

Abstract
Noninvasive imaging in living subjects with positron emission tomography (PET) provides early detection of diseases in humans. For this application, it is necessary to prepare specific molecular imaging probes labeled with positron-emitting radioisotopes such as fluorine-18 for obtaining high-quality PET imaging. In this review, we describe recent trends in the F-18 radiolabeling method for the introduction of no-carrier-added fluorine-18, which was obtained from an 18O(p,n)18F reaction, into a specific molecular site, which in turn is intended to serve as an imaging agent for PET study. These labeling protocols are based on ionic liquid media 18F radiofluorination in the presence of some water, enzymatic 18F fluorination using fluorinase in water solution, non-polar protic alcohol media 18F radiofluorination and its mechanism, and nucleophilic 18F fluorination of an aromatic iodonium salt precursor.
Keywords: Nucleophilic fluorination, Fluorine-18, Ionic liquid, Tert alcohol, Fluorinase, Iodonium salts
Introduction
Positron emission tomography (PET) has been widely used in the medical imaging of molecular and biological processes, and provides promising opportunities to monitor metabolism and detect diseases in humans [1]. It is necessary to prepare specific molecular imaging probes labeled with positron-emitting radioisotopes for obtaining high-quality PET imaging [1, 2]. For this application, in particular fluorine-18 has many desirable characteristics: 18F-Radiolabeled biologically active compounds, such as peptides, neuro-transmitter ligands, and enzyme inhibitor analogues, can be used to trace biochemical processes while maintaining favorable interaction with the target since fluorine-18 can afford minimal steric interference; its relatively long half-life (t1/2 = 110 min) can allow [18F]-labeled radiopharmaceuticals to be produced regionally and shipped for imaging studies to nearby hospitals that are not equipped with cyclotrons for radionuclide production; the [18F]fluorine atom can form stable bonding to carbon at the molecular level; finally, fluorine-18 can be produced easily from cyclotron via irradiation of [18O] water targets with proton beams [3].
However, despite the many desirable characteristics of fluorine-18, only a few [18F]-radiolabeling processes are currently available for introducing fluorine-18 to biologically active compounds rapidly, efficiently, and conveniently [4]. In particular, in case of the preparation of receptor-binding PET radiopharmaceuticals, these processes require highly specific activity [5]. The typical method for introducing a single fluorine-18 at a specific molecular site is the nucleophilic displacement of the corresponding sulfonates and halides by the [18F]fluoride ion at the no-carrier-added level [4–6]. While [18F]fluoride in [18O]H2O generated from a cyclotron has been used traditionally for this purpose, its limited solubility in organic solvents and generally low nucleophilicity mean that vigorous reaction conditions are required for this sort of [18F]-radiolabeling reaction. To facilitate the nucleophilic [18F]-radiolabeling process, various “naked” highly nucleophilic [18F]fluoride anion systems have been developed. The solubility and nucleophilicity of [18F]fluoride anions were improved by the use of bulky counter cations, which often have been generated by phase-transfer-type protocols, such as quaternary ammonium [18F]fluoride derivatives and alkali metal [18F]fluoride/crown-ether complexation. In addition, because [18F]fluoride anions are tightly hydrated and become chemically inert in water, although they are potent nucleophiles in their desolvated state, the azeotropic drying step of the aqueous solution ([18F]F-/[18O]H2O) is also required to remove water from [18F]fluoride anion reagents [7].
As shown in Fig. 1, “naked” highly nucleophilic [18F]fluoride anions, such as tetrabutylammonium [18F]fluoride ([18F]TBAF) and the [18F]KF/Kryptofix[2.2.2] complex, are known to be representative reagents for [18F]-radiolabeling synthesis: [18F]TBAF is generated from tetrabutylammonium hydroxide (TBAOH) or tetrabutylammonium bicarbonate with [18F]F- produced from [18O]H2O; the [18F]KF/Kryptofix[2.2.2] complex is generated from K2CO3 with [18F]F- in H2O. However, while the enhanced nucleophilicity of these “naked” [18F]fluoride anions generally accelerates the rate of [18F]-radiolabeling reactions [6], these reaction systems are generally strongly basic, which restricts their [18F]-radiolabeling utility because, in case of the use of base-sensitive precursor molecules, it engenders various side reactions, such as eliminations, cycloalkylation, etc. [7]. In addition, because it is difficult to get a naked [18F]fluoride anion that is completely anhydrous, naked hydroxide, which is generated from reaction media, leads to hydroxylations that form alcohols as a byproduct [7]. Therefore, many research groups have shown interest in the alternative [18F]-radiolabeling reaction method for mild conditions but the chemo-selective introduction of [18F]fluorine into biologically active compounds for PET imaging [7, 8].
Fig. 1.
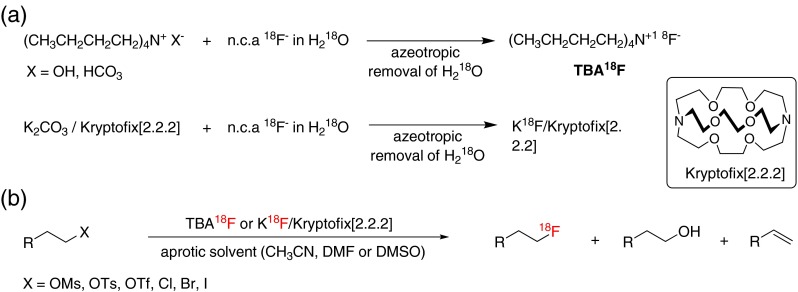
This figure shows the preparation of TBA18F as well as K18F/Kryptofix[2.2.2] as a “naked” fluoride-18 source (a), and nucleophilic 18F fluorination using the conventional method affords a desirable fluoroproduct with the formation of byproducts such as alcohols and alkenes (b)
Ionic Liquid Media [18F] Radiofluorination in the Presence of Some Water
Ionic liquid media [9–11] nucleophilic [18F] radiofluorination using [18F]fluoride ions and Cs2CO3 in the presence of some water was reported as a new method for fluorine-18 labeling in 2003 [12, 13]. In this method, the [18F] fluorination of mesyloxyalkanes using Cs2CO3 to generate Cs[18F]F in ionic liquid, 1-butyl-3-methylimidazolium triflate ([bmim][OTf]), afforded the corresponding [18F] fluoroproduct in 93% radiochemical yield (RCY) after reaction at 120°C for 5–10 min (Fig. 2a). The reaction system was not capped for azeotropic removal of water to proceed simultaneously with the reaction at this reaction temperature, reducing the overall reaction time considerably. This means that [18F]fluoride in water, as obtained from the production target in the cyclotron, can be added directly to the [18F]-radiofluorination reaction media without a dehydration process, which was typically a time-consuming step required in other previous methods. Moreover, even though the RCY dropped slightly, the [18F]-radiofluorination reaction also proceeded efficiently in the presence of larger amounts of water (250 μl) than the optimized amounts of water (50 μl) as shown in Fig. 2b [12]. Jeong et al. [14] reported a successful application in the improved preparation of [18F]FDG, which is the most popular radiopharmaceutical for PET imaging, using this ionic liquid media [18F]-radiofluorination method. However, this [18F]-labeling method could have problems when the [18F]radiopharmaceutical was polar and contained many heteroatoms, because it became difficult to extract it from the ionic liquid phase [15]. To overcome this drawback of ionic liquids, there are some reports about a polymer-supported ionic liquid that can be used for nucleophilic non-radioactive fluorination as a catalyst [15, 16]. However, the application of these catalysts to [18F]-radiofluorination labeling reactions of radiopharmaceuticals has not been reported up to now.
Fig. 2.

This figure shows nucleophilc 18F fluorination in ionic liquid [bmim][OTf], the structure of [bmim][OTf] (a), and the influence on the amount of water in the F-18 labeling reaction (b)
Enzymatic [18F] Radiofluorination Using Fluorinase Enzyme in Water Solution
Since fluorides are tightly hydrated and become “inert” fluorides (Fig. 3), water as a reaction medium cannot be used for nucleophilic fluorination [17]. Perhaps the sole exception to this is enzymatic fluorination in water catalyzed by a fluorinase enzyme, but here, specific hydrogen bonding of the enzyme with the fluoride and the substrate appears to facilitate the formation of C-F bonds [17, 18]. This finding that a fluorination enzyme as a biocatalyst (isolated from the bacterium Streptomyces cattleya) can provide C-F bond formation has opened up some prospects for such an [18F]-radiolabeling method in this area. In 2003, Martarello et al. tried to perform enzymatic [18F] radiofluorination of (S)-adenosyl-L-methionine (SAM) as both a precursor and fluorinase substrate to 5′-[18F]fluoro-5′-deoxyadenosine ([18F]-5′-FDA) using wild-type fluorinase and [18F]fluoride/[18O]water directly supplied from the cyclotron in Tris-HCl buffer solution, but they found that the process afforded [18F]-5′-FDA in only 1% of the overall radiochemical yield [19]. Although this enzymatic approach was technically inapplicable, it showed the possibility of the enzymatic [18F]-radiofluorination method. In a recent report by the same research group, the radiochemical yields for [18F]-5′-FDA production from [18F]fluoride had improved dramatically (up to 95%) by the use of recombinant fluorinase and L-amino acid oxidase (L-AAO) in the same enzymatic [18F]radiofluorination. L-amino acid oxidase acted as a scavenger to remove L-methionine for preventing a reverse reaction in the reaction, consequently increasing the reaction rate and RCY (Fig. 4) [20]. More recently, [18F]-radiolabeled nucleoside derivatives were extensively studied as potential radiopharmaceuticals for PET tumor imaging by Winkler et al. [21]. They reported one-pot [18F] radiofluorination/base-swap biotransformations of (S)-adenosyl-L-methionine to [18F]-5′-deoxy-5′-fluorouridines (via the formation of [18F]-5′-FDA) with four enzymes, fluorinase, L-AAO, purine nucleoside phosphorylase (PNP), and thymidine phosphorylase (TP), in the presence of uracil, potassium phosphate, and [18F]fluoride (Fig. 5). These results demonstrated a new application of enzymatic [18F] radiofluorination for the synthesis of various [18F]-radiolabeled nucleosides. However, the limitation of the precursor as a fluorinase substrate and the relatively long reaction time could be drawbacks in terms of the application of various types of compounds and the generation of large amounts of radioactive radiopharmaceuticals.
Fig. 3.

This figure illustrates a hydrated fluoride in water: “inert” (isolated) fluoride; no nucleophilic activity
Fig. 4.

This figure illustrates the enzymatic [18F]-fluorination using fluorinase in water solution
Fig. 5.
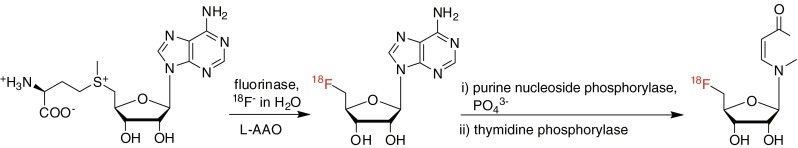
This figure shows the fluorinase-coupled base swaps after enzymatic radiolabeling for the synthesis of [18F]-5′-deoxy-5′-fluorouridines
Non-polar Protic Alcohol Media [18F] Radiofluorination and its Mechanism
It is well known that protic solvents such as water and alcohols are not appropriate for most nucleophilic [18F]-radiofluorination reactions since protic solvents reduce the nucleophilicity of [18F]fluoride by extensive hydrogen bonding and interaction with the partial positive charge of these solvents. Therefore, polar aprotic solvents, such as acetonitrile, N,N-dimethylformamide (DMF), and dimethyl sulfoxide (DMSO), have been widely used as a solvent for these [18F]-radiolabeling reactions. The nucleophilicity of [18F]fluoride in polar aprotic solvents usually is enhanced through the selective solvation of its counter cations by the negative end of the polar aprotic solvent dipole and the lack of a proton for hydrogen bonding, which leave the [18F]fluoride free or “naked” [22]. However, recently, a highly efficient aliphatic nucleophilic fluorination and [18F]-radiofluorination method with alkali metal fluorides or [18F]fluoride using nonpolar protic tert alcohols as a reaction medium has been reported (Table 1) [23]. In this method, the tert-alcohol media, such as tert-butyl alcohol and tert-amyl alcohol, greatly enhanced the reactivity of alkali metal fluorides. In particular, the use of tert alcohols could reduce the formation of typical byproducts (namely, alkenes, alcohols, or ethers) dramatically in the nucleophilic fluorination of base-sensitive compounds, such as 1-(2-mesylethyl)naphthalene and N-5-bromopentanoyl-3,4-dimethoxyaniline (Table 1, entries 2 and 3, respectively) compared with the use of conventional polar aprotic solvents [23–25].
Table 1.
Nucleophilic fluorination and synthesis of 18F-fluorine-labeled PET radiopharmaceuticals using the tert-alcohol solvent system
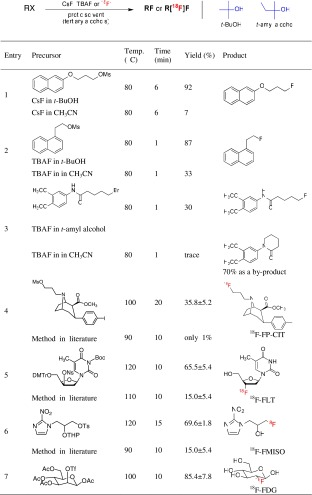
The attributes of this fluorination method play an important role in the efficient automatic preparation of [18F] radiopharmaceuticals for PET molecular imaging studies. Although [18F]FP-CIT is known to be an important radiopharmaceutical for PET imaging of dopamine transporters, it is impossible to use it routinely because it has only been obtained in 1% yield. However, using the tert-alcohol media [18F]-fluorination method, [18F]FP-CIT was produced in approximately 36% yield at 100°C for 20 min in one step (entry 4) [26]. This mass production of [18F]FP-CIT could be used clinically and commercially. This report demonstrated that this new [18F]-radiolabeling method also provided the syntheses of other [18F] radiopharmaceuticals—[18F]FLT (65.5%) [27], [18F]FMISO (69.6%) [28], and [18F]FDG (85.4%)—in high radiochemical yield and purity, and in shorter times compared to conventional syntheses.
Because this finding was in marked contrast with the expectations from the conventional [18F]-radiofluorination mechanism; recently, the mechanism of the nucleophilic fluorination reaction in tert-alcohol solvent has been reported. As shown in Fig. 6, the authors suggested the mechanism: limited solvation of [18F]fluoride coordinated with bulky tert alcohols (named “flexible” fluoride) could make the fluoride an especially good nucleophile in this medium; leaving group reactivity could be enhanced by hydrogen bonding between the tert-alcohol solvent with oxygen atoms of a sulfonate leaving group; side reactions, such as eliminations, hydroxylations, and intramolecular alkylations, could be suppressed by the protic environment, particularly when using base-sensitive precursors [24, 25]. In a more recent paper, it was proven that “flexible” fluoride could be formed in the tert-alcohol media fluorination processing as a result of the preparation of tetrabutylammonium tetra t-butanol coordinated fluoride, TBAF(t-BuOH)4, from commercially available TBAF in t-BuOH. This TBAF(t-BuOH)4 also showed good performance in the nucleophilic fluorination because of its ideal favorable properties as a fluoride source, such as the dehydrated state for anhydrous reaction conditions, low hygroscopicity, good solubility in organic solvents, and good nucleophilicity with low basicity (Fig. 7) [29–31].
Fig. 6.
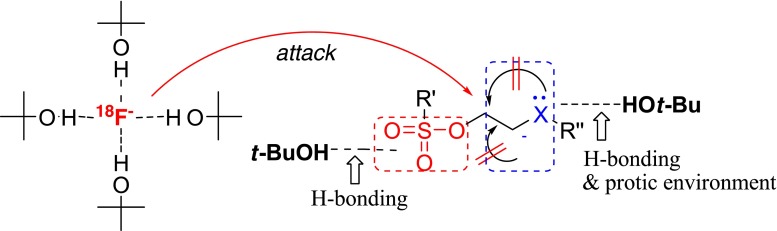
The proposed mechanism of a tert-alcohol medium in nucleophilic aliphatic [18F]-radiofluorination reactions
Fig. 7.

This figure shows the reactivity of TBAF(t-BuOH)4 and TBAF in a nucleophilic fluorination reaction (a) and X-ray crystal structure of “flexible” fluoride—TBAF(t-BuOH)4 (b)
Nucleophilic [18F] Fluorination of Aromatic Iodonium Salt Precursor with No-carrier-added [18F]fluoride
The method for the introduction of fluorine-18 at a specific aromatic molecular site has received much attention in the area of [18F] radiochemistry because of the many biologically active compounds that contain various aromatic rings [32]. Although the nucleophilic [18F]-flurorination reactions of aromatic target molecules with no-carrier-added [18F]fluoride so far have generally been limited to the introduction of 18F on electron-deficient aromatic rings [33, 34], this reaction protocol not only provides highly specific active [18F]fluoroarenes, but also yields high chemo-selectively compared to the electrophilic aromatic [18F] flurorination using [18F]F2 or [18F]acetylhypofluorite [35]. Recently, diaryliodonium salts have received much attention in the direct nucleophilic aromatic [18F] fluorination of various arenes, in particular, electron-rich arenes, because these salts have allowed efficient nucleophilic substitution on arenes as shown in Fig. 8 [36, 37]. Ross et al. [38] developed heteroaromatic iodonium salts, such as aryl-(2-thienyl)iodonium salt derivatives, as precursors for regioselective nucleophilic [18F]flurorination. In this report, the highly electron-rich heteroaromatic 2-thienyl group made the no-carrier-added [18F]fluoride attack the aryl group regioselectively, and consequently only the desired [18F]-radiolabeled arenes could be obtained without any other [18F]-radiolabeled byproducts (Fig. 8b). The attributes of this method play a crucial role in the syntheses of radiopharmaceuticals containing various aromatic rings (via direct aromatic [18F] labeling) for PET imaging studies. For example, Lee et al. [39, 40] reported that [18F]-radiolabeled farglitazar (GI 262570) as a peroxisome proliferator-activated receptor gamma ligand could be synthesized through the nucleophilic [18F] fluorination of the corresponding iodonium salt precursor with [18F]fluoride (Fig. 8c). However, this [18F]-labeling method might often have the limitation of difficulty in preparing the iodonium salt precursor.
Fig. 8.
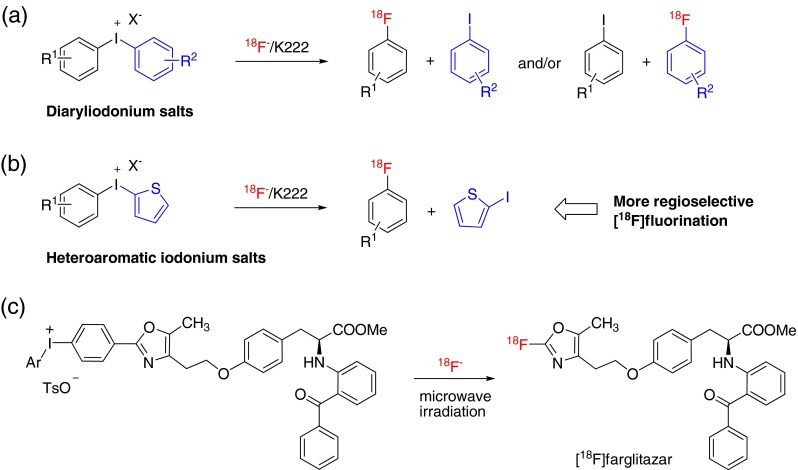
This figure shows the introduction of fluorine-18 into arenes through the reactions of diaryliodonium salt (a) and heteroaromatic iodonium salts such as aryl-(2-thienyl)iodonium salt with no-carrier-added [18F]fluoride (b). This shows that [18F]farglitazar was synthesized from the corresponding iodonium salt precursor (c)
Conclusion
In summary, we have described various recent nucleophilic [18F]-radiolabeling protocols with no-carrier-added [18F]fluoride. The final goal of these novel fluorine-18 radiolabeling protocols is to obtain F-18-labeled radiopharmaceuticals efficiently for their application as PET imaging agents in nuclear medicine and life sciences. This is a good example showing that, since it could become possible to produce [18F]FP-CIT and [18F]FLT automatically and routinely through the new alcohol media [18F]-radiofluorination method, they could be used clinically and commercially in Korea. To increase the role of PET nuclear molecular imaging in medicine and life sciences, it is necessary to find both more reliable radiopharmaceuticals and radiolabeling methods. Many studies on the development of even more efficient protocols (greater selectivity, shorter reaction time, higher radiochemical yield, etc.) for nucleophilic radiolabeling reactions with no-carrier-added [18F]fluoride to produce radiopharmaceuticals for in vivo imaging using PET are in progress in many research groups.
Acknowledgment
This work was supported by Nuclear Research and Development Program of the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean government (MEST) (grant code: 2009-0078422 and 2009-0062472 to D.W.K). We also thank Dr. Vinod H. Jadhav for helpful discussions.
References
- 1.Kilbourn MR, Jarabek PA, Welch MJ. An improved [18O]water target for [18F]fluoride production. Int J Appl Radiat Isot. 1985;36:327–328. doi: 10.1016/0020-708X(85)90099-7. [DOI] [PubMed] [Google Scholar]
- 2.Chi DY. The development of radiopharmaceuticals for human body imaging. J Korean Ind Eng Chem. 2003;14:253–262. [Google Scholar]
- 3.Bolton R. Radiohalogen incorporation into organic systems. J Labelled Compd Radiopharm. 2002;45:485–528. doi: 10.1002/jlcr.575. [DOI] [Google Scholar]
- 4.Gerstenberger MRC, Haas A. Methods of fluorination in organic chemistry. Angew Chem Int Ed Engl. 1981;20:647–667. doi: 10.1002/anie.198106471. [DOI] [Google Scholar]
- 5.Mascaretti OA. Modern methods for the monofluorination of aliphatic organic compounds. Aldrichimica Acta. 1993;26:47–58. [Google Scholar]
- 6.Kilbourn MR. Nucl. Sci. Series, NAS-NS3203. Washington DC: National Academy Press; 1990. [Google Scholar]
- 7.Coenen HH. Synthesis and application of isotopically labeled compounds. Amsterdam: Elsevier; 1989. pp. 433–448. [Google Scholar]
- 8.Chi DY, Kilbourn MR, Katzenellenbogen JA, Welch MJ. A rapid and efficient method for the fluoroalkylation of amines and amides. Development of a method suitable for incoration of the short-lived positron emitting radionuclide fluorine-18. J Org Chem. 1987;52:658–664. doi: 10.1021/jo00380a030. [DOI] [Google Scholar]
- 9.Welton T. Room-temperature ionic liquids, solvents for synthesis and catalysis. Chem Res. 1999;99:2071–2083. doi: 10.1021/cr980032t. [DOI] [PubMed] [Google Scholar]
- 10.Holbrey JD, Seddon KR. (1999) The phase behaviour of 1-alkyl-3-methylimidazolium tetrafluoroborates; ionic liquids and ionic liquids crystals. J Chem Soc Dalton Trans 2133-2139
- 11.Wasserscheid P, Keim W. Ionic liquids–new “solution” for transition metal catalysis. Angew Chem Int Ed Engl. 2000;39:3772–3789. doi: 10.1002/1521-3773(20001103)39:21<3772::aid-anie3772>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Kim DW, Choe YS, Chi DY. A new nucleophilic fluorine-18 labeling method for aliphatic mesylates: reaction in ionic liquids shows tolerance for water. Nucl Med Biol. 2003;30:345–350. doi: 10.1016/S0969-8051(03)00017-9. [DOI] [PubMed] [Google Scholar]
- 13.Kim DW, Song CE, Chi DY. New method of fluorination using potassium fluoride in ionic liquid: significantly enhanced reactivity of fluoride and improved selectivity. J Am Chem Soc. 2002;121:10278–10279. doi: 10.1021/ja026242b. [DOI] [PubMed] [Google Scholar]
- 14.Kim HW, Jeong JM, Lee YS, Chi DY, Chung KH, Lee DS, et al. Rapid synthesis of [18F]FDG without an evaporation step using an ionic liquid. Appl Radiat Isot. 2004;61:1241–1246. doi: 10.1016/j.apradiso.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 15.Kim DW, Chi DY. Polymer-supported ionic liquids: imidazolium salts as catalysts for nucleophilic substitution reactions including fluorinations. Angew Chem Int Ed. 2004;43:483–485. doi: 10.1002/anie.200352760. [DOI] [PubMed] [Google Scholar]
- 16.Kim DW, Hong DJ, Jang KS, Song CE, Chi DY. Structural modification of polymer-supported ionic liquids: ionic resins as catalysts for nucleophilic substitution reactions. Adv Synth Catal. 2006;348:1719–1727. doi: 10.1002/adsc.200606119. [DOI] [Google Scholar]
- 17.O’Hagan D, Schaffrath C, Cobb SL, Hamilton JTG, Murphy CD. Biochemistry: biosynthesis of an organofluorine molecule. Nature. 2002;416:279. doi: 10.1038/416279a. [DOI] [PubMed] [Google Scholar]
- 18.Schaffrath C, Deng H, O’Hagan D. Isolation and characterization of 5′-fluorodeoxyadenosine synthase, a fluorination enzyme from Streptomyces cattleya. FEBS Lett. 2003;547:111–114. doi: 10.1016/S0014-5793(03)00688-4. [DOI] [PubMed] [Google Scholar]
- 19.Martarello L, Schaffrath C, Deng H, Gee AD, Lockhart A, O’Hagan D. The first enzymatic method for C-18F bond formation: the synthesis of 5′-[18F]-fluoro-5′-deoxyadenosine for imaging with PET. J Label Compd Radiopharm. 2003;46:1181–1189. doi: 10.1002/jlcr.779. [DOI] [Google Scholar]
- 20.Deng H, Cobb SL, Gee AD, Lockhart A, Martarello L, McGlinchey RP et al (2006) Fluorinase mediated C-18F bond formation, an enzymatic tool for PET labelling. Chem Commun 652–654 [DOI] [PubMed]
- 21.Winkler M, Domarkas J, Schweiger LF, O’Hagan D. Fluorinase-coupled base swaps: synthesis of [18F]-5′-deoxy-5′-fluorouridines. Angew Chem Int Ed. 2008;47:10141–10143. doi: 10.1002/anie.200804040. [DOI] [PubMed] [Google Scholar]
- 22.Reichardt C. Solvents and solvent effects in organic chemistry. New York: Wiley-VCH; 2002. [Google Scholar]
- 23.Kim DW, Ahn DS, Oh YH, Lee S, Kil HS, Oh SJ, et al. A new class of SN2 reactions catalyzed by protic solvents: facile fluorination for isotopic labeling of diagnostic molecules. J Am Chem Soc. 2006;128:16393–16397. doi: 10.1021/ja0646895. [DOI] [PubMed] [Google Scholar]
- 24.Kim DW, Jeong HJ, Lim ST, Sohn MH, Katzenellenbogen JA, Chi DY. Facile nucleophilic fluorination reaction using tert-alcohols as a reaction medium: significantly enhanced reactivity of alkali metal fluorides and improved selectivity. J Org Chem. 2008;73:957–962. doi: 10.1021/jo7021229. [DOI] [PubMed] [Google Scholar]
- 25.Kim DW, Jeong HJ, Lim ST, Sohn MH, Chi DY. Facile nucleophilic fluorination by synergistic effect between polymer-supported ionic liquid catalyst and tert-alcohol. Tetrahedron. 2008;64:4209–4214. doi: 10.1016/j.tet.2008.02.094. [DOI] [Google Scholar]
- 26.Chaly T, Dhawan V, Kazumata K, Antonini A, Margouleff C, Dahl JR, et al. Radiosynthesis of [18F]N-3-fluoropropyl-2-beta-carbomethoxy-3-beta-(4-iodophenyl)nortropane and the first human study with positron emission tomography. Nucl Med Biol. 1996;23:999–1004. doi: 10.1016/S0969-8051(96)00155-2. [DOI] [PubMed] [Google Scholar]
- 27.Oh SJ, Mosdzianowski C, Chi DY, Kim JY, Kang SH, Ryu JS, et al. Fully automated synthesis system of 3′-deoxy-3′-[18F]fluorothymidine. Nucl Med Biol. 2004;31:803–809. doi: 10.1016/j.nucmedbio.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Oh SJ, Chi DY, Mosdzianowski C, Kim JY, Kil HS, Kang SH, et al. Fully automated synthesis of [18F]fluoromisonidazole using a conventional [18F]FDG module. Nucl Med Biol. 2005;32:899–905. doi: 10.1016/j.nucmedbio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Kim DW, Jeong HJ, Lim ST, Sohn MH. Tetrabutylammonium tetra(tert-butyl alcohol)-coordinated fluoride as a facile fluoride source. Angew Chem Int Ed Engl. 2008;47:8404–8406. doi: 10.1002/anie.200803150. [DOI] [PubMed] [Google Scholar]
- 30.Pilcher AS, Ammon HL, DeShong P. Utilization of tetrabutylammonium triphenylsilyldifluoride as a fluoride source for nucleophilic fluorination. J Am Chem Soc. 1995;117:5166–5167. doi: 10.1021/ja00123a025. [DOI] [Google Scholar]
- 31.Sun H, DiMagno SG. Anhydrous tetrabutylammonium fluoride. J Am Chem Soc. 2005;127:2050–2051. doi: 10.1021/ja0440497. [DOI] [PubMed] [Google Scholar]
- 32.Namavari M, Bishop A, Satyamurthy N, Bida G, Barrio JR. Regioselective radiofluorodestannylation with [18F]F2 and [18F]CH3COOF; a high yield synthesis of 6-[18F]fluoro-L-dopa. Appl Radiat Isot. 1992;43:989–996. doi: 10.1016/0883-2889(92)90217-3. [DOI] [PubMed] [Google Scholar]
- 33.Hamacher K, Hamkens W. Remote controlled one-step production of 18F labeled butyrophenone neuroleptics exemplified by the synthesis of n.c.a. [18F]N-methylspiperone. Appl Radiat Isot. 1995;46:911–916. doi: 10.1016/0969-8043(95)00185-G. [DOI] [Google Scholar]
- 34.Katsifis A, Hamacher K, Schnitter J, Stocklin G. Synthesis of fluorine-18-labelled 5- and 6-fluoro-2-pyridinamine. Appl Radiat Isot. 1993;44:1015–1020. doi: 10.1016/0969-8043(93)90005-U. [DOI] [Google Scholar]
- 35.Hess E, Sichler S, Kluge A, Coenen HH. Synthesis of 2-[18F]fluoro-L-tyrosine via regiospecific fluoro-de-stannylation. Appl Radiat Isot. 2002;57:185–191. doi: 10.1016/S0969-8043(02)00091-X. [DOI] [PubMed] [Google Scholar]
- 36.Pike VW, Aigbirhio FI (1995) Reactions of cyclotron-produced [18F]fluoride with diaryliodonium salts-a novel single-step route to no-carrier-added [18F]fluoroarenes. J Chem Soc Chem Commun 2215–2216
- 37.Shah A, Pike VW, Widdowson DA. The synthesis of [18F]fluoroarenes from the reaction of cyclotron-produced [18F]fluoride ion with diaryliodonium salts. J Chem Soc Perkin Trans. 1998;1:2043–2046. doi: 10.1039/a802349b. [DOI] [Google Scholar]
- 38.Ross TL, Ermert J, Hocke C, Coenen HH. Nucleophilic 18F-fluorination of heteroaromatic iodonium salts with no-carrier-added [18F]fluoride. J Am Chem Soc. 2007;129:8018–8025. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 39.Lee BC, Lee KC, Lee H, Mach RH, Katzenellenbogen JA. Strategies for the labeling of halogen-substituted peroxisome proliferator-activated receptor gamma ligands: potential positron emission tomography and single photon emission computed tomography imaging agents. Bioconjugate Chem. 2007;18:514–523. doi: 10.1021/bc060191g. [DOI] [PubMed] [Google Scholar]
- 40.Lee BC, Dence CS, Zhou H, Parent EE, Welch MJ, Katzenellenbogen JA. Fluorine-18 labeling and biodistribution studies on peroxisome proliferator-activated receptor-gamma (PPARg) ligands: potential positron emission tomography (PET) imaging agents. Nucl Med Biol. 2009;36:147–153. doi: 10.1016/j.nucmedbio.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]