HTDQ with a 12-thymine spacer at the 5′ end (HTDQ-12T). (A) Static image of a gradient particle incubated with 100 pM thrombin and 10 nM PAb-T. Longer thymine spacers lead to higher signal. (B) Bar graph showing the fold increase in signal when HTDQ-12T (orange) and HTDQ-6T (green) regions are compared to the HTDQ region, over a range of thrombin concentrations (10 pM, 50 pM, 100 pM, 500 pM). All particles were incubated with 10 nM PAb-T.
Detection in Complex Matrices
In order to assess signal recovery in complex media, samples need to accommodate exogenous alpha-thrombin. This criterion ruled out blood plasma, which would form a fibrin clot upon introduction of alpha-thrombin.30 Although the hydrogel particles have been shown to be compatible with complex biological mixtures such as blood-derived fluids, serum was not a suitable medium for these assays due to conflicting data regarding endogenous alpha-thrombin levels in serum, and the potential of inhibited alpha-thrombin complexes dissociating over time, releasing the protein into serum and leading to inaccurate analysis.16, 30, 45–47 Additionally, serum thrombin concentration would not be reflective of blood concentration of the protein in a clinical situation. We chose instead to evaluate the gel particle performance in two custom assays. In an interference assay, the matrix effect of serum or plasma was mimicked by the introduction of highly abundant blood proteins. In a specificity assay, target discrimination was examined using two proteolytically and structurally similar products of alpha-thrombin (beta-thrombin and gamma-thrombin) provided in stoichiometric ratio with alpha-thrombin.48 Both assays were conducted in the presence of 100 pM alpha-thrombin.
The complex mixture in the interference assay was comprised of human serum albumin (HSA) and Immunoglobulin G (IgG) 50X in excess of the target and prothrombin 10X in excess of the target. HSA and IgG are highly abundant in blood and may interfere with a detection assay.28 Meanwhile, prothrombin is the immediate precursor to alpha-thrombin and is always present in blood, making it crucial to ensure that it does not cross react.48 No additional nonspecific binding was observed on the particles as a result of these interfering proteins, and the reaction efficiency was unaffected for both HTQ-3′ and PAb-T labeled particles (Table 1).
Table 1.
Percent recovery in specificity and interference assays performed in the presence of 100 pM alpha-thrombin. HTQ-3′ labeled particles demonstrate full recovery in both the specificity and interference assay, but PAb-T labeled particles are unable to reliably recover signal in the specificity assay despite showing high recovery in the interference assay.
| Reporter | Assay | Percent Recovery |
|---|---|---|
| HTQ-3′ | Specificity | 106 ± 6 |
| HTQ-3′ | Interference | 109 ± 13 |
| PAb-T | Specificity | 43 ± 68 |
| PAb-T | Interference | 91 ± 20 |
In the specificity assay, we found that the HTDQ probe had a tendency to cross-react with these proteolytic products of alpha-thrombin, but that HTQ-3′ labeled alpha-thrombin with a high degree of specificity and provided full recovery of signal. However, PAb-T did not have the same stringency in labeling and reacted with all forms of thrombin bound to the probe, leading to large non-specific signals. Due to high variation in these signals, assays that employed PAb-T were unable to recover alpha-thrombin with reliability (Table 1).
The results from the specificity assay therefore show a clear advantage of using HTQ-3′, which was able to discriminate target in the close proximity of structurally similar proteins. HTQ-3′s specificity is likely due to the more iterative and evolutionary nature of the aptamer selection process. Additionally, the results of this assay highlight the importance of supplying probe in great excess of the target. Even though beta-thrombin and gamma-thrombin were bound to HTDQ, the capture efficiency of alpha-thrombin was unaffected since available probe sites did not saturate during the assay.
Conclusions
In this study, we have shown the feasibility of integrating encoded microgel particles with an aptamer-based capture and detection scheme for sensitive and reliable protein detection. We note clear advantages for conducting the assay on a gel substrate in which the biomolecules have greater flexibility, less steric hindrance, and a well hydrated environment. We are able to optimize buffer systems that are compatible with target multiplexing, and we bypass problems such as dissociation of ligands over time by using a rapid and high throughput microfluidic scanner to analyze our particles. Unlike demonstrations with planar substrates, we show that adding spacers in our probe has minimal effect on the detection capability of our particles. We additionally show that high abundance serum proteins do not cause significant assay interference, and that aptamers integrated into this platform may offer distinct advantages in terms of specificity in a media such as serum. Our ability to successfully interface aptamer-based protein detection with the SFL platform allows for the future development of molecular diagnostics and biological discovery using a range of biomolecules.
Supplementary Material
Acknowledgments
This work was supported by the Ragon Institute of MGH, MIT & Harvard, and the NIH T32 GM08334 Interdepartmental Biotechnology Training Grant.
References
- 1.Nguyen T, Hilton JP, Lin Q. Microfluid Nanofluid. 2009;6:347–362. [Google Scholar]
- 2.Mosing RK, Bowser MT. J Sep Sci. 2007;30:1420–1426. doi: 10.1002/jssc.200600483. [DOI] [PubMed] [Google Scholar]
- 3.Collett JR, Cho EJ, Ellington AD. Methods. 2005;37:4–15. doi: 10.1016/j.ymeth.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Cho EJ, Lee JW, Ellington AD. Annu Rev Anal Chem. 2009;2:241–264. doi: 10.1146/annurev.anchem.1.031207.112851. [DOI] [PubMed] [Google Scholar]
- 5.Breaker RR. Nature. 2004;432:838–845. doi: 10.1038/nature03195. [DOI] [PubMed] [Google Scholar]
- 6.Navani NK, Li YF. Curr Opin Chem Biol. 2006;10:272–281. doi: 10.1016/j.cbpa.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Iliuk AB, Hu L, Tao WA. Anal Chem. 2011;83:4440–4452. doi: 10.1021/ac201057w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hasegawa H, Taira KI, Sode K, Ikebukuro K. Sensors. 2008;8:1090–1098. doi: 10.3390/s8021090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho E, Collett J, Szafranska A, Ellington A. Anal Chim Acta. 2006;564:82–90. doi: 10.1016/j.aca.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 10.Birtwell S, Morgan H. Integr Biol. 2009;1:345–362. doi: 10.1039/b905502a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Q, Zhu J, Van Eyk JE. Clinical Chemistry. 2010;56:314–318. doi: 10.1373/clinchem.2009.135087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunker SE, Cederquist KB, Keating CD. Nanomedicine. 2007;2:695–710. doi: 10.2217/17435889.2.5.695. [DOI] [PubMed] [Google Scholar]
- 13.Broder GR, Ranasinghe RT, She JK, Banu S, Birtwell SW, Cavalli G, Galitonov GS, Holmes D, Martins HFP, MacDonald KF, Neylon C, Zheludev N, Roach PL, Morgan H. Anal Chem. 2008;80:1902–1909. doi: 10.1021/ac7018574. [DOI] [PubMed] [Google Scholar]
- 14.Pregibon DC, Toner M, Doyle PS. Science. 2007;315:1393–1396. doi: 10.1126/science.1134929. [DOI] [PubMed] [Google Scholar]
- 15.Chapin SC, Appleyard DC, Pregibon DC, Doyle PS. Angew Chem. 2011;50:2289–2293. doi: 10.1002/anie.201006523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Appleyard DC, Chapin SC, Doyle PS. Anal Chem. 2010;83:193–199. doi: 10.1021/ac1022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Appleyard DC, Chapin SC, Srinivas RL, Doyle PS. Nat Protoc. 2011 doi: 10.1038/nprot.2011.400. In Press. [DOI] [PubMed] [Google Scholar]
- 18.Dendukuri D, Gu SS, Pregibon DC, Hatton TA, Doyle PS. Lab Chip. 2007;7:818–828. doi: 10.1039/b703457a. [DOI] [PubMed] [Google Scholar]
- 19.Zubtsov DA, Savvateeva EN, Rubina AY, Pan’kov SV, Konovalova EV, Moiseeva OV, Chechetkin VR, Zasedatelev AS. Anal Biochem. 2007;368:205–213. doi: 10.1016/j.ab.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 20.Sorokin NV, Chechetkin VR, Pan’kov SV, Somova OG, Livshits MA, Donnikov MY, Turygin AY, Barsky VE, Zasadetalav AS. J Biomol Struct Dyn. 2006;24:57–66. doi: 10.1080/07391102.2006.10507099. [DOI] [PubMed] [Google Scholar]
- 21.Tasset DM, Kubik MF, Steiner W. J Mol Biol. 1997;272:688–698. doi: 10.1006/jmbi.1997.1275. [DOI] [PubMed] [Google Scholar]
- 22.Tracy RP. Chest. 2003;124:49S–57S. doi: 10.1378/chest.124.3_suppl.49s. [DOI] [PubMed] [Google Scholar]
- 23.Akiyama A, Ikedaa K, Kondoa H, McGeer PL. Neurosci Lett. 1992;146:152–154. doi: 10.1016/0304-3940(92)90065-f. [DOI] [PubMed] [Google Scholar]
- 24.Xi G, Reiser G, Keep RF. J Neurochem. 2002;84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x. [DOI] [PubMed] [Google Scholar]
- 25.Lao YH, Peck K, Chen LC. Anal Chem. 2009;81:1747–1754. doi: 10.1021/ac801285a. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Liu B. Langmuir. 2009;25:12787–12793. doi: 10.1021/la901703p. [DOI] [PubMed] [Google Scholar]
- 27.Centi S, Tombelli S, Minunni M, Mascini M. Anal Chem. 2007;79:1466–1473. doi: 10.1021/ac061879p. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Q, Lu X, Yuan CG, Li XF, Le XC. Anal Chem. 2009;81:7484–7489. doi: 10.1021/ac900961y. [DOI] [PubMed] [Google Scholar]
- 29.Tennico YH, Hutanu D, Koesdjojo MT, Bartel CM, Remcho VT. Anal Chem. 2010;82:5591–5597. doi: 10.1021/ac101269u. [DOI] [PubMed] [Google Scholar]
- 30.Bini A, Minunni M, Tombelli S, Centi S, Mascini M. Anal Chem. 2007;79:3016–3019. doi: 10.1021/ac070096g. [DOI] [PubMed] [Google Scholar]
- 31.Song M, Zhang Y, Li T, Wang Z, Yin J, Wang H. J Chromatogr A. 2009;1216:873–878. doi: 10.1016/j.chroma.2008.11.085. [DOI] [PubMed] [Google Scholar]
- 32.Xie S, Walton SP. Biosens Bioelectron. 2010;25:2663–2668. doi: 10.1016/j.bios.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu S, Zhang X, Luo W, Wang Z, Guo X. Angew Chem. 2011;50:2496–2502. doi: 10.1002/anie.201006469. [DOI] [PubMed] [Google Scholar]
- 34.Xiang Y, Zhang Y, Qian X, Chai Y, Wang J, Yuan R. Biosens Bioelectron. 2010;25:2539–2542. doi: 10.1016/j.bios.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Lee HJ, Corn RM. Anal Chem. 2007;79:1082–1088. doi: 10.1021/ac061849m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baldrich E, Restrepo A, O’Sullivan CK. Anal Chem. 2004;76:7053–7063. doi: 10.1021/ac049258o. [DOI] [PubMed] [Google Scholar]
- 37.Balamurugan S, O A, McCarley RL, Soper SA, Spivak DA. Anal Chem. 2008;80:9630–9634. doi: 10.1021/ac8009559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stadtherr K, Wolf H, Lindner P. Anal Chem. 2005;77:3437–3443. doi: 10.1021/ac0483421. [DOI] [PubMed] [Google Scholar]
- 39.Dandy DS, Wu P, Grainger DW. Proc Natl Acad Sci U S A. 2007;104:8223–8228. doi: 10.1073/pnas.0606054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pregibon D, Doyle P. Anal Chem. 2009;81:873–4881. doi: 10.1021/ac9005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho M, K Y, Han S, Min K, Rahman MA, Shim Y, q Ban C. BMB Rep. 2007;41:126–130. doi: 10.5483/bmbrep.2008.41.2.126. [DOI] [PubMed] [Google Scholar]
- 42.Smith KA. Science. 1988;240:1169–1176. doi: 10.1126/science.3131876. [DOI] [PubMed] [Google Scholar]
- 43.Peck D, Crawford ED, Ross KN, Stegmaier K, Golub TR, Lamb J. Genome Biol. 2006;7:-. doi: 10.1186/gb-2006-7-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chowdhury F, Williams A, Johnson P. J Immunol Methods. 2009;340:55–64. doi: 10.1016/j.jim.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 45.Yang H, Ji J, Liu Y, Kong J, Liu B. Electrochem Commun. 2009;11:38–40. [Google Scholar]
- 46.Ferguson WS, Finlay TH. Arch Biochem Biophys. 1983;220:301–208. doi: 10.1016/0003-9861(83)90414-9. [DOI] [PubMed] [Google Scholar]
- 47.Chapin SC, Doyle PS. Anal Chem. 2011;83:7179–7185. doi: 10.1021/ac201618k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fenton JW, Fasco MJ, Stackrow AB. J Biol Chem. 1976;252:3587–3598. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.