

Abstract
A loop-mediated isothermal amplification (LAMP) assay for open reading frame 1 (ORF1) of the glutamine synthetase gene of Neisseria gonorrhoeae was able to tolerate urea concentrations of ≤1.8 M, compared with a PCR assay that was functional at concentrations of <100 mM. The LAMP assay was as sensitive as the PCR assay while being faster and simpler to perform.
TEXT
Gonorrhea is the second most common sexually transmitted infection in the developed world (1), with 106 million incidences of gonorrhea infection reported yearly worldwide (2), representing a significant global health burden.
The use of nucleic acid amplification testing (NAAT) for gonorrhea is becoming more widespread. NAAT offers unparalleled specificities and sensitivities, with PCR assays typically boasting sensitivities and specificities of >95% and 100%, respectively, compared with sensitivities of ∼68% obtained via culture (3).
Loop-mediated isothermal amplification (LAMP) is a novel nucleic acid amplification method that was designed to amplify target nucleic acids under isothermal conditions in a highly specific and rapid manner (4). The LAMP reaction requires a constant temperature of ∼63°C and can be easily carried out in a water bath (5). LAMP assays enable more rapid diagnoses, offer improved sensitivities (6, 7), and have higher tolerances for inhibitors that are present in a number of clinical samples, including feces and blood, than do PCR-based tests (8).
Diagnosing genital Neisseria gonorrhoeae infection by NAAT is carried out from a variety of sample types, including invasive swab samples and urine specimens. Urea, present in urine, is a known inhibitor of PCRs at concentrations of >50 mM (9). Urea prevents the noncovalent bonding of polymerase enzymes and interferes with primer annealing, hence inhibiting amplification (10). Diagnostically, this leads to an increase in the likelihood of false-negative results and a reduction in assay sensitivity (11). Although the concentration of urea in adult human urine is highly variable, a previous study (9) found the average concentration of urea to be ∼300 mM, which is above the minimum concentration needed for PCR inhibition. The most simple methods for nucleic acid extraction, such as the preparation of a crude lysate, Chelex-based methods, and proteinase K digestion, are unsuitable for processing urine samples for PCR due to their inability to remove the inhibitory urea. Therefore, the use of more complex extraction methods is recommended (12).
In the present study, the minimum concentration of urea that was inhibitory to LAMP was determined in order to test the suitability of the LAMP assay for testing urine samples with simple extraction methods. The detection of N. gonorrhoeae DNA from spiked urine samples was used to test this principle.
N. gonorrhoeae (strain ATCC 19424) was cultured on chocolate agar (blood agar base [Sigma-Aldrich, United Kingdom] and defibrinated horse blood [TCS, United Kingdom]) at 37°C in 5% CO2.
LAMP primers were designed for open reading frame 1 (ORF1) of the glutamine synthetase (glnA) gene (GenBank accession number M84113) of N. gonorrhoeae. This region offers excellent diagnostic sensitivity as a PCR target for this organism (13). The LAMP primers were designed using PrimerExplorer version 4 (see http://primerexplorer.jp/e/). The ORF1 LAMP primer sequences were F3 (5′-TGG TCG GTG CTT CAA AGT G-3′), B3 (5′-GCA CGT CCA CCA ATC CAT T-3′), FiP (5′-CAA ACA CGC CAA AGC CCT GAA CGC ACG AGG CGT TTG TAG G-3′), and BiP (5′-TGT AGT AGA GCG CGG TAT CGG ACG GTC AAA ACC TGT TCG CA-3′). PCR primers specific to the same target were designed using Primer3 (see http://bioinfo.ut.ee/primer3-0.4.0/). The ORF1 PCR forward and reverse primer sequences were 5′-GAC GTA TTT CAT ATC TTG GG-3′ and 5′-GGT GAA CAT TTT GGA AG-3′, respectively. Primer specificities were checked using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
DNA extractions from N. gonorrhoeae cell suspensions were carried out using a DNA minikit (Qiagen, Inc., USA). DNA quantification was carried out using a Spectrostar plate reader in conjunction with an LVis plate (BMG Labtech, United Kingdom). The corresponding genome copy number was calculated from the weight of the N. gonorrhoeae genome, which is 2.45 fg (2.2 × 106 bp × 665 Da/bp × 1.67 × 10−24 g/Da) (14).
PCRs were set up with 25 μl of RedTaq reaction mix (Sigma-Aldrich, United Kingdom), 0.5 pmol forward primer, 0.5 pmol reverse primer, and 5 μl DNA sample. Reaction mixtures were then made up to 50 μl with molecular-grade water. The reaction conditions were 94°C for 5 min of initial denaturation, followed by 35 cycles at 94°C for 30 s, 47°C for 30 s, and 72°C for 30 s, and then final extension at 72°C for 5 min. Thermal cycling was carried out in an ABI 2720 instrument (Applied Biosystems, United Kingdom). The reaction product was a 139-bp fragment.
LAMP reactions were set up using reagents from a Mast Isoplex DNA amplification kit (Mast Group Ltd., United Kingdom; under license from Eiken Chemical Co., Japan) according to the manufacturer's instructions. The final primer concentrations were 0.2 μM for F3 and B3 and 1.6 μM for BiP and FiP. Reactions were monitored in real time using an ESEQuant tube scanner (Qiagen, Inc., United Kingdom). Reactions were terminated by heating them to 80°C for 1 min. Detection of the reaction products from the PCR and LAMP reactions was carried out by electrophoresis, using 2% (wt/vol) agarose gels with UV transillumination. For endpoint visual detection, the 1 μl of intercalating dye was replaced with 1 μl of calcein (a fluorescent detection reagent; Eiken Chemical Co., Japan).
As a small-scale test of the specificities of the ORF1 LAMP and PCRs, reaction mixtures containing 0.1 μg of DNA each from Chlamydia trachomatis (lab strain, serovar L2), Trichomonas vaginalis (strain ATCC 30001), and Pseudomonas aeruginosa (strain ATCC 15692) were set up for each assay. These organisms are all causes of urethritis. No amplifications were seen, confirming the high levels of primer specificity.
The limit of detection of both the N. gonorrhoeae ORF1 PCR and LAMP assays was found to be 20 copies of the N. gonorrhoeae genome (Table 1). Real-time monitoring showed that the time to amplification was shorter with an increased starting concentration of the template, with the reaction containing 2 × 105 copies showing the earliest amplification and crossing the manually set threshold at 17 min (Table 1). The latest time to amplification (27.3 min) was seen in the reaction containing 20 copies. The times to amplification were within 2 min in all 3 replications of the experiment.
TABLE 1.
Sensitivities of ORF1 LAMP and PCR assays
| N. gonorrhoeae genome copy no. | LAMP time to amplification (min)a | PCR result |
|---|---|---|
| 2 × 105 | 17 | + |
| 2 × 104 | 18.6 | + |
| 2 × 103 | 20.7 | + |
| 2 × 102 | 25 | + |
| 20 | 27.3 | + |
| 2 | No amplification | − |
| 0 | No amplification | − |
Average times to amplification of ORF1 LAMP reactions containing 2 × 105, 2 × 104, 2 × 103, 2 × 102, 20, or 2 copies of the N. gonorrhoeae genome. Time to amplification was taken as the time point that the fluorescent signal exceeded a manually set threshold line on the Tube Scanner Studio software package (Qiagen). A LAMP reaction containing 5 μl of water instead of DNA was used as a negative control. ORF1 PCRs were carried out from the same DNA dilutions, and the products were separated using electrophoresis and then were visualized.
To determine the minimum concentration of urea that was inhibitory to the ORF1 LAMP and the PCR assays, the LAMP reactions and PCRs, including 0.1 μg of N. gonorrhoeae DNA, were formulated with 1 mM, 10 mM, 100 mM, 1 M, and 2 M concentrations of urea. Additional ORF1 LAMP reactions were carried out with 1 M, 1.2 M, 1.4 M, 1.6 M, 1.8 M, and 2 M urea to define the sensitivities of the reactions at these higher levels. Molecular-grade urea powder (Sigma, United Kingdom) was dissolved at 30°C in molecular-grade water to 8.5 M and then sterilized by filtration, diluted appropriately, and added to the LAMP reaction in place of water. The ORF1 PCR tolerated a urea concentration of 10 mM, whereas a concentration of 100 mM proved to be inhibitory (Fig. 1A). The LAMP assay detected N. gonorrhoeae DNA at urea concentrations of ≤1.8 M (Fig. 1B), demonstrating the increased robustness of the LAMP assay to urea-mediated inhibition.
FIG 1.
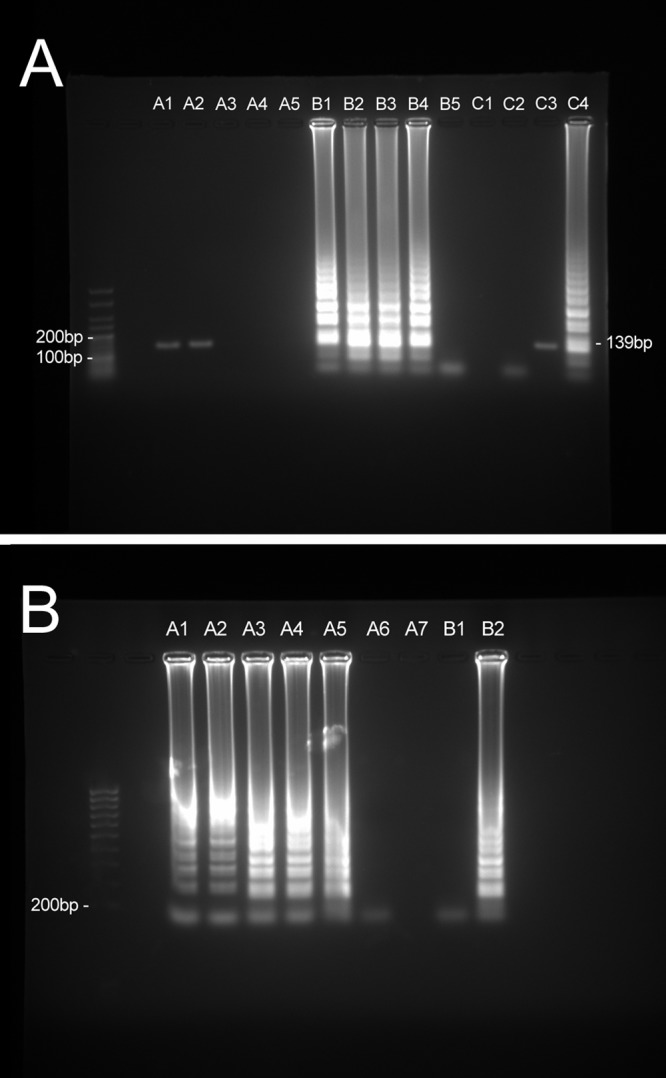
(A) Tolerance of the ORF1 PCR assay and ORF1 LAMP assay to urea: agarose gel electrophoresis of ORF1 PCR (lanes A1 through A5) and LAMP (lanes B1 through B5) reaction products from reactions with urea concentrations of 1 mM (lanes A1 and B1), 10 mM (lanes A2 and B2), 100 mM (lanes A3 and B3), 1 M (lanes A4 and B4), and 2 M (A5 and B5). Positive reactions contained 0.1 μg of N. gonorrhoeae DNA. PCR (lane C3) and LAMP (lane C4) reactions containing no urea were carried out as positive controls. PCR (lane C1) and LAMP (lane C2) reactions containing 5 μl of water instead of DNA were used as negative controls. (B) Agarose gel electrophoresis of reaction products from LAMP reactions with urea concentrations of 1 M (lane A1), 1.2 M (lane A2), 1.4 M (lane A3), 1.6 M (lane A4), 1.8 M (lane A5), and 2 M (lane A6). (A7) Blank. Positive reactions contained 0.1 μg of N. gonorrhoeae DNA. (B1) A LAMP reaction containing 5 μl of water instead of DNA was used as a negative control. (B2) A LAMP reaction containing no urea was used as a positive control.
The application of LAMP for the direct detection of N. gonorrhoeae DNA from urine samples was demonstrated by carrying out ORF1 LAMP reactions on 5-μl samples of both molecular-grade water and human urine spiked with 2 × 103 copies, 2 × 102 copies, 20 copies, 2 copies, and 0 copies of the N. gonorrhoeae genome. The reaction mixtures contained 1 μl of calcein to enable visual detection (15). The urine used was taken from one individual over a 12-h period, stored at 6°C, and used no later than 24 h after excretion. The sensitivity of the LAMP reaction was not reduced by the presence of urine, with a minimum of 20 copies detectable in urine or water. Positive reactions underwent an orange-to-green color change, which was unambiguous in natural light. The negative reactions remained orange regardless of whether urine or water was used.
The ORF1 LAMP assay detected the minimum copy number detectable by PCR and required less time. The inclusion of calcein allowed for the direct detection of ≥20 genome copies from urine samples after 1 h and without requiring any specialist equipment. The ability of the LAMP reaction to withstand higher levels of urea than those found in human urine enables the detection of target nucleic acids from urine samples that either have not undergone a nucleic extraction process or have undergone a very simple process such as heat treatment. This makes the ORF1 LAMP assay, in conjunction with calcein, well suited to the sensitive detection of N. gonorrhoeae directly from urine samples in a resource-poor setting.
ACKNOWLEDGMENT
This work was supported by the Technology Strategy Board, United Kingdom (project TP60-132).
Footnotes
Published ahead of print 12 March 2014
REFERENCES
- 1.Newman LM, Moran JS, Workowski KA. 2007. Update on the management of gonorrhea in adults in the United States. Clin. Infect. Dis. 44:S84–S101. 10.1086/511422 [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2012. Global action plan to control the spread and impact of antimicrobial resistance in Neisseria gonorrhoeae. World Health Organization, Geneva, Switzerland [Google Scholar]
- 3.Luijt DS, Bos PAJ, van Zwet AA, Van Voorst Vader PC, Schirm J. 2005. Comparison of COBAS AMPLICOR Neisseria gonorrhoeae PCR, including confirmation with N. gonorrhoeae-specific 16S rRNA PCR, with traditional culture. J. Clin. Microbiol. 43:1445–1447. 10.1128/JCM.43.3.1445-1447.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28:E63. 10.1093/nar/28.12.e63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Njiru ZK, Mikosza AS, Matovu E, Enyaru JC, Ouma JO, Kibona SN, Thompson RC, Ndung'u JM. 2008. African trypanosomiasis: sensitive and rapid detection of the sub-genus Trypanozoon by loop-mediated isothermal amplification (LAMP) of parasite DNA. Int. J. Parasitol. 38:589–599. 10.1016/j.ijpara.2007.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaneko H, Iida T, Aoki K, Ohno S, Suzutani T. 2005. Sensitive and rapid detection of herpes simplex virus and varicella-zoster virus DNA by loop-mediated isothermal amplification. J. Clin. Microbiol. 43:3290–3296. 10.1128/JCM.43.7.3290-3296.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakao R, Stromdahl E, Magona J, Faburay B, Namangala B, Malele I, Inoue N, Geysen D, Kajino K, Jongejan F, Sugimoto C. 2010. Development of loop-mediated isothermal amplification (LAMP) assays for rapid detection of Ehrlichia ruminantium. BMC Microbiol. 10:296. 10.1186/1471-2180-10-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Francois P, Tangomo M, Hibbs J, Bonetti EJ, Boehme CC, Notomi T, Perkins MD, Schrenzel J. 2011. Robustness of a loop-mediated isothermal amplification reaction for diagnostic applications. FEMS Immunol. Med. Microbiol. 62:41–48. 10.1111/j.1574-695X.2011.00785.x [DOI] [PubMed] [Google Scholar]
- 9.Khan G, Kangro HO, Coates PJ, Heath RB. 1991. Inhibitory effects of urine on the polymerase chain reaction for cytomegalovirus DNA. J. Clin. Pathol. 44:360–365. 10.1136/jcp.44.5.360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hedman J, Radstrom P. 2013. Overcoming inhibition in real-time diagnostic PCR. Methods Mol. Biol. 943:17–48. 10.1007/978-1-60327-353-4_2 [DOI] [PubMed] [Google Scholar]
- 11.Chernesky MA, Jang D, Sellors J, Luinstra K, Chong S, Castriciano S, Mahony JB. 1997. Urinary inhibitors of polymerase chain reaction and ligase chain reaction and testing of multiple specimens may contribute to lower assay sensitivities for diagnosing Chlamydia trachomatis infected women. Mol. Cell Probes 11:243–249. 10.1006/mcpr.1997.0109 [DOI] [PubMed] [Google Scholar]
- 12.Behzadbehbahani PEK, Vallely APJ, Cleator GM. 1997. Detection of BK virus in urine by polymerase chain reaction: a comparison of DNA extraction methods. J. Virol. Methods 67:161–166. 10.1016/S0166-0934(97)00101-8 [DOI] [PubMed] [Google Scholar]
- 13.Chaudhry U, Saluja D. 2002. Detection of Neisseria gonorrhoeae by PCR using orf1 gene as target. Sex. Transm. Infect. 78:72. 10.1136/sti.78.1.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geraats-Peters CW, Brouwers M, Schneeberger PM, van der Zanden AG, Bruisten SM, Weers-Pothoff G, Boel CH, van den Brule AJ, Harmsen HG, Hermans MH. 2005. Specific and sensitive detection of Neisseria gonorrhoeae in clinical specimens by real-time PCR. J. Clin. Microbiol. 43:5653–5659. 10.1128/JCM.43.11.5653-5659.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomita N, Mori Y, Kanda H, Notomi T. 2008. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 3:877–882. 10.1038/nprot.2008.57 [DOI] [PubMed] [Google Scholar]