

Abstract
Rapid and accurate tests are currently needed to identify individuals with high levels of Loa loa microfilaria (mf), so that these individuals may be excluded from mass ivermectin administration campaigns against onchocerciasis and lymphatic filariasis being conducted in areas where Onchocerca volvulus, Wuchereria bancrofti, and L. loa are coendemic. To address this need, colorimetric loop-mediated isothermal amplification (LAMP) assays targeting the L. loa-specific gene sequences LLMF72 and LLMF342 were developed for the detection and quantification of L. loa microfilaremia. Both LAMP assays were highly specific (100%) for L. loa infection compared to the absence of infection or infection with related filarial pathogens. The LLMF72-based LAMP assay showed greater analytic sensitivity (limit of detection, 0.1 pg/ml of genomic DNA [gDNA] and/or 5 mf/ml) than the LLMF342-based LAMP assay (10 pg/ml of gDNA and/or 50 mf/ml), and its analytic sensitivity was similar to that of LLMF72-based quantitative PCR (qPCR). A high level of correlation was observed between microfilaria counts as determined by LLMF72-based qPCR and time to positivity by the LAMP assay, and performance measures of sensitivity, specificity, and positive and negative predictive values were similar for both assays when applied to field-collected clinical samples. By simply varying the run time, the LAMP assay was able to accurately distinguish individuals at risk for serious adverse events (SAEs) after exposure to ivermectin, using thresholds of >5,000 mf/ml and >30,000 mf/ml as indicators of increasing levels of risk. In summary, LLMF72 LAMP represents a new molecular diagnostic tool that is readily applicable as a point-of-care method for L. loa microfilarial detection and quantification in resource-limited countries where L. loa infection is endemic.
INTRODUCTION
Loa loa, the causative agent of loiasis, is a parasitic nematode transmitted to humans by the tabanid Chrysops fly (1), with transmission confined to the rainforest and some savannah areas of West and Central Africa (2). Although the overwhelming majority of L. loa-infected individuals are clinically asymptomatic, Calabar swelling (transient, localized angioedema), and the subconjunctival migration of an adult worm (“eyeworm”) are the most common clinical manifestations (3–5). Rarely, nephropathy, cardiomyopathy, retinopathy, neuropsychiatric complications, and encephalopathy (6–12) can occur as a consequence of chronic infection. Among the 8 filarial infections of humans, L. loa infection has largely been neglected as a public health concern in Africa, though it has gained prominence of late because of the neurologic serious adverse events (SAEs) that have occurred, occasionally leading to death, in highly microfilaremic individuals following exposure to ivermectin (IVM) given during mass drug administration (MDA) for control of Onchocerca volvulus and/or Wuchereria bancrofti in regions where these filarial species and L. loa are coendemic. (13, 14).
Although the pathogenesis of the neurologic SAEs is still not well understood (8, 15), data clearly demonstrate a relationship between the pretreatment L. loa microfilaria (mf) density (14, 16) and the risk of SAEs, with levels of 5,000 mf/ml and 30,000 mf/ml felt to be the most significant thresholds (13), though other factors may also be involved (17–19). Consequently, some IVM-based MDA programs for onchocerciasis (Ov) have been delayed, and lymphatic filariasis (LF) control programs (using IVM-albendazole) have been put on hold in large parts of Central Africa where L. loa infection is also endemic (20).
In areas where L. loa infection is endemic, the routine method for diagnosis and quantification of L. loa is based on calibrated microscopic examination of midday blood samples for microfilariae, a process that is neither point of care nor high throughput. Indeed, it is felt to be impractical for use as a widespread screening tool (21). Alternative serological (22, 23) and molecular (24–26) tests have been developed, but to date, only real-time PCR methodology has been able to combine a high degree of sensitivity and specificity with the ability to accurately quantify L. loa mf levels (26, 27). These methods, however, require a centralized, well-equipped laboratory and relatively expensive reagents.
Loop-mediated isothermal amplification (LAMP) has emerged as a potential alternative to DNA amplification techniques requiring thermocycling and automated fluorescence description (28–30). LAMP relies on an autocycling strand displacement DNA synthesis performed at a single temperature (30, 31). The by-product of the LAMP reaction is magnesium pyrophosphate that accumulates as the reaction progresses and that can be monitored using turbidity measurements or visually using a variety of intercalating dyes (e.g., SYBR green I, calcein, hydroxy naphthol blue [HNB]). In particular, adding HNB dye to the reaction tube before amplification has improved the capacity of the LAMP method to be used as a point-of-care diagnostic test, since it has made the assay easier to use in resource-limited settings (32, 33). Recently, LAMP technology has been applied with high accuracy to detect infection with a wide array of pathogens, including bacteria (34, 35), viruses (36), and parasites (28, 37, 38).
In the present study, we have used two L. loa-specific single-copy target genes, LLMF72 and LLMF342, to design LAMP primers for detection of L. loa mf in human blood and have developed methods that have a sensitivity and specificity similar to those of the previously described quantitative PCR (qPCR) assay targeting the LLMF72 gene (26). Furthermore, we have identified LAMP conditions that allow for colorimetric determination of microfilarial levels above or below specified thresholds such that our LAMP method now has the potential to be a practical point-of-care tool for identifying people at risk for SAEs when given IVM.
MATERIALS AND METHODS
Samples.
The source of genomic DNA from L. loa, W. bancrofti, Brugia malayi, and Mansonella perstans microfilariae (mf) has been described previously (26). For some experiments, 100,000 purified L. loa mf were placed in a fixed volume (1 ml) of distilled water and serially diluted to create standards for quantification. DNA was then extracted and used for assessment of the sensitivity of the assays. Finally, to evaluate the performance of the L. loa loop-mediated isothermal amplification (LAMP) assay compared to the standard quantitative PCR (qPCR) method, 93 midday dried blood spots on filter paper (Whatman no. 1 filter paper) previously collected in Cameroon (26) were assessed. These blood spots were obtained by pricking the fingers of volunteers living in a region where L. loa infection is endemic. Briefly, capillary blood was spotted onto Whatman filter paper, dried, and stored at 4°C until their use for DNA extraction.
DNA extraction for spiked mf and field-collected blood spot samples.
Blood spots were punched using a disposable 6-mm biopsy punch (Acuderm, Inc., Ft. Lauderdale, FL, USA). Two punched blood spots (10 μl dried blood each) were immersed in 400 μl of water in a Precellys lysing tube (Peqlab, Wilmington, DE, USA) containing glass and ceramic beads. The tube content was homogenized at 6,500 relative centrifugal force (rcf) for 5 min using a Precellys24 bead beating machine (Peqlab, Wilmington, DE, USA). The homogenized samples were then heated for 30 min at 99°C while shaking in a thermomixer (Eppendorf, North America, USA). Finally, the samples were centrifuged at 15,700 rcf for 10 min, and the supernatant was collected for use. DNA was also isolated from water spiked with 100,000 purified L. loa mf/ml using the method described above.
Primer design.
L. loa mf-specific LAMP primers were designed to target the L. loa-specific gene sequences LLMF72 (GenBank accession no. HM753552.1) and LLMF342 (intronic region 1,900 to 2,200 of an L. loa contig 3.498; GenBank accession no. ADBU02000498.1) using Primer Explorer v4 (http://primerexplorer.jp/elamp4.0.0/index.html). A set of six specific primers comprising two outer primers, two inner or internal primers, and two loop primers were designed for each targeted gene. Details of the sequences and targets of the designed primers are listed in Table 1. All of the primers (Eurofins MWG Operon, Huntsville, AL, USA) were purified by high-performance liquid chromatography (HPLC). Their specificity was further confirmed using the BLAST algorithm (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
TABLE 1.
Characteristics and sequences of LAMP primers
| Target gene | Primera | Primer sequence | Length (no. of nucleotides) | % GC |
|---|---|---|---|---|
| LLMF72 | F3 | AGATTTGACGGCAACGGAAG | 20 | 50 |
| B3 | GCGTCAGTTTCGTGTTGTGA | 20 | 50 | |
| FIP | CCGGAATCAGAGGAACGCTTGATCAACGTCAGAAATCAGCCA | 42 | 50 | |
| BIP | GCACAGCAGAGTCTTCTAGTGGCGTTGATGACGCTCCCAA | 40 | 55 | |
| LF | GGTGATGTAAAAGCAGGCTGT | 21 | 47.6 | |
| LB | TAAGTTTTCCAGGAACTGCACC | 22 | 45.5 | |
| LLMF342 | F3 | CAGCAGAGTGACTTACGT | 18 | 50 |
| B3 | AAAGCGATCGATTTGTCAAT | 20 | 35 | |
| FIP | CCGAGAATGTTGAGAGCATAGTAATGACGGTGAACAGTTACACA | 44 | 43.2 | |
| BIP | AAGCTCATCAGGAATTATCCTTCTGAGAAAAATGACGGTTACGAAAT | 47 | 36.2 | |
| LF | TGCTTAATTGGTGCCTTGAAGC | 22 | 45.5 | |
| LB | TAGTTATCTCGTATACGGCGGTCA | 24 | 45.8 |
F3, forward outer primer; B3, backward outer primer; FIP, forward internal primer; BIP, backward internal primer; LF, loop forward primer; LB, loop backward primer.
LAMP assay.
The LAMP reaction was performed using a Loopamp DNA amplification kit designed by the Eiken Chemical Company (Eiken Chemical Co., Ltd., Tokyo, Japan). Briefly, the reaction was carried out in a total volume of 25 μl (23 μl of the reaction mixture and 2 μl of the DNA template) in PCR microtubes (United Scientific Products, San Leandro, CA, USA). The reaction mixture contained 50 pmol each of the FIP (forward internal primer) and BIP (backward internal primer), 5 pmol each of the F3 (forward outer) and B3 (backward outer) primers, 25 pmol each of the LB (loop backward) and LF (loop forward) primers in 12.5 μl of a 2× buffer provided in the kit. Ultrapure water (7 μl), 1 μl of 1 mM hydroxy naphthol blue (HNB) (Sigma-Aldrich, Inc., St. Louis, MO, USA), 1 μl of the large fragment of Bst DNA polymerase (Eiken Chemical Co., Ltd., Tokyo, Japan), and 2 μl of the DNA template were added to complete the reaction mixture. Positive (L. loa mf genomic DNA, 100 pg/ml) and negative (water) controls were included in each experiment, and precautions were taken to prevent cross-contamination (all experiments were performed at an AirClean 600 PCR workstation [AirClean Systems, Creedmoor, NC, USA] with UV radiation for sterilization). All reactions were performed at 65°C (isothermal condition) for up to 60 min. The LAMP assay was stopped by incubation at 80°C for 5 min, inactivating the Bst DNA polymerase. The amplification efficiency was measured by the HNB color change or by monitoring turbidity using a real-time turbidimeter (Eiken Chemical Co., Ltd., Tokyo, Japan). A sample was considered positive for L. loa mf DNA if an obvious color change from purple to blue or an increase in turbidity was observed, compared to the negative control. All samples were run in duplicate. If the duplicates varied (one positive, one negative), the samples were rerun in triplicate.
Real-time PCR assay.
Quantitative PCR (qPCR) was performed as previously described (26); all samples were also tested for the ability to amplify a control plasmid DNA that was added to the DNA samples prior to DNA extraction to ensure a lack of inhibition (27).
Statistical analysis of results.
Statistical analyses, including specificity and sensitivity calculations and correlations (Spearman rank) were performed using GraphPad Prism 6.0 (GraphPad Software, Inc., San Diego, CA, USA).
RESULTS
Analytical specificity and sensitivity of Loa loa LAMP assay.
The species specificity of the LAMP assays was assessed using a real-time turbidimeter (Fig. 1A) with the LLMF72 set of primers. As can be seen, these primers fail to amplify genomic DNA from B. malayi, W. bancrofti, O. volvulus, and M. perstans, whereas they amplified genomic DNA (gDNA) from L. loa easily and did so in a dose-dependent manner. The primers derived from LLMF72 and LLMF342 also failed to amplify genomic DNA from any of the five Plasmodium species capable of infecting humans (data not shown).
FIG 1.
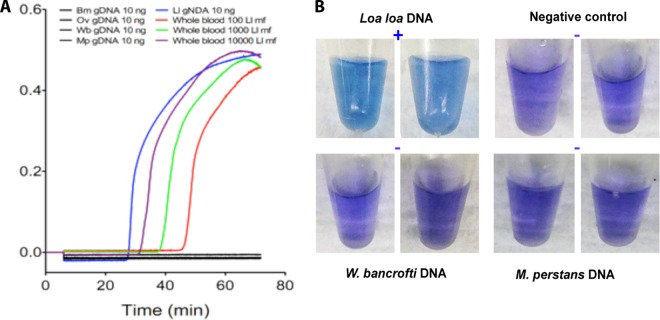
Specificity of LAMP assays for detection of Loa loa DNA. (A) Real-time turbidometry of the LAMP assay targeting the L. loa-specific gene sequence LLMF72 using genomic DNA from B. malayi (Bm), W. bancrofti (Wb), O. volvulus (Ov), or M. perstans (Mp) (all in black), or L. loa (Ll) (in blue) or DNA extracted from normal whole blood spiked with 10,000 (purple), 1,000 (green), or 100 (red) intact L. loa microfilariae (mf). (B) Colorimetric results of LAMP assay using LLMF72 primers on genomic DNA from L. loa, W. bancrofti, and M. perstans, or water as a negative control.
To allow for a simplified detection method that could be visualized by the naked eye, colorimetric detection of the LAMP assays using a HNB dye was performed next (Fig. 1B). For both LAMP assays targeting the L. loa-specific gene sequences LLMF72 and LLMF342 (not shown), specificity for L. loa could be seen in that only the L. loa DNA samples were blue (positive), whereas samples with each of the other related filarial species were purple (negative).
To assess the analytic sensitivity of the LAMP assays, the limit of DNA detection was determined by testing serial dilutions of genomic DNA obtained from different numbers of L. loa microfilariae (mf) (Fig. 2). With the LLMF72 assay, the smallest amount of detectable gDNA was 0.1 pg/ml (Fig. 2A), whereas for the LLMF342 assay, the limit of detection was 10 pg/ml (Fig. 2A). When both sets of primers were used together (Fig. 2A) in an attempt to improve the sensitivity even further, we found little to no improvement over the analytic sensitivity of LLMF72 alone. Similarly, when these assays were performed on DNA extracted from different concentrations of mf, we found that the limits of detection were 5 mf/ml for LLMF72 primer, 50 mf/ml for LLMF342 primer, and 5 mf/ml for the two sets of primers together (Fig. 2B).
FIG 2.
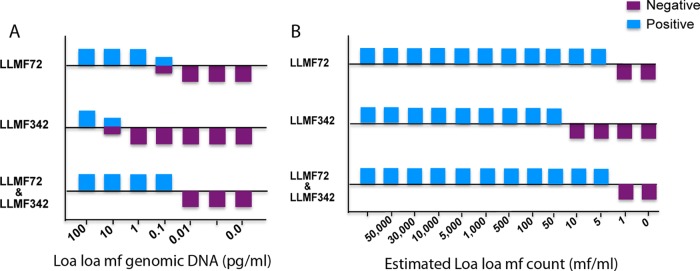
Analytic performance of colorimetric L. loa LAMP assays. (A and B) Serial dilutions of genomic DNA (A) or DNA extracted from serial dilutions of water spiked with intact L. loa microfilariae (B) were used as the template in LAMP assays with LLMF72 or LLMF342 primer sets or with multiplexed primer sets (LLMF72 and LLMF342 together). HNB was added to the reaction mixture.
Time-dependent threshold assessments for performing semiquantitative LAMP assays.
Because the goal of this project was to provide a potentially point-of-care method of amplification, detection, and quantitation, we next assessed whether our LAMP assay could be used in a semiquantitative manner by exploring how varying the reaction time affected the results of the colorimetric LAMP assays when performed on DNA obtained from different concentrations of L. loa mf. For each of the LLMF72, LLMF342, and LLMF72-LLMF342 LAMP assays, the color change in the reaction tubes from purple (negative) to blue (positive) was monitored every 5 min. The time to positivity was then plotted as a function of mf concentration (Fig. 3). For DNA samples prepared from L. loa mf concentrations of 30,000 mf/ml and above, the earliest color change was observed 15 min after initiation for LLMF72-based assays, 25 min for LLMF342-based assays, and 20 min for combined LLMF72-LLMF342-based assays. At the limits of detection for L. loa mf concentration (5 mf/ml in LLMF72 and LLMF72-LLMF342 assays and 10 mf/ml in LLMF342 LAMP), a color change occurred after 30 min for the LLMF72 assay and after 40 min for LLMF342 and the combined LLMF72-LLMF342 assays (Fig. 3). In addition, over the range of mf concentration dilutions, there was a high degree of correlation observed between the time to LAMP reaction positivity (minutes) and the cycle number (threshold cycle [CT] value) obtained by qPCR for both the LLMF72 assay (Fig. 4A; r = 0.96 and P < 0.0001) and LLMF342 assay (Fig. 4B; r = 0. 97 and P < 0.0001).
FIG 3.

Semiquantitative application of colorimetric L. loa LAMP assays based on time to positivity. The time to color change (in minutes) from violet to blue using primers specific for LLMF72, LLMF342, or both together was determined for samples prepared with different concentrations of L. loa mf.
FIG 4.

Correlation between colorimetric L. loa LAMP time to positivity and quantitative PCR (qPCR) cycle number. (A and B) The time to LLMF72 (A) or LLMF342 (B) LAMP assay positivity for samples prepared from different L. loa mf DNA concentrations is correlated with qPCR cycle number when run on the same samples. The Spearman r and P value for each correlation are indicated.
On the basis of our observations with varying the LAMP assay run time, we identified run times that would allow for naked-eye determination of whether the L. loa mf concentration in a sample was above or below specified threshold levels (such as those that might be used to estimate the risk of neurologic SAEs after exposure to ivermectin). The run times identified for L. loa mf by examination of the LLMF72 assay were 15 min for a threshold of >30,000 mf/ml, 20 min for a threshold of >5,000 mf/ml, and 25 min for a threshold of 100 mf/ml. This difference in time to positivity based on the amount of DNA was also observed in the LLMF342 and LLMF72-LLMF342 assays, though the run time for which a positive test corresponded to the specified threshold concentrations of 30,000, 5,000, and 100 mf/ml differed slightly in the various assays.
Loa loa LLMF72 LAMP performance compared to qPCR.
To further assess the performance of the LAMP assay, a formal comparison was made between LLMF72-based qPCR and colorimetric LAMP using field-collected dried blood spots obtained from 93 individuals living in regions of Cameroon where L. loa infection is endemic. Of the 93 samples tested, 50 were positive and 43 were negative by qPCR. All qPCR-positive samples were also positive by LAMP, and there were 3 additional qPCR-negative samples that were positive or indeterminate (not purple but not completely blue) in the LAMP assay (presumably false-positive results). Thus, considering qPCR to be the “gold standard,” the LAMP assay has a sensitivity of 100.0% (95% confidence interval [95% CI], 92.9% to 100.0%), a specificity of 93.0% (95% CI, 80.9% to 98.5%), a positive predictive value of 94.3% (95% CI, 94.3% to 98.8%), and a negative predictive value of 100.0% (95% CI, 91.2% to 100.0%).
DISCUSSION
Because coincident L. loa infection has had a severely negative impact on control (MDA) programs for onchocerciasis and lymphatic filariasis in West and Central Africa, there is a consensus that point-of-care diagnostics are needed for L. loa infection in regions of the world where Onchocerca volvulus, Wuchereria bancrofti, and L. loa infections are coendemic to achieve global elimination goals for O. volvulus and W. bancrofti. Current qPCR-based methods are highly sensitive, specific, and quantitative but require sophisticated equipment and are costly. Thus, we developed a LAMP assay and demonstrated its potential for use as a point-of-care diagnosis tool in field settings.
Both LLMF72- and LLMF342-based LAMP assays were highly specific for L. loa with respect to other related filarial parasites (W. bancrofti, M. perstans, B. malayi, and O. volvulus). This result was expected based on de novo bioinformatics assessments previously performed in a slightly different context (26). Moreover, its analytic sensitivity was as high as that of qPCR (Fig. 4). The LAMP assay was also highly comparable to the “gold standard” qPCR in terms of clinical sensitivity and specificity. In addition, its predictive positive (94.3%) and negative (100.0%) values support its use for clinical diagnostic purposes.
Although LAMP assays (like qPCR) can be quantitated in real time using a sophisticated turbidimeter (38), having the ability to perform the assays at the point of care or with minimal instrumentation led us to monitor our assays using a colorimetric readout through the use of HNB dye, previously shown to be useful in endpoint LAMP assays (32). Thus, the HNB-based LAMP assay provides a potential point-of-care method of rapid amplification and easy detection of L. loa DNA that, when standardized, can accurately distinguish the levels of mf that are correlated with increased risk for SAEs (>30,000 mf/ml; LAMP assay positive at 15 min) from the levels for individuals who might not be at risk (<5,000 mf/ml; LAMP assay positive at 20 min). By following the time to positivity of our LAMP assays, we were able to make these assays semiquantitative and useful for specified threshold detection of mf burden. This has important practical implications for MDA efforts in regions where onchocerciasis, lymphatic filariasis, and L. loa infection are endemic and where identifying individuals at increased risk of neurologic SAEs after ivermectin treatment (14, 39) is of paramount usefulness.
LAMP has other significant advantages over current methods for the molecular diagnosis of infection. First, the Bst polymerase catalyzing the LAMP reaction is less likely to be inhibited than is Taq polymerase (40, 41). Second, the colorimetric readout that occurs in as little as 15 min means that the LAMP assay results can be obtained without the use of sophisticated instrumentation. Finally, the ability to perform high-throughput analysis of samples in 96-well microplates (32, 42) and to run the assay in a heated water bath or other constant temperature heating instrument provide advantages that should allow LAMP to move out of the laboratory and to be performed in field conditions (43).
Precedence for point-of-care LAMP-based diagnostic assays in low-resource settings has been reported in two recent studies involving detection of malaria parasites in clinical samples in rural Uganda (37, 44). Compared to a single well-nested PCR, the LAMP-based malaria diagnostic assay importantly lowers the detection threshold and thereby opens new avenues for diagnosis, surveillance, and screening in elimination strategies (37). Additionally, LAMP assays offer advantages of speed and accuracy compared to currently available methods when used as primary diagnostic tools for evaluation of febrile travelers returning to resource-rich countries (44).
In the present study, compared to the reference qPCR assay, the LLMF72 LAMP assay has shown extremely good performance in samples run concurrently in the 2 types of assays (LAMP assays targeting the L. loa-specific gene sequences LLMF72 and LLMF342) obtained from an area of Cameroon where L. loa infection is endemic. One potential limitation of our L. loa LAMP assays is that sensitivity of any molecular assay could be compromised by variability in the target gene sequence across regions or over time as the result of selective pressures. The single-copy genes used in the present study, however, do not appear to be under selective pressure (based on small surveys of L. loa from multiple countries [data not shown]), and thus, these sequences appear to be invariant.
One additional limitation to LAMP assays as a point-of-care test is their cost, which at $5.31/test for the Loopamp DNA amplification kit used in our study is still too high for routine use in resource-limited countries. The use of ready-made reaction mixtures prepared with individual reagents ($0.28/test) may be a more accessible solution (45). Therefore, future product improvements should include scaling up throughput and lowering costs to address screening requirements in areas where L. loa infection is endemic.
In conclusion, we have developed LAMP assays capable of estimating L. loa mf burden. The assays are highly sensitive and species specific and can be used with a wide variety of DNA templates (gDNA, extracted DNA, boiled fresh whole blood, or blood spot samples). When applied to clinical samples, the LAMP assays performed similarly to reference qPCR with a greatly reduced cost and time. Therefore, LAMP assays represent a powerful alternative to PCR for diagnosis of L. loa infection in resource-limited countries, though it may require additional adjustments for field-ready point-of-care use. Nevertheless, they will ultimately benefit global health programs aimed at elimination of filarial infections.
ACKNOWLEDGMENTS
We thank Joseph Kubofcik for help with the qPCR methods and Sasi Bennuru for suggestions throughout.
P.M.D., D.L.F., J.K., and T.B.N. conceived and designed the experiments. P.M.D., J.A.H., and D.L.F. performed the experiments. P.M.D. and T.B.N. analyzed the data. P.M.D., D.L.F., and J.K. contributed reagents/materials/analysis tools. P.M.D. wrote the paper. D.L.F., J.K., and T.B.N. reviewed the paper.
The Division of Intramural Research (DIR) of the NIAID provided all of the funds for this study.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 2 April 2014
REFERENCES
- 1.Ratmanov P, Mediannikov O, Raoult D. 2013. Vectorborne diseases in West Africa: geographic distribution and geospatial characteristics. Trans. R. Soc. Trop. Med. Hyg. 107:273–284. 10.1093/trstmh/trt020 [DOI] [PubMed] [Google Scholar]
- 2.Kelly-Hope LA, Bockarie MJ, Molyneux DH. 2012. Loa loa ecology in central Africa: role of the Congo River system. PLoS Negl. Trop. Dis. 6:e1605. 10.1371/journal.pntd.0001605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akue JP, Nkoghe D, Padilla C, Moussavou G, Moukana H, Mbou RA, Ollomo B, Leroy EM. 2011. Epidemiology of concomitant infection due to Loa loa and Mansonella perstans in Gabon. PLoS Negl. Trop. Dis. 5:e1329. 10.1371/journal.pntd.0001329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takougang I, Meli J, Lamlenn S, Tatah PN, Ntep M. 2007. Loiasis-a neglected and under-estimated affliction: endemicity, morbidity and perceptions in eastern Cameroon. Ann. Trop. Med. Parasitol. 101:151–160. 10.1179/136485907X154511 [DOI] [PubMed] [Google Scholar]
- 5.Eballe AO, Epee E, Koki G, Owono D, Mvogo CE, Bella AL. 2008. Intraocular live male filarial Loa loa worm. Clin. Ophthalmol. 2:965–967 [PMC free article] [PubMed] [Google Scholar]
- 6.Cruel T, Arborio M, Schill H, Neveux Y, Nedelec G, Chevalier B, Teyssou R, Buisson Y. 1997. Nephropathy and filariasis from Loa loa. Apropos of 1 case of adverse reaction to a dose of ivermectin. Bull. Soc. Pathol. Exot. 90:179–181 (In French.) [PubMed] [Google Scholar]
- 7.Gobbi F, Boussinesq M, Mascarello M, Angheben A, Gobbo M, Rossanese A, Corachan M, Bisoffi Z. 2011. Case report: loiasis with peripheral nerve involvement and spleen lesions. Am. J. Trop. Med. Hyg. 84:733–737. 10.4269/ajtmh.2011.10-0458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmes D. 2013. Loa loa: neglected neurology and nematodes. Lancet Neurol. 12:631–632. 10.1016/S1474-4422(13)70139-X [DOI] [PubMed] [Google Scholar]
- 9.Toussaint D, Danis P. 1965. Retinopathy in generalized Loa loa filariasis. A clinicopathological study. Arch. Ophthalmol. 74:470–476. 10.1001/archopht.1965.00970040472007 [DOI] [PubMed] [Google Scholar]
- 10.Andy JJ, Bishara FF, Soyinka OO, Odesanmi WO. 1981. Loasis as a possible trigger of African endomyocardial fibrosis: a case report from Nigeria. Acta Trop. 38:179–186 [PubMed] [Google Scholar]
- 11.Lukiana T, Mandina M, Situakibanza NH, Mbula MM, Lepira BF, Odio WT, Kamgno J, Boussinesq M. 2006. A possible case of spontaneous Loa loa encephalopathy associated with a glomerulopathy. Filaria J. 5:6. 10.1186/1475-2883-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackenzie C, Geary T, Prichard R, Boussinesq M. 2007. Where next with Loa loa encephalopathy? Data are badly needed. Trends Parasitol. 23:237–238. 10.1016/j.pt.2007.04.007 [DOI] [PubMed] [Google Scholar]
- 13.Chippaux JP, Boussinesq M, Gardon J, Gardon-Wendel N, Ernould JC. 1996. Severe adverse reaction risks during mass treatment with ivermectin in loiasis-endemic areas. Parasitol. Today 12:448–450. 10.1016/0169-4758(96)40006-0 [DOI] [PubMed] [Google Scholar]
- 14.Gardon J, Gardon-Wendel N, Demanga N, Kamgno J, Chippaux JP, Boussinesq M. 1997. Serious reactions after mass treatment of onchocerciasis with ivermectin in an area endemic for Loa loa infection. Lancet 350:18–22. 10.1016/S0140-6736(96)11094-1 [DOI] [PubMed] [Google Scholar]
- 15.Boussinesq M, Kamgno J, Pion SD, Gardon J. 2006. What are the mechanisms associated with post-ivermectin serious adverse events? Trends Parasitol. 22:244–246. 10.1016/j.pt.2006.04.006 [DOI] [PubMed] [Google Scholar]
- 16.Boussinesq M, Gardon J, Gardon-Wendel N, Chippaux JP. 2003. Clinical picture, epidemiology and outcome of Loa-associated serious adverse events related to mass ivermectin treatment of onchocerciasis in Cameroon. Filaria J. 2(Suppl 1):S4. 10.1186/1475-2883-2-S1-S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamgno J, Boussinesq M, Labrousse F, Nkegoum B, Thylefors BI, Mackenzie CD. 2008. Encephalopathy after ivermectin treatment in a patient infected with Loa loa and Plasmodium spp. Am. J. Trop. Med. Hyg. 78:546–551 [PubMed] [Google Scholar]
- 18.Kamgno J, Pion SD, Mackenzie CD, Thylefors B, Boussinesq M. 2009. Loa loa microfilarial periodicity in ivermectin-treated patients: comparison between those developing and those free of serious adverse events. Am. J. Trop. Med. Hyg. 81:1056–1061. 10.4269/ajtmh.2009.09-0356 [DOI] [PubMed] [Google Scholar]
- 19.Pion SD, Filipe JA, Kamgno J, Gardon J, Basanez MG, Boussinesq M. 2006. Microfilarial distribution of Loa loa in the human host: population dynamics and epidemiological implications. Parasitology 133:101–109. 10.1017/S0031182006000035 [DOI] [PubMed] [Google Scholar]
- 20.World Health Organization. 2010. Progress report 2000–2009 and strategic plan 2010–2020 of the global programme to eliminate lymphatic filariasis: halfway towards eliminating lymphatic filariasis. WHO/HTM/NTD/PCT/2010.6. World Health Organization, Geneva, Switzerland [Google Scholar]
- 21.Molyneux DH. 2009. Filaria control and elimination: diagnostic, monitoring and surveillance needs. Trans. R. Soc. Trop. Med. Hyg. 103:338–341. 10.1016/j.trstmh.2008.12.016 [DOI] [PubMed] [Google Scholar]
- 22.Burbelo PD, Ramanathan R, Klion AD, Iadarola MJ, Nutman TB. 2008. Rapid, novel, specific, high-throughput assay for diagnosis of Loa loa infection. J. Clin. Microbiol. 46:2298–2304. 10.1128/JCM.00490-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klion AD, Vijaykumar A, Oei T, Martin B, Nutman TB. 2003. Serum immunoglobulin G4 antibodies to the recombinant antigen, Ll-SXP-1, are highly specific for Loa loa infection. J. Infect. Dis. 187:128–133. 10.1086/345873 [DOI] [PubMed] [Google Scholar]
- 24.Jimenez M, Gonzalez LM, Carranza C, Bailo B, Perez-Ayala A, Muro A, Perez-Arellano JL, Garate T. 2011. Detection and discrimination of Loa loa, Mansonella perstans and Wuchereria bancrofti by PCR-RFLP and nested-PCR of ribosomal DNA ITS1 region. Exp. Parasitol. 127:282–286. 10.1016/j.exppara.2010.06.019 [DOI] [PubMed] [Google Scholar]
- 25.Toure FS, Kassambara L, Williams T, Millet P, Bain O, Georges AJ, Egwang TG. 1998. Human occult loiasis: improvement in diagnostic sensitivity by the use of a nested polymerase chain reaction. Am. J. Trop. Med. Hyg. 59:144–149 [DOI] [PubMed] [Google Scholar]
- 26.Fink DL, Kamgno J, Nutman TB. 2011. Rapid molecular assays for specific detection and quantitation of Loa loa microfilaremia. PLoS Negl. Trop. Dis. 5:e1299. 10.1371/journal.pntd.0001299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fink DL, Fahle GA, Fischer S, Fedorko DF, Nutman TB. 2011. Toward molecular parasitologic diagnosis: enhanced diagnostic sensitivity for filarial infections in mobile populations. J. Clin. Microbiol. 49:42–47. 10.1128/JCM.01697-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cao L, Cheng R, Yao L, Yuan S, Yao X. 2014. Establishment and application of a loop-mediated isothermal amplification method for simple, specific, sensitive, and rapid detection of Toxoplasma gondii. J. Vet. Med. Sci. 76:9–14. 10.1292/jvms.13-0275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Wang G, Zhang D, Yin H, Wang M. 2013. Detection of acute toxoplasmosis in pigs using loop-mediated isothermal amplification and quantitative PCR. Kor. J. Parasitol. 51:573–577. 10.3347/kjp.2013.51.5.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28:E63. 10.1093/nar/28.12.e63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagamine K, Hase T, Notomi T. 2002. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probes 16:223–229. 10.1006/mcpr.2002.0415 [DOI] [PubMed] [Google Scholar]
- 32.Goto M, Honda E, Ogura A, Nomoto A, Hanaki K. 2009. Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxy naphthol blue. Biotechniques 46:167–172. 10.2144/000113072 [DOI] [PubMed] [Google Scholar]
- 33.Nie K, Zhao X, Ding X, Li XD, Zou SM, Guo JF, Wang DY, Gao RB, Li XY, Huang WJ, Shu YL, Ma XJ. 2013. Visual detection of human infection with influenza A (H7N9) virus by subtype-specific reverse transcription loop-mediated isothermal amplification with hydroxynaphthol blue dye. Clin. Microbiol. Infect. 19:E372–E375. 10.1111/1469-0691.12263 [DOI] [PubMed] [Google Scholar]
- 34.Aizawa Y, Oishi T, Tsukano S, Taguchi T, Saitoh A. 2014. Clinical utility of loop-mediated isothermal amplification for rapid diagnosis of Mycoplasma pneumoniae in children. J. Med. Microbiol. 63:248–251. 10.1099/jmm.0.068288-0 [DOI] [PubMed] [Google Scholar]
- 35.Soli KW, Kas M, Maure T, Umezaki M, Morita A, Siba PM, Greenhill AR, Horwood PF. 2013. Evaluation of colorimetric detection methods for Shigella, Salmonella, and Vibrio cholerae by loop-mediated isothermal amplification. Diagn. Microbiol. Infect. Dis. 77:321–323. 10.1016/j.diagmicrobio.2013.09.009 [DOI] [PubMed] [Google Scholar]
- 36.Wang G, Shang Y, Wang Y, Tian H, Liu X. 2013. Comparison of a loop-mediated isothermal amplification for orf virus with quantitative real-time PCR. Virol. J. 10:138. 10.1186/1743-422X-10-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hopkins H, Gonzalez IJ, Polley SD, Angutoko P, Ategeka J, Asiimwe C, Agaba B, Kyabayinze DJ, Sutherland CJ, Perkins MD, Bell D. 2013. Highly sensitive detection of malaria parasitemia in a malaria-endemic setting: performance of a new loop-mediated isothermal amplification kit in a remote clinic in Uganda. J. Infect. Dis. 208:645–652. 10.1093/infdis/jit184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poole CB, Tanner NA, Zhang Y, Evans TC, Jr, Carlow CK. 2012. Diagnosis of brugian filariasis by loop-mediated isothermal amplification. PLoS Negl. Trop. Dis. 6:e1948. 10.1371/journal.pntd.0001948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boussinesq M, Gardon J. 1998. Challenges for the future: loiasis. Ann. Trop. Med. Parasitol. 92(Suppl 1):S147–S151 [PubMed] [Google Scholar]
- 40.Kaneko H, Kawana T, Fukushima E, Suzutani T. 2007. Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J. Biochem. Biophys. Methods 70:499–501. 10.1016/j.jbbm.2006.08.008 [DOI] [PubMed] [Google Scholar]
- 41.Nkouawa A, Sako Y, Li T, Chen X, Wandra T, Swastika IK, Nakao M, Yanagida T, Nakaya K, Qiu D, Ito A. 2010. Evaluation of a loop-mediated isothermal amplification method using fecal specimens for differential detection of Taenia species from humans. J. Clin. Microbiol. 48:3350–3352. 10.1128/JCM.00697-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rijken MJ, Papageorghiou AT, Thiptharakun S, Kiricharoen S, Dwell SL, Wiladphaingern J, Pimanpanarak M, Kennedy SH, Nosten F, McGready R. 2012. Ultrasound evidence of early fetal growth restriction after maternal malaria infection. PLoS One 7:e31411. 10.1371/journal.pone.0031411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han ET. 2013. Loop-mediated isothermal amplification test for the molecular diagnosis of malaria. Expert Rev. Mol. Diagn. 13:205–218. 10.1586/erm.12.144 [DOI] [PubMed] [Google Scholar]
- 44.Polley SD, Gonzalez IJ, Mohamed D, Daly R, Bowers K, Watson J, Mewse E, Armstrong M, Gray C, Perkins MD, Bell D, Kanda H, Tomita N, Kubota Y, Mori Y, Chiodini PL, Sutherland CJ. 2013. Clinical evaluation of a loop-mediated amplification kit for diagnosis of imported malaria. J. Infect. Dis. 208:637–644. 10.1093/infdis/jit183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poon LL, Wong BW, Ma EH, Chan KH, Chow LM, Abeyewickreme W, Tangpukdee N, Yuen KY, Guan Y, Looareesuwan S, Peiris JS. 2006. Sensitive and inexpensive molecular test for falciparum malaria: detecting Plasmodium falciparum DNA directly from heat-treated blood by loop-mediated isothermal amplification. Clin. Chem. 52:303–306 [DOI] [PubMed] [Google Scholar]