

Abstract
The inhibition of the class A SHV-1 β-lactamase by 7-(tert-butoxycarbonyl)methylidenecephalosporin sulfone was examined kinetically, spectroscopically, and crystallographically. An 1.14Å X-ray crystal structure shows that the stable acyl-enzyme, which incorporates an eight-membered ring, is a covalent derivative of Ser70 linked to the 7-carboxy group of 2-H-5,8-dihydro-1,1-dioxo-1,5-thiazocine-4,7-dicarboxylic acid. A cephalosporin-derived enzyme complex of this type is unprecedented and the rearrangement leading to its formation may offer new possibilities for inhibitor design. The observed acyl-enzyme derives its stability from the resonance stabilization conveyed by the β-aminoacrylate (i.e. vinylogous urethane) functionality as there is relatively little interaction of the eight-membered ring with active site residues. Two mechanistic schemes are proposed, differing in whether, subsequent to acylation of the active site serine and opening of the β-lactam, the resultant dihydrothiazine fragments on its own, or is assisted by an adjacent nucleophilic atom, in the form of the carbonyl oxygen of the C7 tert-butyloxycarbonyl group. This compound was also found to be a submicromolar inhibitor of the class C ADC-7 and PDC-3 β-lactamases.
INTRODUCTION
The accelerating evolution and dissemination of β-lactamase-mediated bacterial resistance1 to front-line β-lactam antibiotics, and ability of Gram-negative pathogens to couple such resistance with porin alterations and efflux,2 necessitate the search for more effective, broader spectrum, β-lactamase inhibitors.3 Such inhibitors can be co-administered with β-lactam antibiotics in highly efficacious combinations to treat resistant infections.4 Current commercial inhibitors include penicillin sulfones, sulbactam (1) and tazobactam (2), together with clavulanic acid (3). Newer inhibitors are in clinical development.5 In the cases of both the β-lactam antibiotics, and β-lactam-derived β-lactamase inhibitors, understanding the mechanistic fate of the initial acyl-enzyme can facilitate design of more efficient drugs.
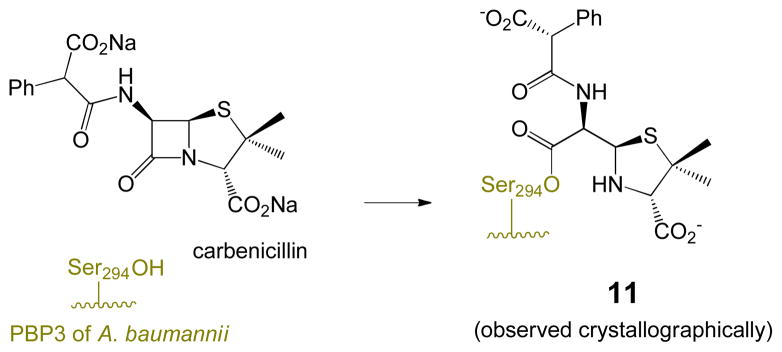
Key to activity of the penicillin sulfones is fragmentation of the dioxothiazolidine ring,6 which occurs subsequent to acylation of the active site serine as shown in Scheme 1. This ring-opening is characteristic of the sulfones, since numerous structures7 of antibiotic complexes (i. e. penicillin sulfides) with penicillin-binding proteins (PBPs) illustrate that the non-oxidized thiazolidine remains intact, as shown in Scheme 2, a complex of carbenicillin with PBP3 of Pseudomonas aeruginosa. This is presumably due to the weaker basicity and resulting enhanced leaving group ability of the sulfinate (pKa methanesulfinic acid ≈ 3) of the sulfones relative to the thiolate (pKa methanethiol ≈ 10) of the sulfides. The resultant imininium ion (generated in the E configuration) can tautomerize to a resonance-stabilized β-aminoacrylate or undergo a nucleophilic capture with a second active-site serine as shown in Scheme 1. The β-aminoacrylates (6 and 7), show high planarity of the N-C=C-C=O, suggesting resonance interaction of the nitrogen electrons with the carbonyl, thus enhancing hydrolytic stability.
Scheme 1.

Scheme 2.
As commercial inhibitors rely on active site functionality for recognition and inactivation processes, including both tautomerization8 and nucleophilic capture,9 they are limited to inactivation of specific class A β-lactamases and are susceptible to point mutations, leading to ‘inhibitor-resistant’ mutants.10 Shortcomings of current inhibitors, proliferation of AmpC producing strains, and appearance of class D β-lactamases, necessitate development of inhibitors with a broader inhibitory spectrum.
The inhibitory spectrum of penicillin sulfones can be expanded by incorporating an alkylidene group at C6.11 One such modification, shown in Scheme 3, involves an appropriately positioned heteroatom (usually contained in an attached heterocycle attached to the terminus of the exocyclic double bond) enabling intramolecular capture of the incipient iminium ion. This intramolecular assistance/capture avoids reliance on active-site residues to complete the rearrangement, thus broadening the inhibitory spectrum. The final (crystallographically observed11c) stabilized acyl-enzyme is a resonance-stabilized β-aminoacrylate in the form of indolizine-2-carboxylate 16.
Scheme 3.

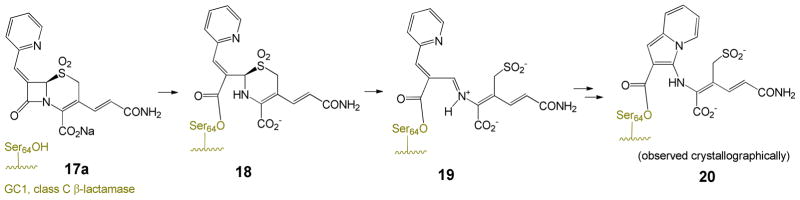
By contrast, cephalosporins have been much less utilized in β-lactamase inhibitor design. The 7-alkylidenecephalosporin sulfones (17) were observed to be β-lactamase inhibitors in 199512 and later developed to broaden their inhibitory spectrum.13 A complex of inhibitor 17a with the extended-spectrum class C β-lactamase GC1 was examined crystallographically14 (Scheme 4).
Scheme 4.
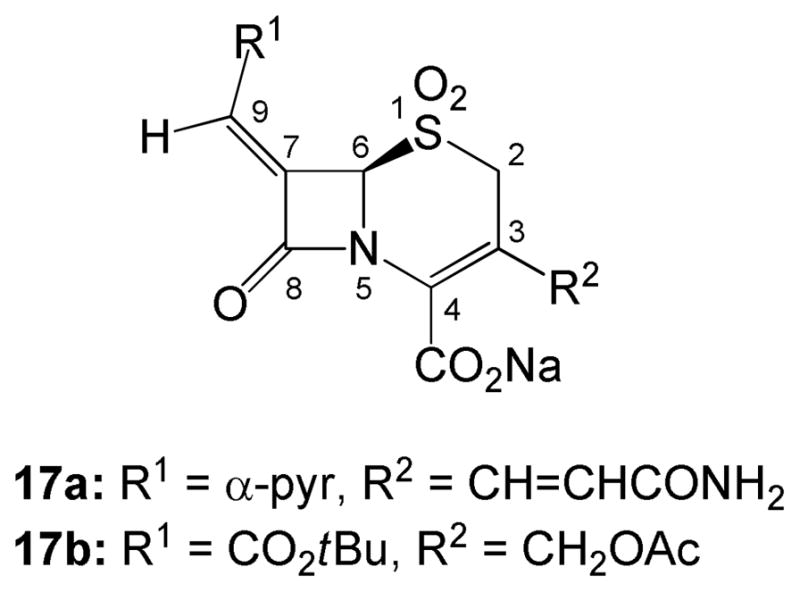
In the initial publication regarding 7-alkylidenecephalosporin sulfones, one potent inhibitor, 17b in Figure 2 (also referred to as DCM-1-10), lacked a nitrogen-heterocycle attached to the 7-alkylidene moiety. Although demonstrating potent and specific inhibition of the TEM-2 β-lactamase, the activity of 17b resembled the inhibitory spectrum of commercial inhibitors, and the mechanism was not investigated. In recognition that this compound did not fit inhibitory motifs previously observed, and that understanding mechanism can lead to improved inhibitors, we decided to undertake a crystallographic study of inhibitor 17b with the class A SHV-1 β-lactamase.
Figure 2.
7-Alkylidenecephalosporins
MATERIALS AND METHODS
Expression and purification
wt-SHV-1, ADC-7, and PDC-3 were expressed and purified as previously described15,16,17 For protein expression and β-lactamase characterization, the blaOXA-24 gene was cloned into the pET24a (+) vector (Novagen, Madison, WI) according to the following method. Using the Gene-Amp XL PCR kit (Applied Biosystems), high-fidelity amplification of blaOXA-24 without leader peptide sequence from the OXA-24/pIM-1-RA clone designed by Héritier et al 18 was performed with primers OXA-24FOR (5′CATATGTCTATTAAAACTAAATCTGA3′) and OXA-24REV (5′GGATCCTTAAATGATTCCAAGA3′). The cycling conditions used were 95 °C for 30 s, 55 °C for 1 min, and 72 °C for 1 min for 25 cycles, after which there was final extension at 72 °C for 10 min. A restriction digest of the pET24a (+) vector was done using NdeI and BamHI. The amplification product was purified using the QIAquick gel extraction kit (Venlo, Netherlands) and digested using NdeI and BamHI. This product was ligated to the digested pET24a (+) vector and electroporated into E. coli DH10B. The resulting construct was sequenced with pET24a (+) primers T7 promoter primer and T7 terminator primer. After sequencing verification, the construct was transformed into E. coli BL21(DE3) cells for protein expression.
OXA-24 β-lactamase was prepared after induction with isopropyl-β-D-thiogalactopyranoside (IPTG; Sigma Chemical Company, St. Louis, MO). A five hundred milliliter culture was induced at an optical density at 600 nm of 0.6 (final IPTG concentration, 0.2 mM). The culture was harvested 3 h after induction, centrifuged, and the pellet frozen at −20 °C overnight. The following day, the cell pellet was resuspended in 50 mM Tris buffer, pH 7.4. β-lactamase was released by using stringent periplasmic fractionation and initially purified by preparative isoelectric focusing as described previously.19 Additional purification was performed by high pressure liquid chromatography with a Sephadex Hi Load 26/60 column (GE Healthcare, Piscataway, NJ). Protein was eluted with 50 mM sodium phosphate buffer, pH 7.2. The purity of each preparation was assessed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. Protein samples were resolved on a 5% stacking, 12% separating SDS-polyacrylamide gel and stained with Coomassie brilliant blue R250 (Fisher, Pittsburgh, PA). Protein concentrations were determined by the Bio-Rad protein assay with bovine serum albumin (Sigma) as the standard.
Synthesis
DCM-1-10 was synthesized as previously described.12
Crystallization and Soaking
wt-SHV-1 crystals were grown using the vapor diffusion method in 24-well sitting drop trays (Hampton Research) using a 500 μL reservoir solution of 21 – 30% PEG600 and 0.1 M HEPES pH 6.8–7.8. A total of 5 mg/mL protein was combined with Cymal-6 (final concentration 0.56 mM, Hampton Research) then mixed with the reservoir solution at a 1:1 ratio to a final drop size of 5 μL. Inhibitor soaks were performed in well solution plus 50 mM inhibitor for 30 min. Crystals were subsequently cryoprotected in well solution supplemented with 20% 2-methyl-2,4-pentanediol (well solution for inhibitor soaked crystals also contained 50 mM inhibitor) plus immersion in liquid nitrogen.
Data Collection and Refinement
Data was collected at the Stanford Synchrotron Radiation Lightsource, Menlo Park, CA. wtSHV-1 crystallized in space group P212121 and diffraction images were integrated and scaled using HKL2000 software.20 The structure was solved using isomorphous replacement (using PDB ID: 1SHV). The structure was resolved to 1.14Å and REFMAC5 was used to carry out all- (waters and protein) anisotropic refinement to yield a structure with final R/Rfree of 0.13/0.16. Coot21 was used for manual refinement and model building and yielded a model with 97.31% (217), 1.79% (4) and 0.90% (2) of residues in Ramachandran preferred, allowed and outlying regions, respectively. At 1.14Å resolution, the two outliers (residues M69 and Y105) are confidently placed within strong 1.5σ Fo-Fc density despite their less than ideal dihedral angles. After initial refinement and addition of water and Cymal-6 molecules, electron maps clearly indicated the presence of a covalently-bound DCM-1-10 species in the active site. DCM-1-10 topology files were generated using an online server, PRODRG, and DCM-1-10 was included in subsequent rounds of refinement.
Kinetics
Steady state kinetics were performed on an Agilent 8453 diode array spectrophotometer (Palo Alto, CA) in 10mM phosphate-buffered saline (pH 7.4). Vmax and Km were determined from initial steady-state velocities for nitrocefin (NCF), Δε482 =17,400 M−1cm−1. The kinetic parameters were obtained using iterative non-linear least-squares fit of the data to the Henri-Michaelis equation using Origin 8.0 (OriginLab, Northampton, MA) according to Equation 1:
| (1) |
We determined the Ki for the inhibitors by measuring initial steady-state velocities in the presence of a constant concentration of enzyme with increasing concentrations of inhibitor against the indicator substrate NCF (100 μM). Assuming a competitive mode of inhibition under these conditions, initial velocity (v0) measurements immediately after mixing yield a Kiapp as represented by Equation 2:
| (2) |
Kiapp values were corrected for nitrocefin affinity (Km = 20μM) according to Equation 3:
| (3) |
IC50, defined as the inhibitor concentration resulting in a reduction of NCF (100μM) hydrolysis by 50%, was determined by measurements of initial velocities after 5 min pre-incubation of enzyme with inhibitor. The first-order rate constant for enzyme inactivation, kinact, was determined by monitoring the reaction time courses in the presence of increasing concentrations of inactivators. A fixed concentration of enzyme, nitrocefin, and increasing concentrations of inactivator were used in each assay. The kobs for inactivation was determined graphically as the reciprocal of the ordinate of the intersection of the straight lines obtained from the initial, v0, and final, vf, steady-state velocities. Each kobs was plotted versus inhibitor concentration, I, and fit to Equation 4 to determine kinact and kinact/KI (the second order rate constant for reaction of free enzyme with free inhibitor to give inactive enzyme):
| (4) |
Turnover numbers (tn) or partition ratios (kcat/kinact) were determined as the ratio of inhibitor concentration to enzyme concentration that was necessary to decrease the enzyme activity by 95%. The turnover numbers were determined after a 24 h incubation with increasing concentrations of the inhibitor. Incubations were done in a final volume of 300 μl and 25 μl of this reaction mixture were added to a 1 mL final volume to determine the residual enzyme activity using 100 μM nitrocefin. The tn was used to determine the ratio of inhibitor to enzyme in a 45-min experiment in which steady state velocities were measured at set time points following the combination of enzyme and inhibitor.
The apparent first order rate constant for reactivation of the β-lactamase, kobs react, was determined based on a previously published method.22 Briefly, 1.39 μM SHV-1 was mixed with 0.124 mM DCM-1-10 in a total volume of 80 μL for 30 min at RT. This mixture was placed on a Macro Spin Column G10 (The Nest Group, Southborough, MA) and spun in an Eppendorf 5415D table-top centrifuge at 2000 rpm at RT for 30 s to remove unbound DCM-1-10. Reactivation of enzyme was monitored after a 30 min incubation of the 80 μL flowthrough at rt. A 5 μL aliquot was removed and the transient recovery of activity monitored in the presence of 100μM nitrocefin as an indicator substrate.
Ultraviolet difference (UVD) spectroscopy
Based on a previously published method23 UV difference spectra (absorption spectra of 3.5 μM DCM-1-10) was measured from λ = 220 – 350 nm during a 60 min observation period.
Raman crystallography
SHV-1 crystals were obtained as described previously.24 The Raman microscopy system has been described.25,26,27 An SHV-1 crystal was first transferred from the mother liquor solution to a 4 μL drop of 2 mM HEPES. A 647 nm, 90 mW Kr+ laser beam (Innova 70 C, Coherent, Palo Alto, CA) was focused on the protein crystal in a 4 μL hanging drop using the 20× objective of the Raman microscope. During data collection, spectra were acquired for 1 s and 100 accumulations were averaged for each time point. After obtaining spectra of the apo SHV-1 crystals, inhibitors were added to the drop to achieve a final volume of 5 μL and a final inhibitor concentration of 5 mM. Spectra were then acquired every 2–3 min after addition of an inhibitor. To obtain difference spectra, an apo SHV-1 spectrum was subtracted from the enzyme/inhibitor spectra at varying time intervals following addition of inhibitor.
Calculations
Ab initio quantum mechanical calculations were performed on CWRU’s cluster facility to predict the Raman spectra of DCM-1-10 and model intermediate compounds using Gaussian 03.28 Calculations were performed at the DFT level using the 6-31+G(d) basis set. DFT calculations were performed with Becke’s three parameter hybrid method using the correlation functional of Lee, Yang, and Parr (B3LYP). The vibrations giving rise to the most intense calculated peaks could be visualized using “GaussView”, revealing which molecular vibrations contribute to the peaks.
RESULTS
Crystallographic data show DCM-1-10 covalently bound to SHV-1 at the catalytic Ser70 in the form of an eight-membered thiazocine ring. A new C-S bond is formed between the sulfur and C9 of the alkylidene moiety. Both 2Fo-Fc (contoured at 1.0σ) and Fo-Fc (contoured at 3.0σ) electron density maps indicate the presence of this C-S bond (Figure 3; all atoms in the 8-membered ring had Fo-Fc density above 4.25σ contour level. Both electron density maps indicate strong bifurcated C4 carboxylate density, sulfone oxygen density and trifurcated C7 tert-butyl ester density. The C3 acetoxy group is not as well resolved but there is strong 2Fo-Fc and Fo-Fc density for the C-O-C oxygen.
Figure 3.

Fo-Fc electron density map contoured at 3.0σ (A) and 2Fo-Fc contoured at 1.0σ of DCM-1-10 bound to wtSHV-1
Despite the presence of the C8 tert-butyl ester, the deacylation water is present, in the same location as in apo wtSHV-1. DCM-1-10 was best modeled at 70% active site occupancy. Also modeled is a HEPES buffer fragment at 30% occupancy, located where previously observed in SHV-2.29 The position of the HEPES fragment partially overlaps with the position of DCM-1-10 and both molecules cannot therefore occupy the active site simultaneously. Although the resolution is not high enough to visualize double-bond characteristics, the torsion angles within the 8-membered ring of DCM-1-10 suggest the presence of two double bonds as drawn in Scheme 7a. The first double bond yields a torsion angle of −4.2° (for atoms N5-C6-C7-Cx) (Figure 4). The torsion angle around the same bond for atoms N5-C6-C7-C8 is −160° (Figure 4) which thus deviates somewhat more from −180° suggesting that atom C8, part of the carbonyl bond, is somewhat out of the plane. The second double bond in the 8-membered ring yields a torsion angle of −15.5° (for atoms N5-C4-C3-C2).
Schemes 7a and 7b.

Figure 4.
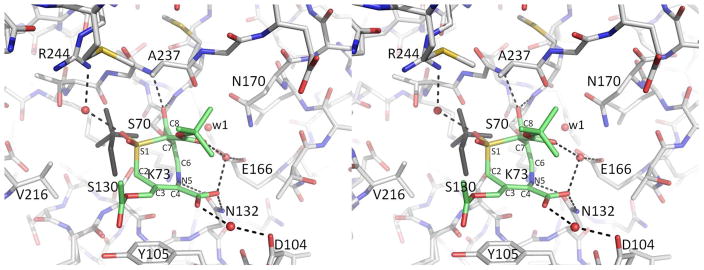
Interactions of DCM-1-10 in the active site of SHV-1. Hydrogen bonds between DCM-1-10 (green carbon atoms) and SHV-1 (grey carbon atoms) are depicted as dashed lines. Water mediated hydrogen bonds are also depicted. The sulfone containing HEPES fragment (30% occupancy) is depicted in dark grey. The deacylation water molecule is labeled as w1.
DCM-1-10 makes few hydrogen-bonding contacts with active-site residues. The carbonyl oxygen occupies the oxyanion hole making hydrogen bonds with the backbone nitrogens of residues A237 and S70. One of the C4 carboxylate oxygens is within hydrogen bonding distance of residue N132; one of the sulfone oxygens makes a water-mediated hydrogen bond with R244. The N5 nitrogen makes a somewhat long hydrogen bond with the oxygen of side chain N132 (3.32Å). Additionally, several van der Waal’s interactions take place: between DCM-1-10’s tert-butyl moiety and residue A237 and between C2 and S130. Lastly, there is a π-stacking interaction between the C3-C4 bond and residue Y105.
Kinetics
Using qualitative measurements, we first determined the spontaneous hydrolysis rate of 3.5 μM DCM-1-10 in 10 mM PBS, pH 7.4. UVD spectroscopy was performed to follow the reaction for 60 minutes. Figure 5 shows that there is a measurable rate of spontaneous hydrolysis in PBS (ΔO.D.λ=228 = 0.007). UVD spectroscopy was performed on the hydrolysis of DCM-1-10 in 100 mM NaOH (Supplemental Figure S1) which allowed us to determine Δελ=228 (11,730 M−1·cm−1). A plot of time vs. absorbance (λ = 228 nm, Supplemental Figure S2) permitted us to determine the pseudo first order rate constant of spontaneous hydrolysis (3.0 × 10−5 s−1). In direct terms, there is a spontaneous rate of hydrolysis of 0.0002 μM · s−1.
Figure 5.

UV difference spectroscopy of 3.5 μM DCM-1-10 alone.
First, a general mechanism for irreversible β-lactamase inhibition can be summarized as Equation 5a.
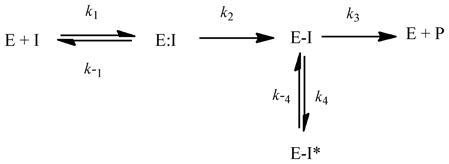 |
(5a) |
Here, E:I represents the Michaelis complex, E-I the acyl-enzyme, E-I* the tautomer, and P represents product.
DCM-1-10 was next characterized kinetically using the target enzyme SHV-1 and results are summarized in Table 1a. Several kinetic parameters were unable to be measured because of the high concentration of DCM-1-10 required. However, we were able to determine IC50, kobs react, Ki and tn.
Table 1a.
Kinetic parameters for DCM-1-10, tazobactam and clavulanic acid for comparison. Some values not measureable (NM) because high concentrations of inhibitor were required. kreact measured after a 30 min incubation of 1:90 enzyme:inhibitor ratio.
| Inhibitor | IC50 (μM) | kinact (s−1) | kobs react (s−1) 30min | Ki (μM) | KI (μM) | kinact/KI (μM−1s−1) | tn (24h) |
|---|---|---|---|---|---|---|---|
| Tazobactam | 0.030 ± 0.003 | 0.11 ± 0.01 | 0.040 ± 0.001 | 0.100 ± 0.025 | 0.16 ± 0.03 | 0.69 ± 0.14 | 5 |
| Clavulanate | 0.19 ± 0.02 | 0.040 ± 0.004 | > 0.2 | 0.14 ± 0.01 | 2.6 ± 0.3 | 0.020 ± 0.003 | 40 |
| DCM-1-10 | 0.8 ± 0.1 | 0.001a | 0.0006 ± 0.0001 | >20 | NM | NM | 7 |
Determined (± 10% error) based on analysis of first-order exponential in Figure 6.
Given these considerations, the observed IC50 (0.8 ± 0.1 μM) is 4-fold higher than clavulanate (0.19 ± 0.02 μM) and 27-fold higher than tazobactam (0.030 ± 0.003 μM). Additionally, the Ki is about 166-fold greater than tazobactam and clavulanate. However, the 24-hour turnover number for DCM-1-10 (tn = 7) is comparable to tazobactam (tn = 5) and kobs react is 67-fold lower than tazobactam and at least 333-fold lower than clavulanate. A timed inactivation of 140 nM SHV-1 (E) with 980 nM DCM-1-10 (I) (I:E = 7:1) shows 95% inactivation of SHV-1 within 45 min (Figure 6).
Figure 6.

Timed course inactivation of 140 nM SHV-1 with 980 nM DCM-1-10 (SHV-1:DCM-1-10 = 1:7) and 100 nM ADC-7 with 4.5 μM DCM-1-10 (ADC-7:DCM-1-10 = 1:45).
With regards to class C (PDC-3 and ADC-7) β-lactamases, we found that DCM-1-10 readily inhibits ADC-7 and PDC-3 with IC50 values that are sub-micromolar (Table 1b). In contrast, OXA-24 (class D) is not inhibited by DCM-1-10 (IC50 > 100 μM, Table 1b). Interestingly, we also discovered that DCM-1-10 acts more like a substrate for both OXA-24 and PDC-3 (tn = 3400 and 2000, respectively, Table 1b). These measurements merit further analysis. As the inhibition of ADC-7 was very notable (IC50 = 130 nM) we performed a timed inactivation of ADC-7 using I:E of 45:1 based on tn. We observed that inactivation was rapid compared to SHV-1. Further studies are planned to determine the details of this process and the mechanism.
Table 1b.
Kinetic parameters for DCM-1-10 with ADC-7, PDC-3, and OXA-24 β-lactamases.
| Enzyme | IC50 (μM) | tn (24h) |
|---|---|---|
| ADC-7 | 0.13 ± 0.01 | 45 |
| PDC-3 | 0.74 ± 0.08 | 2000 |
| OXA-24 | > 100 | 3400 |
Mass Spectrometry
To establish the nature of the inactivation product, ESI-MS was performed with a Q-Star quadrupole time-of-flight mass spectrometer equipped with a nanospray source. We inactivated SHV-1 with a ten-fold excess of DCM-1-10. The incubation (inactivation) time was 15 min (900 s). The deconvoluted spectra are presented (Figures 7A–B). The ESI-MS measurement (28,872 ± 4 amu) was in agreement with the theoretical mass of the SHV-1 β-lactamase (28,872 amu). Covalent attachment of DCM-1-10 to the SHV-1 β-lactamase was demonstrated in the spectra. During the time period studied, we did not find evidence of the fragmentation of DCM-1-10 (403 ± 4 amu).
Figure 7.
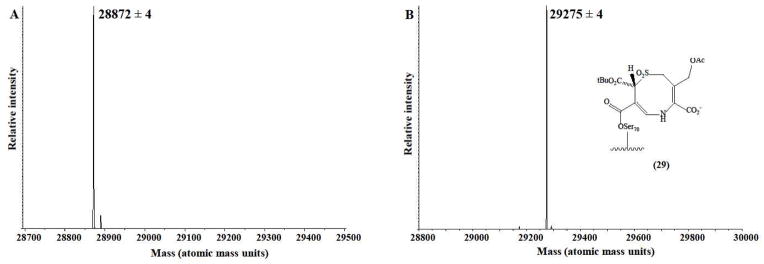
Mass spectra of SHV-1 alone (A) and with DCM-1-10 showing the formation of compound 29 from Scheme 7a (B).
Raman evidence for ring formation
The Raman data for DCM-1-10 reacting in a single crystal of WT SHV-1 supports the conclusion that a stable eight-membered ring is formed as an acyl-enzyme. The crystal of SHV-1 was suspended in a hanging drop of holding solution and a concentrated solution of DCM-1-10 was injected into the hanging drop resulting in a final concentration of 5 mM. The DCM-1-10 soaks into the crystal and reacts rapidly. The Raman difference spectrum [enzyme + DCM-1-10] minus [enzyme] is stable within the dead time of our experiment and we only detected the stable product (an acyl-enzyme). This is shown in the bottom trace in Figure 8.
Figure 8.

Raman difference spectra of wt SHV-1 single crystal reaction with DCM-1-10. The top spectrum is unreacted DCM-1-10 (5 mM in aqueous solution, pH 7.5). The bottom one is 10 min after DCM-1-10 soak in (5 mM, pH 7.5). The peak assignments are shown in Tables 2 and 3.
Figure 8 compares the Raman spectrum of free aqueous DCM-1-10 with that of the product within crystal. In order to assign the features in Figure 8, “Gaussian” quantum chemical calculations were carried out. Assignments for free DCM-1-10 are shown in Table 2. These are the characteristic lactam C=O stretch at 1787 cm−1 and the C=C stretch from the group within the six-membered thiazine ring at 1650 cm−1. The calculations indicate that the C-S-C symmetric stretch from the sulfone group is at 796 cm−1.
Table 2.
Major peak assignment for unreacted DCM-1-10 from Gaussian calculations.
| |||
|---|---|---|---|
| Calculation results of unreacted DCM-1-10 | |||
| Experimental group (cm−1) | Calculated Group (cm−1) | Calculated Raman intensity | Peak assignment |
| 1787 | 1770 | 90 | C=O of β-lactam ring |
| 1725 | 1730 | 292 | C=O stretch from C3 acetoxy |
| 1705 | 1710 | 155 | Symmetric stretch from tert-butyl ester carbonyl |
| 1650 | 1670 | 2390 | C=C stretch at C7 position |
| 796 | 825 | 50 | -C-S-C- stretch of dihydrothiazine ring |
The Raman spectrum seen in Figure 8 (top) undergoes major changes when reaction is complete (Figure 8, bottom). Disappearance of the 1787 cm−1 lactam C=O occurs following acylation of the active site serine and opening of the β-lactam. The 1725 cm−1 C=O stretch peak provides support that the acetoxy group Ac-O remains attached to the C3′ position of the thiazocine ring.31,32 Based on the x-ray analysis, we undertook calculations on the eight-membered thiazocine predicted to be the product (29) in reaction Scheme 7a. Calculations (Table 3) show that the 1657 cm−1 profile has contributions from the two double bonds in the ring (Table 3). They also indicate that the peak at 1320 cm−1 can be assigned to N-H bend and the peak at 797 cm−1 can be assigned to the symmetric stretch of the C-S-C bonds within the ring. These assignments taken together constitute a body of evidence that supports the presence of the eight-membered ring. The broad intensity with several sub-maxima between 863 and 932 cm−1 is tentatively assigned to ring deformation modes that occur in this region.30
Table 3.
Major peak assignment for eight-membered intermediate 29 (Scheme 7a) from Gaussian calculations
| |||
|---|---|---|---|
| Calculation results of intermediate 29 (Scheme 7a) | |||
| Experimental group (cm−1) | Calculated Group (cm−1) | Calculated Raman intensity | Peak assignment |
| 1725 (unresolved) | 1730 | 30 | C=O stretch from C3 acetoxy, tert-butyl ester carbonyl, Ser70 ester carbonyl |
| 1650 | 1680 | 311 | C6=C7 stretch of dihydrothiazocine ring |
| 1320 | 1353 | 50 | N5-H bending |
| 797 | 825 | 50 | -C-S-C- stretch |
DISCUSSION
While the key to the inhibition of the penicillin sulfones is formation of iminium ion 5, the larger 6-membered dihydrothiazine ring of cephalosporins is less prone to an analogous fragmentation, potentially explaining the reduced inhibitory activity of most cephalosporin sulfones. Additionally, in cephalosporins with a 3′-leaving group (such as acetate), there is elimination of that group after enzyme acylation, leading to exocyclic alkenes such as 22 and 24, that are not mechanistically well-suited to fragmentation of the dihydrothiazine, as summarized in Schemes 5 and 6.31,32 Indeed, all current PDB files of acyl-enzymes resulting from 3-(acetoxymethyl)cephalosporin antibiotics with various PBPs possess a C3-exocyclic double bond, resulting from loss of the C3′-acetoxy group. Inhibitory cephalosporin sulfones possessing such a leaving group are expected to partition between loss of the 3′-group and fragmentation, as shown in Scheme 6, thereby reducing their inhibitory potency.
Scheme 5.
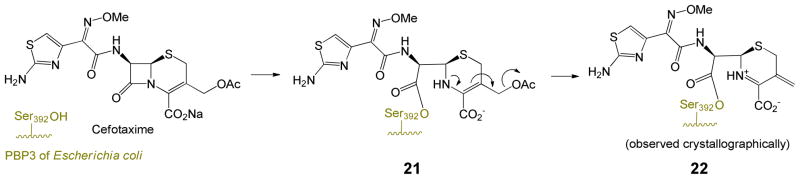
Scheme 6.

A proposed mechanism for the formation of the observed dihydro-1,5-thiazocine is shown in Scheme 7a. An analogous transformation of the 6-methylidenpenem BRL42715 to a dihydro-1,4-thiazepine33 is shown in Scheme 7b. The hydrolytic stability of the acyl-enzyme 29 is attributed to the resonance stabilization of the carbonyl group of the ester with the nitrogen lone pair of the β-aminoacrylate. Several features deserve comment.
Both transpositions are formally a 1,3-allylic transposition of a sulfur atom: a sulfinate (cephem) or a thiolate (penem).34 The sulfinate has maintained its positional orientation on the β-face of the antibiotic, approaching the exocyclic double bond from the re face, thereby generating a new stereogenic center (at C8 of the dihydrothiazocine) in the R configuration. This is unlikely to have occurred in a concerted fashion (i.e. directly from 26 to 29) since concerted suprafacial 1,3-sigmatropic rearrangements are disallowed thermally.35 Thus, the transition states in both cases involve a two-step process with an intermediate iminium ion, formed, due to the geometric constraints of the 6-membered dihydrothiazine ring, initially in the E configuration, which then closes, via an intramolecular Michael addition,36 to generate an eight- or seven-membered ring, respectively. The initial ring openings (26⇆27 and 31⇆32) are likely reversible, as shown. In formation of 29, the Michael addition could have occurred at C7 of the cephem to form the seven-membered ring, but does not, probably due to the double activation (for attack at C9) of the C=C by the acyl-enzyme carbonyl and the protonated iminium ion versus the single activation (for attack at C7) by the tert-butyloxycarbonyl.
Particularly in the formation of the seven-membered ring (i.e. 32 → 34), this transition state would be highly strained, as supported by the fact that trans-cycloheptene is unstable at room temperature.37 Isomerization of iminium ions 27 and 32 from the E to Z configuration, however, would remove this barrier as shown in Schemes 7a and 7b.
Mechanistic studies on the E/Z isomerization of imines indicate that configurational inversion can occur by two mechanisms,38 each involving the N-protonated conjugate acid of the imine (Figure 9). As shown in Scheme 8, the first mechanism (upper) entails protonation of the imine, followed by rotation about the C-N bond, and deprotonation (Scheme 8a), while the second involves protonation, followed by nucleophilic addition to the iminium ion, and then elimination and deprotonation, as shown in Scheme 8b. Based on the observed pKa values of 7.5 to 8 for structurally analogous iminium ions,39 it is likely that these intermediate imines (27, 28, 32, 33) are protonated. Additionally, as observed with other substituted imines,40 the conjugate acid may be further stabilized through intramolecular hydrogen bonding to the C4 (or C3) carboxylate group, as shown in Figure 9.
Figure 9.
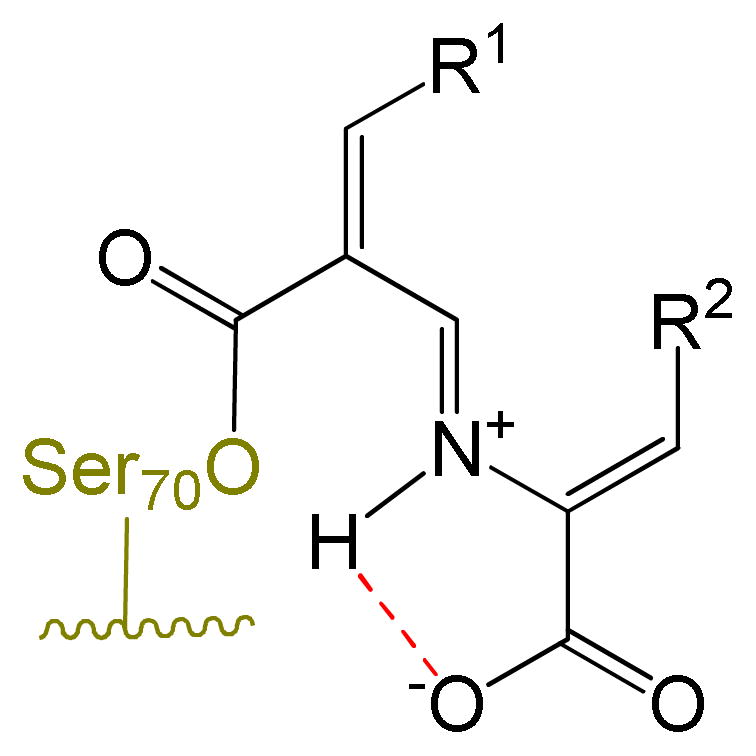
Schemes 8a and 8b.
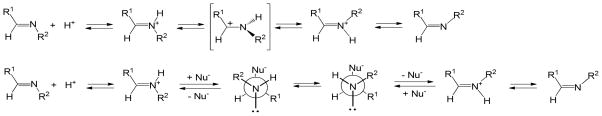
General mechanisms of Z/E configurational inversion of imines.38
One structural feature that both inhibitors (i.e. 17b and 30) have in common is two double bonds flanking the N-C-S linkage shown (purple) in Scheme 9. After fragmentation each double bond can contribute to delocalization of either positive charge on carbon or the lone pair electron density on nitrogen. Acting together, these double bonds may diminish the degree of double bond character between the bridgehead carbon (i.e. C6 of the cephem, or C5 of the penem, respectively) and the nitrogen atom, thereby reducing the barrier to inversion of the configuration of the iminium ion. Additionally, the Δ-3,4 (cephem) or Δ-2,3 (penem) double bond (in the Z configuration) can serve to hold the sulfur in the appropriate location for intramolecular Michael attack and possibly provide optimal geometry for intramolecular H-bonding to the imine (Figure 9). Each inhibitor possesses a weakly nucleophilic heteroatom appropriately positioned on the terminus of the C7 (cephem) or C6 (penem) double bond to undergo addition/elimination to the intermediate iminium ion, potentially facilitating inversion of configuration as shown in Scheme 10.
Scheme 9.
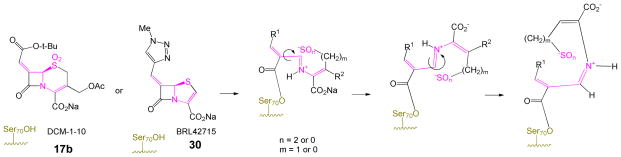
Scheme 10.

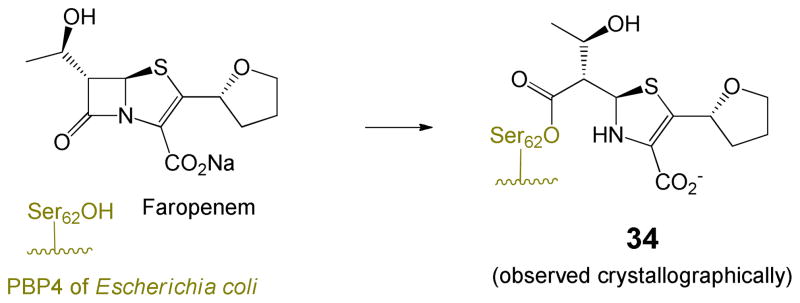
A question arises as to why 6-alkylidenepenems (e.g. 30), where the sufur is in the sulfide oxidation state, undergo fragmentation of the dihydrothiazole after acylation. Numerous structures of acyl-enzymes produced from penam sulfides (vide supra) all have the thiazolidine intact. While it may be argued that the endocyclic double bond of the dihydrothiazole produces additional strain, and lowers the pKa of the thiolate (now an enethiolate) thereby leading to fragmentation, it is important to note that the only two PDB structures of penems lacking the C6 exocyclic double bond41 indicate that the dihydrothiazole does not fragment as illustrated by acyl-enzymes derived from 6-(hydroxyethyl)penems shown in Scheme 11. It is logical to conclude that the exocyclic double bond at C6 of the penem plays a crucial role in facilitating fragmentation and stabilizing the intermediate iminium species.
Scheme 11.
A final option is shown in Scheme 12. In this mechanism, the fragmentation itself is effected not by formation of the iminium ion, but by direct attack of the nucleophilic heteroatom on the intact five and six membered rings, respectively. The alkylidene functionality provides anchimeric assistance to the departure of the sulfur leaving group, providing access to an intermediate that can fragment to the iminium salts in either the Z or E configuration. This fragmentation would need to occur at a rate faster than loss of a proton from the tert-butyl ester to generate the butenolide. This is reasonable since fragmentation to the linear intermediate requires no base and nitrogen-stabilized carbocations (i.e. iminium ions 27, 28, 32, 33) are expected to be highly stabilized.42
Scheme 12.

CONCLUSION
Together with the penicillins and carbapenems, cephalosporins represent a major class of β-lactam antibiotic. As such, it is notable that no commercial β-lactamase inhibitor possesses the cephem scaffold. This research documents an unprecedented rearrangement of a cephalosporin sulfone, leading to a stable acylenzyme. While the inhibitor is, by contemporary standards, of modest spectrum and potency, the discovery of a new inhibitor of unprecedented mechanism may have significance in the design of new chemotherapeutic agents.
Supplementary Material
Figure 1.
Current commercial β-lactamase inhibitors.
Acknowledgments
JDB acknowledges support from the Robert A. Welch Foundation (grant N-0871). FvdA acknowledges support of the NIH through grant AI062968. RAB acknowledges support from NIH under award numbers R01AI072219, R01AI063517, and R01AI100560 and by funds and/or facilities provided by the Louis Stokes Cleveland Department of Veterans Affairs Medical Center and the VISN 10 Geriatric Research, Education and Clinical Care Center (VISN 10) of the Department of Veterans Affairs. PRC acknowledges support from NIH through grant GM54072.
Footnotes
The authors declare no competing financial interest.
Crystallographic data and refinement statistics (Table S1), UV difference spectroscopy of hydrolysis of DCM-1-10 in 100 mM NaOH (Figure S1), and exponential curve fit of time vs absorbance to determine first order rate constant for spontaneous hydrolysis of DCM-1-10 (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.(a) Canton R, Akova M, Carmeli Y, Giske CG, Glupczynski Y, Gniadkowski M, Livermore DM, Miriagou V, Naas T, Rossolini GM, Samuelsen O, Seifert H, Woodford N, Nordmann P. Clin Microbiol Infect. 2012;18:413. doi: 10.1111/j.1469-0691.2012.03821.x. [DOI] [PubMed] [Google Scholar]; (b) Nordmann P, Naas T, Poirel L. Emerg Inf Dis. 2011;17:1791. doi: 10.3201/eid1710.110655. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pfeifer Y, Cullik A, Witte W. Int J Med Microbiol. 2010;300:371. doi: 10.1016/j.ijmm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 2.(a) Poole K. Front Microbiol. 2011;2:1. doi: 10.3389/fmicb.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zavascki AP, Carvalhaes CG, Picao RC, Gales AC. Expert Rev Anti Infect Ther. 2010;8:71. doi: 10.1586/eri.09.108. [DOI] [PubMed] [Google Scholar]
- 3.Biondi S, Long S, Panunzio M, Qin WL. Curr Med Chem. 2011;18:4223. doi: 10.2174/092986711797189655. [DOI] [PubMed] [Google Scholar]
- 4.(a) Lee N, Kwok-Yung Y, Kumana CR. Drugs. 2003;63:1511. doi: 10.2165/00003495-200363140-00006. [DOI] [PubMed] [Google Scholar]; (b) Adam D. J Int Med Res. 2002;30(Suppl 1):10A. doi: 10.1177/14732300020300S103. [DOI] [PubMed] [Google Scholar]; (c) Lister PD. Pharmacotherapy. 2000;20:213S. doi: 10.1592/phco.20.14.213s.35045. [DOI] [PubMed] [Google Scholar]
- 5.Bush K. Curr Opin Pharmacol. 2012;12:527. doi: 10.1016/j.coph.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 6.(a) Brenner DG, Knowles JR. Biochemistry. 1984;23:5833. doi: 10.1021/bi00319a024. [DOI] [PubMed] [Google Scholar]; (b) Imtiaz U, Billings EM, Knox JR, Mobashery S. Biochemistry. 1994;33:5728. doi: 10.1021/bi00185a009. [DOI] [PubMed] [Google Scholar]; (c) Kuzin AP, Nukaga M, Nukaga Y, Hujer A, Bonomo RA, Knox JR. Biochemistry. 2001;40:1861. doi: 10.1021/bi0022745. [DOI] [PubMed] [Google Scholar]; (d) Padayatti PS, Helfand MS, Totir MA, Carey MP, Hujer AM, Carey PR, Bonomo RA, van den Akker F. Biochemistry. 2004;43:843. doi: 10.1021/bi035985m. [DOI] [PubMed] [Google Scholar]
- 7.For a few recent examples, see: PDB ID: 3OCL: complex of carbenicillin with PBP3 of P. aeruginosa Sainsbury S, Bird L, Rao V, Shepherd SM, Stuart DI, Hunter WN, Owens RJ, Ren J. J Mol Biol. 2011;405:173. doi: 10.1016/j.jmb.2010.10.024.PDB ID: 3UDI: complex of penicillin G with PBP1a of Acinetobacter baumannii: Han S, Caspers N, Zaniewski RP, Lacey BM, Tomaras AP, Feng X, Geoghegan KF, Shanmugasundaram V. J Am Chem Soc. 2011;133:20536. doi: 10.1021/ja208835z.
- 8.Kalp M, Buynak JD, Carey PR. Biochemistry. 2009;48:10196. doi: 10.1021/bi901416t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas VL, Golemi-Kotra D, Kim C, Vakulenko SB, Mobashery S, Shoichet BK. Biochemistry. 2005;44:9330. doi: 10.1021/bi0502700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Totir MA, Padayatti PS, Helfand MS, Carey MP, Bonomo RA, Carey PR, van den Akker F. Biochemistry. 2006;45:11895. doi: 10.1021/bi060990m.Li M, Conklin BC, Taracila MA, Hutton RA, Skalweit MJ. Antimicrob Agents Chemother. 2012;56:5678. doi: 10.1128/AAC.00711-12.and references cited therein. (c) For a review of β-lactamase inhibitors including inhibitor resistance see: Buynak JD. Beta- Lactamases. In: Frere J-M, Uversky VN, editors. Molecular Anatomy and Physiology of Proteins series. Chapter 9. Nova Science Publishers Inc; New York: 2012. pp. 217–258.
- 11.(a) Chen YL, Chang CW, Hedberg K, Guarino K, Welch WM, Kiessling L, Retsema JA, Haskell SL, Anderson M, Manousos M, Barrett JF. J Antibiot. 1987;40:803. doi: 10.7164/antibiotics.40.803. [DOI] [PubMed] [Google Scholar]; (b) Buynak JD, Rao AS, Doppalapudi VR, Adam G, Petersen PJ, Nidamarthy SD. Bioorg Med Chem Lett. 1999;9:1997. doi: 10.1016/s0960-894x(99)00325-x. [DOI] [PubMed] [Google Scholar]; (c) Bou G, Santillan E, Sheri A, Beceiro A, Sampson JM, Kalp M, Bethel CR, Distler AM, Drawz SM, Pagadala SRR, van den Akker F, Bonomo RA, Romero A, Buynak JD. J Am Chem Soc. 2010;132:13320. doi: 10.1021/ja104092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buynak JD, Wu K, Bachmann B, Khasnis D, Hua L, Nguyen HK, Carver CL. J Med Chem. 1995;38:1022. doi: 10.1021/jm00006a022. [DOI] [PubMed] [Google Scholar]
- 13.(a) Buynak JD, Doppalapudi VR, Adam G. Bioorg Med Chem Lett. 2000;10:853. doi: 10.1016/s0960-894x(00)00098-6. [DOI] [PubMed] [Google Scholar]; (b) Buynak JD, Vogeti L, Doppalapudi VR, Solomon GM, Chen H. Bioorg Med Chem Lett. 2002;12:1663. doi: 10.1016/s0960-894x(02)00205-6. [DOI] [PubMed] [Google Scholar]; (c) Buynak JD. Curr Med Chem. 2004;11:1951. doi: 10.2174/0929867043364847. [DOI] [PubMed] [Google Scholar]
- 14.Cricklow GV, Nukaga M, Doppalapudi VR, Buynak JD, Knox JR. Biochemistry. 2001;40:6233. doi: 10.1021/bi010131s. [DOI] [PubMed] [Google Scholar]
- 15.Hujer KM, Hamza NS, Hujer AM, Perez F, Helfand MS, Bethel CR, Thomson JM, Anderson VE, Barlow M, Rice LB, Tenover FC, Bonomo RA. Antimicrob Agents Chemother. 2005;49:2941. doi: 10.1128/AAC.49.7.2941-2948.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drawz SM, Taracila M, Caselli E, Prati F, Bonomo RA. Protein Sci. 2011;20:941. doi: 10.1002/pro.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodkey EA, Drawz SM, Sampson JM, Bethel CR, Bonomo RA, van den Akker F. J Am Chem Soc. 2012;134:16798. doi: 10.1021/ja3073676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heritier C, Poirel L, Aubert D, Nordmann P. Antimicrob Agents Chemother. 2003;47:268. doi: 10.1128/AAC.47.1.268-273.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin S, Thomas M, Shlaes DM, Rudin SD, Knox JR, Anderson V, Bonomo RA. Biochem J. 1998;333:395. doi: 10.1042/bj3330395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Meth Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Padayatti PS, Sheri A, Totir MA, Helfand MS, Carey MP, Anderson VE, Carey PR, Bethel CR, Bonomo RA, Buynak JD, van den Akker F. J Am Chem Soc. 2006;128:13235. doi: 10.1021/ja063715w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sulton D, Pagan-Rodriguez D, Zhou X, Liu Y, Hujer AM, Bethel CR, Helfand MS, Thomson JM, Anderson VE, Buynak JD, Ng LM, Bonomo RA. J Biol Chem. 2005;42:35528. doi: 10.1074/jbc.M501251200. [DOI] [PubMed] [Google Scholar]
- 24.Kalp M, Totir MA, Buynak JD, Carey PR. J Am Chem Soc. 2009;131:2338. doi: 10.1021/ja808311s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carey PR. Annu Rev Phys Chem. 2006;57:527. doi: 10.1146/annurev.physchem.57.032905.104521. [DOI] [PubMed] [Google Scholar]
- 26.Carey PR. Chem Rev. 2006;106:3043. doi: 10.1021/cr0502854. [DOI] [PubMed] [Google Scholar]
- 27.Che T, Bonomo RA, Shanmugam S, Bethel CR, Pusztai-Carey M, Buynak JD, Carey PR. J Am Chem Soc. 2012;134:11206. doi: 10.1021/ja303168n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frisch MJT, GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03; revision C.02. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 29.Nukaga M, Mayama K, Hujer AM, Bonomo RA, Knox JR. J Mol Biol. 2003;328:289. doi: 10.1016/s0022-2836(03)00210-9. [DOI] [PubMed] [Google Scholar]
- 30.Lin-Vien DC, NB, Fateley WG, Grasselli JG. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules. Chapter 2. ACADEMIC PRESS; San Diego: 1991. [Google Scholar]
- 31.Faraci WS, Pratt RF. Biochemistry. 1985;24:903. doi: 10.1021/bi00325a014. [DOI] [PubMed] [Google Scholar]
- 32.For crystallographic data, see: PDB ID 3VSL (complex of cefotaxime with MRSA PBP3) Yoshida H, Kawai F, Obayashi E, Akashi S, Roper DI, Tame JRH, Park SY. J Mol Biol. 2012;423:351. doi: 10.1016/j.jmb.2012.07.012.PDB ID 1IYP (complex of cefotaxime with TOHO 1 extended spectrum class A β-lactamase): Shimamura T, Ibuka A, Fushinobu S, Wakagi T, Ishiguro M, Ishii Y, Matsuzawa H. J Biol Chem. 2002;277:46601. doi: 10.1074/jbc.M207884200.
- 33.(a) Bulychev A, Massova I, Lerner SA, Mobashery S. J Am Chem Soc. 1995;117:4797. [Google Scholar]; (b) Nukaga M, Abe T, Venkatesan AM, Mansour TS, Bonomo RA, Knox JR. Biochemistry. 2003;42:13152. doi: 10.1021/bi034986b. [DOI] [PubMed] [Google Scholar]; (c) Michaux C, Charlier P, Frere JM, Wouters J. J Am Chem Soc. 2005;127:3262. doi: 10.1021/ja0426241. [DOI] [PubMed] [Google Scholar]
- 34.For examples of other such 1,3-transpositions of sulfonyl groups, see: Bur SK. Top Curr Chem. 2007;274:125.Bordwell FG, Pagani GA. J Am Chem Soc. 1975;97:118.Ogura K, Iihama T, Kiuchi S, Kajiki T, Koshikawa O, Takahashi K, Iida H. J Org Chem. 1986;51:700.
- 35.Woodward RB, Hoffmann R. Angew Chem Internat Ed. 1969;8:781. [Google Scholar]
- 36.Michael additions of sulfinate anions are well-known: Posner T. Ber Dtsch Chem Ges. 1902;35:799.Kohler EP, Reimmer M. Am Chem J. 1904;31:163.Gilman H, Cason LF. J Am Chem Soc. 1950;72:3469.Pinnick HW, Reynolds MA. J Org Chem. 1979;44:160.
- 37.(a) Squillacote ME, Defellipis J, Shu Q. J Am Chem Soc. 2005;127:15983. doi: 10.1021/ja055388i. [DOI] [PubMed] [Google Scholar]; (b) Wallraff GM, Boyd RH, Michl J. J Am Chem Soc. 1983;105:4550. [Google Scholar]
- 38.Johnson JE, Morales NM, Gorczyca AM, Dolliver DD, McAllister MA. J Org Chem. 2001;66:7979. doi: 10.1021/jo010067k. [DOI] [PubMed] [Google Scholar]
- 39.(a) Kayser RH, Pollack RM. J Am Chem Soc. 1977;99:3379. doi: 10.1021/ja00837a076. [DOI] [PubMed] [Google Scholar]; (b) Buist GJ, Lucas HJ. J Am Chem Soc. 1957;79:6157. [Google Scholar]
- 40.Pollack RM, Kayser RH, Damewood JR. J Am Chem Soc. 1977;99:8232. [Google Scholar]
- 41.PDB ID: 2EXA, complex of faropenem with PBP4 of E. coli: Kishida H, Unzai S, Roper DI, Lloyd A, Park SY, Tame JRH. Biochemistry. 2006;45:783. doi: 10.1021/bi051533t.PDB ID: 1B12, complex of penem-derived inhibitor with Type 1 signal peptidase from E. coli: Paetzel M, Dalbey RE, Strynadka NCJ. Nature. 1998;396:186. doi: 10.1038/24196.
- 42.For an example of a substitution reaction involving anchimeric assistance by a tert-butyl ester, without loss of the tert-butyl group, see: Walsh S, Pasternak A, Cato B, Finke PE, Frie J, Fu Q, Kim D, Pio B, Shahripour A, Shi Z-C, Tang H. Preparation of piperidine derivatives as inhibitors of renal outer medullary potassium channel. 2013039802. PCT Int Appl WO. 2013:48.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.