

Abstract
The R2 protein of class I ribonucleotide reductase generates and stores a tyrosyl radical essential for ribonucleotide reduction and, thus, DNA synthesis. X-ray structures of the protein have enabled detailed mechanistic suggestions, but no structural information has been available for the active radical-containing state of the protein. Here we report on methods to generate the functional tyrosyl radical in single crystals of R2 from Escherichia coli (Y122•). We further report on subsequent high-field EPR experiments on the radical-containing crystals. A full rotational pattern of the spectra was collected and the orientation of the g-tensor axes were determined, which directly reflect the orientation of the radical in the crystal frame. The EPR data are discussed in comparison with a 1.42-Å x-ray structure of the met (oxidized) form of the protein, also presented in this paper. Comparison of the orientation of the radical Y122• obtained from high-field EPR with that of the reduced tyrosine Y122-OH reveals a significant rotation of the tyrosyl side chain, away from the diiron center, in the active radical state. Implications for the radical transfer connecting the diiron site in R2 with the substrate-binding site in R1 are discussed. In addition, the present study demonstrates that structural and functional information about active radical states can be obtained by combined x-ray and high-field EPR crystallography.
Ribonucleotide reductases (RNRs) catalyze the reduction of ribonucleotides to deoxyribonucleotides and are thus essential for DNA synthesis. Class I RNRs consist of two homodimeric proteins: R1, which contains the active site and binding site for allosteric regulators, and R2, which generates and harbors the tyrosyl radical Y122• (Escherichia coli numbering) needed for catalysis. The catalytic reaction in R1 is believed to be initiated by a reversible radical transfer from Tyr 122 in R2 to an active-site cysteine in R1.
The tyrosyl radical on the R2 subunit is generated by means of the reductive cleavage of molecular oxygen at a diiron center. Crystal structures of E. coli R2 are available for both the reduced diferrous (FeIIFeII-Y122-OH) form (1) and the diferric, met, form lacking the radical (FeIIIFeIII-Y122-OH) (2). The structural data have, together with kinetic data and theoretical calculations, served as the basis for the formulation of proposals for the mechanism of radical generation and radical migration in the overall RNR reaction (3–9). However, no structure has been available for the radical-containing form, hence, the orientation and location of the active radical Y122• have not been known.
The diiron center is in the active enzyme in the diferric form and couples antiferromagnetically to an S = 0 ground state (3–5, 10). Detailed high-field EPR experiments have been performed on Y122• in frozen R2 solutions (11–15). The obtained g-tensor values were found to be indicators for the polarity of the radical environment. In particular, it was found that the tyrosyl radical is hydrogen-bonded in RNR of mouse and herpes simplex virus (12, 14), whereas in E. coli it is not (11–15).
In protein solution, both the in vivo activation, reacting the diferrous protein with molecular oxygen, and reactivation of the radical by reacting the met form of R2 with H2O2 (shunt pathway) have been used to generate the tyrosyl radical, and thereby restore catalytic activity (16, 17). Here we report on generation of the radical in the crystal form of the protein, by using both methods, with an estimated yield of 10%. This resulting amount is too low for obtaining an x-ray structure of the radical form, but is sufficient for high-field EPR studies. Analysis of the obtained data enabled determination of a significant conformational change (reorientation) of the radical Y122• as compared with the reduced Y122-OH residue.
Materials and Methods
Protein Expression and Purification.
WT E. coli R2 protein was expressed and purified according to previously published protocols (18). To produce the apoprotein (WT R2 without Fe) for the reconstitution experiment the iron ions were removed before gel filtration by using the chelation protocol of Sjöberg et al. (19).
Crystallization, X-Ray Data Collection, and Refinement.
The WT proteins and apoprotein were crystallized according to the previously published protocol (20). To obtain the high-resolution x-ray data set, crystals were moved to a cryo solution (mother liquor containing 23% glycerol) for a few minutes, picked up in rayon loops (Hampton Research, Riverside, CA) and flash-frozen in liquid nitrogen. X-ray diffraction data extending from 40 to 1.42 Å were collected on a Mar345 imaging plate detector at the Swiss–Norwegian beamline at the European Synchrotron Radiation Facility (Grenoble, France). Processing and scaling of the data were performed using the hkl software suite (21). The structure was refined by using cns (22) with the structure of the reduced R2 protein (1) as a starting model. After rigid body refinement and simulated annealing, waters were added by using warp (23). Refer to Table 1 for data collection and refinement statistics information.
Table 1.
X-ray data collection and refinement statistics
| Data collection | |
| Space group | P212121 |
| Cell parameters a, b, c, Å | 73.84, 84.98, 114.32 |
| Resolution, Å (outer shell) | 40-1.42 (1.47-1.42) |
| No. of observations | 603,231 |
| Unique reflections | 123,371 |
| Rsym* (outer shell) | 0.064 (0.288) |
| Completeness, % (outer shell) | 90.8 (91.2) |
| Refinement statistics | |
| Rcryst,† % | 16.0 |
| Rfree,‡ % (5% of data) | 18.5 |
| Nonhydrogen atoms | 6,463 |
| Solvent molecules | 910 |
| rms deviations | |
| Bonds, Å | 0.013 |
| Angles, ° | 1.51 |
| Ramachandran plot, % of residues | |
| Most favored | 93.9 |
| Allowed | 6.1 |
| Generously allowed and disallowed | 0.0 |
| Estimated coordinate error, Å | |
| From Luzzati plot | 0.13 |
| From SigmaA | 0.09 |
Rsym = ∑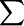 j∑h|〈Ihj − Ih〉|/∑j∑hIjh where Ihj is the jth observation of reflection h.
j∑h|〈Ihj − Ih〉|/∑j∑hIjh where Ihj is the jth observation of reflection h.
Rcryst = ∑∥Fobs − Fcalc∥/|∑obs|, where Fobs and Fcalc are the observed and calculated structure-factor amplitudes, respectively.
Rfree is equivalent to Rcryst for a 5% subset of reflections not used in the refinement.
Radical Generation in R2 Crystals.
In the oxidized FeIIIFeIII-Y122-OH (met) WT R2 protein crystals, the radical was generated via the so-called shunt pathway (17). Crystals were soaked in cryo solution supplemented with 10 mM H2O2 at room temperature for 1 h. To be able to collect both EPR and x-ray data from the same crystal to verify the crystal orientation obtained from the EPR analysis, crystals were mounted in a loop extending from a W-band EPR tube and then flash-frozen in liquid nitrogen. Care was taken to ensure that the rotation axis (EPR tube axis) did not coincide with one of the crystal axes. After EPR measurements the EPR tube with the crystal was mounted on a rotating anode x-ray source, and a few images were exposed to determine the orientation of the crystal with respect to the capillary (laboratory frame). In crystals of the apoprotein, the radical was generated by the normal reconstitution reaction (16). Crystals were soaked in cryo solution supplemented with 5 mM FeCl2/10 mM sodium ascorbate for 30 min and then flash-frozen in liquid nitrogen. As the apoprotein crystals were too small for the collection of good single-crystal EPR data, six small crystals were inserted with arbitrary orientations into one W-band EPR tube.
W-Band (94-GHz) EPR Spectroscopy.
Continuous-wave EPR experiments were performed on a commercial Elexsys 680 spectrometer (Bruker, Rheinstetten, Germany) operating at 94 GHz. Spectra were recorded at 80 K. Low microwave power, typically only a few microwatts, was used to avoid saturation effects. For determination of precise g-tensor values the microwave frequency was measured by a frequency counter, which is integrated in the spectrometer. The magnetic field was calibrated with a g-standard (Li in LiF, g = 2.002293 ± 0.000002; ref. 24) at two different frequencies (typically 94.0 and 94.3 GHz). All spectra were recorded in the “persistent mode” of the superconducting magnet by using the room temperature coils for the field sweep [maximum sweep width, 800 G (1 G = 0.1 mT)], thereby ensuring a high linearity and stability of the magnetic field. The spectrometer design uses a W-band EPR resonator that ensures a high sensitivity in particular for small samples such as protein crystals.
Solutions of active freshly prepared R2 of E. coli were filled into the EPR quartz sample tube (0.87 mm o.d., 0.7 mm i.d.) and frozen in liquid nitrogen. The cold samples were transferred into the spectrometer cryostat. H2O2-treated R2 single crystals (approximate size 0.7 × 0.3 × 0.2 mm3) were frozen together with the W-band capillary in liquid nitrogen. Orientation-dependent spectra were recorded by rotation about the capillary axis perpendicular to the magnetic field in 7.5° steps.
Spectra Analysis.
The EPR spectra from the frozen solutions were analyzed with a software for simulation and fitting of EPR spectra for S = 1/2 systems with anisotropic g- and hyperfine (hf)-tensors with arbitrary relative orientations using second-order perturbation theory, which is described in (ref. 25 and references therein). Spectra from the crystals were simulated with a home-written program (using octave) that calculates the resonant field positions for the tyrosyl radicals for all sites in the crystal (space group P212121), and for all measured orientations to first-order perturbation theory, which is a good approximation at the applied high field (3.35 T). The resulting histogram is convoluted with a derivative Gaussian line-shape function, finally yielding the derivative EPR spectra. The program further includes a simplex downhill algorithm in the multidimensional parameter space combined with a simulated annealing procedure (26) to minimize the sums of mean-square deviations between experimental and simulated spectra. The angular dependent resonant field positions for the spectra were calculated based on the spin Hamilton operator
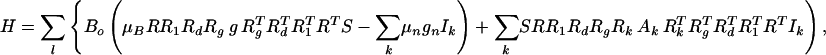 |
where Bo is the magnetic field, μB is the Bohr magneton, g is the electronic g tensor, μn and gn are the nuclear magneton and g factor, S is the electron spin operator, and Ak and Ik are the hf tensor and spin operator for nucleus k. R and RT denote rotation matrices and their transposed counterparts. These describe the orientation of the g-tensor axes with respect to the crystal frame (Rg), the rotations generating from a given site (l = 1) the three other sites (l = 2, 3, 4) within the crystal (180° rotations about the crystal axes a, b, and c, for the space group P212121), (Rl, l = 1–4), the rotation transforming protein chain A into chain B (Rd), and finally the orientation (R) of the crystal axes in the laboratory frame where the magnetic field Bo defines the x axis, and the rotation is performed along the EPR tube (z axis). Rk describes the orientation of the respective hf tensor Ak in the g-tensor axis system. The first sum in Eq. 1 runs over l site (l = 1–4), the next two sums run over k magnetic nuclei in the radical.
Results
High-Resolution Structure of Met R2.
The overall structure of the protein is, as expected, very similar to the previously determined met structure (refs. 27 and 28; PDB ID code ) with an rms deviation of 0.4 Å for the α carbons and 0.8 Å for all atoms. The geometry of the metal center in the 1.42-Å model is, however, far better defined. The major difference is that in the original met structure, both of the metals in the diiron center were assigned as being six-coordinate, whereas in the high-resolution structure, Fe1 is five-coordinate, with one vacant coordination position, but it maintains an octahedral geometry. This difference arises from a slight change in the conformation of D84, where, instead of being a bidentate ligand, it makes a monodentate interaction with Fe1 and the free carboxylate oxygen making a H-bond to the water coordinated to Fe2, and a second weak H-bond to Y122-OH, as seen in Fig. 1.
Fig 1.

Coordination geometry and H-bonds (dashed lines) in the metal center of the 1.42-Å structure of met R2. Distances are in Å.
Frozen-Solution W-Band EPR on Y122• of E. coliR2.
The 94-GHz continuous wave EPR spectrum of Y122• (Fig. 2) from frozen solutions of WT E. coli R2 is shown in Fig. 4 (top black trace). It exhibits a good resolution for the g components and for the hf structure, indicating the absence of hf and g strain, which would be expected in case of a heterogeneous environment (29). The g tensor and proton hf tensor principal values and the relative orientations of their axes were obtained from a fit to the EPR spectrum (Fig. 4, top red trace), and are collected and compared with previously published data for Y122• and for tyrosyl radicals in RNR of other organisms in Table 2 and Fig. 4 (see legend). The obtained values agree very well with published data (11–15, 30).
Fig 2.

Tyrosyl radical with numbering scheme and g-tensor principal axes, which are collinear with the molecular axes (33, 34, 37).
Fig 4.

The 94-GHz EPR spectra (black experiment, red simulation) of Y122• in R2 frozen solution (top trace) and of Y122• generated with H2O2 in a R2 single crystal. The crystal was rotated about an axis with arbitrary orientation with respect to the unit cell axes in 7.5° steps (see text). Colored cosine patterns indicate the angular dependence of the calculated effective g values for all four sites in the unit cell (space group P212121) for both protein chains A and B (see text). The same g- and hf-tensor parameters were obtained from simulations of the frozen solution and single-crystal spectra. For g values see Table 2; g-tensor axes are given in Table 3. hf-tensor values from simulation are as follows: −0.97(6) mT, −0.28(6) mT, and −0.74(6) mT for Ax,y,z [positions 3 and 5, numbers in parentheses are errors in the last digit; x and y axes are rotated by +24° (position 3) and −24° (position 5) with respect to the g-axes], and 2.10(6) mT, 1.85(6) mT, and 1.96(6) mT for Ax,y,z (Hβ1), in good agreement with refs. 14, 30, and 38. Single-component linewidth is 0.6 mT. Experimental conditions are as follows: temperature = 80 K, microwave power 2 μW, modulation amplitude 0.2 mT, modulation frequency 100 kHz, and acquisition time ≈6 min per trace.
Table 2.
g-tensor principal values for Y122• in RNR R2 single-crystal and frozen solutions of E. coli protein, compared with other organisms
| Tyrosyl radical/organism | gx | gy | gz | Ref. |
|---|---|---|---|---|
| Y122•/RNR E. coli*† R2 single crystal | 2.00912 | 2.00455 | 2.00219 | This work |
| Y122•/RNR E. coli*† R2 frozen solution | 2.00912 | 2.00454 | 2.00219 | This work |
| Y122•/RNR E. coli† | 2.00912 | 2.00457 | 2.00225 | 11, 30 |
| Y122•/RNR E. coli† | 2.0091 | 2.0046 | 2.0021 | 14 |
| Y105•/RNR Salmonella typhimurium† | 2.0089 | 2.0043 | 2.0021 | 13 |
| Y177•/RNR mouse‡ | 2.0076 | 2.0044 | 2.0021 | 14 |
| Y177•/RNR mouse‡ | 2.0076 | 2.0043 | 2.0022 | 12 |
| Y132•/RNR herpes simplex 1‡ | 2.0076 | 2.0043 | 2.0022 | 12 |
W-Band EPR on Y122• of E. coli in R2 Crystals.
Several small apoprotein crystals were reconstituted with ferrous iron and molecular oxygen. These crystals were too small for recording orientation-dependent spectra from one single crystal. However, superimposed 94-GHz EPR spectra of six such crystals in one EPR capillary were measured in two different orientations in comparison with the spectrum of Y122• in frozen solution. The spectra of these “superimposed six arbitrary orientation” crystals (Fig. 3) exhibited a range of effective g values and a partially resolved hf pattern in a range typical for tyrosyl radicals. The spectra were orientation-dependent, showing that it is possible to generate the radical Y122• in R2 single crystals by the natural reconstitution reaction.
Fig 3.

The 94-GHz EPR spectra (superposition from six small crystals with arbitrary orientations) of Y122• generated by reconstitution of the diiron center in small crystals of apo-R2 compared with the spectrum in frozen solution (see text). The two different orientations show a pronounced angular dependence of the spectra, indicating crystal order.
Orientation-dependent EPR spectra at 94 GHz were recorded from a larger H2O2-treated R2 single crystal in 7.5° steps. The spectra (Fig. 4) showed superimposed patterns from g shifts and hf splittings from the eight sites in one crystal, and were analyzed by simultaneous fits for all measured orientations with the program based on Eq. 1 described above. Thereby, a similar strategy was applied as in previous single-crystal EPR studies (31, 32). The principal values of the g tensor, of the hf tensors of the two ring protons (Fig. 2, positions 3 and 5), of the hf tensor of the β-proton of the side chain, and the relative orientations of the tensor principal axes with respect to the g-tensor axis system (rotation of +24° and −24° about z for protons 3 and 5) were taken from the simulations of the 94-GHz EPR spectrum of Y122• in R2 frozen solutions (see Fig. 4 legend). The rotation matrices Rl and Rd were known from the crystal space group (P212121), and from the high-resolution x-ray structure, and were an integrated fixed part of the program. This approach reduced the number of free parameters to six Euler angles describing the orientation of the crystal axes in the laboratory frame, matrix R, and the orientation of the g-tensor axes in the crystal frame, matrix Rg.
Both matrices were obtained by using a combined simplex and simulated annealing fit procedure applied simultaneously to all measured spectra. The simulated annealing procedure was necessary to avoid trapping of the minimization algorithm in one of the local minima, which are present as a result of the resolved hf structure in the spectra. Finally, the g-tensor values and the hf-tensor values of the ring protons and the β-proton of the side chain were treated as free fit parameters. No change with respect to the frozen solution values was observed within the experimental error margins (see Table 2 and Fig. 4 legend). Several simulated annealing and fit procedures using different starting parameters always yielded the same orientations of the g-tensor axes of Y122• within less than ±1°, with respect to the crystal axes a, b, and c, which are collected and compared with the orientation of the reduced tyrosine residue Y122-OH from the high-resolution x-ray structure in Table 3. The finally obtained best simultaneous fit to all spectra and the positions of the effective g values for each of the eight sites (cosine patterns) are also shown in Fig. 4. The generally occurring fourfold ambiguity concerning the assignment of the g-tensor axes in the case of the space group P212121 is removed by the known twofold C2 rotation axis transforming protein chain A into chain B as previously shown for another protein crystal (32). We furthermore confirmed the orientation of the crystal in the EPR tube by recording x-ray diffraction patterns of the same crystal for which the EPR data were collected.
Table 3.
Orientation of the g-tensor axes of Y122• in protein chain A of R2 single crystals of E. coli
| Crystal unit cell axes | gx* | gy* | gz* | C2 axis† |
|---|---|---|---|---|
| a | −0.5742 (57) | 0.1993 (45) | −0.7940 (35) | 0.3045 |
| b | 0.3624 (23) | −0.8078 (25) | −0.4649 (37) | 0.9183 |
| c | −0.7341 (51) | −0.5547 (45) | 0.3917 (49) | 0.2531 |
| ϕ (gi/Y122)‡ | 10° | 8° | 5° | – |
Direction cosines for the respective g-tensor principal axis in the crystal frame. Numbers in parentheses are maximum deviations obtained from a series of fit procedures with different starting orientations and they correspond to an error of ±1°. The orientation of the g-tensor for chain B is obtained by 180° rotation about the C2 axis, column 5.
C2 axis that transforms protein chains A and B.
The obtained g-tensor axes, which have been shown to be collinear with the molecular axes (refs. 33 and 34; see Fig. 2), clearly show a rotation of the radical Y122• with respect to the reduced tyrosine Y122-OH by 10°, 8°, and 5° with an error of ±1° for the corresponding x, y, and z axes (see Fig. 5 and Table 3). Interestingly, a reasonably good agreement with the experimental spectra is obtained even when only one protein chain (A) is used in the simulations. In this case, however, the weak lines on the high-field side of the spectra between 120° and 150°, assigned to chain B (Fig. 4) are not reproduced by the simulations. The optimum agreement between simulations and experimental spectra is obtained for relative contributions of 60–70% from chain A and 30–40% from chain B. We estimate the total yield of radicals in the crystals to be ≈0.1 radical per monomer.
Fig 5.

(Upper) Fobs − Fcalc electron density for the reduced tyrosine Y122-OH and orientation of the radical (green) from the EPR data (11° rotation about Cα–Cβ and −3° rotation about Cβ–C1 bond, which agrees within 2° with the g-tensor axes in yellow). Estimated error ±1° for the molecular axes of Y122-OH from the x-ray data and for the g-tensor axes from the EPR data. (Lower) Diiron site showing some of the iron ligands, the μ-oxo-bridge, and the terminal water at Fe1 in red, and the array of the hydrogen bonded amino acid network (dashed red line) within R2. The orientation of the radical Y122-O• is in green.
Discussion
In this work, we have shown that the tyrosyl radical Y122• can be generated in R2 crystals in the following two ways: The shunt pathway, by treatment with H2O2, was used for large crystals (0.7 × 0.3 × 0.2 mm3) of met R2. The different relative radical yield obtained for chain A (60–70%) and chain B (30–40%) is most likely because of the greater accessibility to soaking for subunit A as compared with subunit B, something that has previously been observed for this crystal form (35). Interestingly, the maximum radical yield determined from reconstituted frozen solutions of R2 is also reported to have a nonstoichiometric value of ≈1.5 per protein dimer (36). Of particular interest with respect to the enzymatic reaction is that the radical also can be generated in crystals of the apoprotein by the reconstitution reaction with ferrous iron and molecular oxygen. This finding shows that the protein in the crystal is in the native state and able to undergo the structural changes needed for the reaction. It is also a promising result toward generating and trapping diiron or radical reaction intermediates in crystals of WT or mutant R2s for further x-ray and EPR studies.
The g-tensor principal values of the E. coli tyrosyl radical obtained from the single-crystal spectra of a H2O2-treated crystal were the same as in the frozen solution (Table 2). This result shows that H2O2 treatment of the met R2 crystals did not change the chemical environment of the radical, e.g., by hydrogen bonding to a possible reaction product. It is well known that the gx-value of a tyrosyl radical is shifted from ≈2.0091 [hydrophobic environment, E. coli (37, 38)] to values of ≈2.0076 for the case of a more polar environment (refs. 12, 14, and 37; see Table 2). The hf values of the β1-proton of the side chain (see Fig. 4 legend) measured in the R2 crystal were the same as in freshly prepared frozen R2 solutions. This finding shows that no structural change is introduced to the side chain of the radical in the crystal by the shunt-pathway reaction, because the isotropic part of this coupling is strongly dependent on the side-chain geometry (10–14), according to Aiso(Hβ) = ρ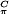 (B′ + B" cos2θ), where ρ
(B′ + B" cos2θ), where ρ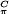 is the spin density at the adjacent π-carbon atom, B′ and B" are empirical constants, and θ is the dihedral angle of the β-proton. From the measured value Aiso(Hβ1) ≈ 2.0 mT, and by using a spin density at C1 of 0.38–0.39 (38, 39) and B" values between 5.24 and 5.7 mT (refs. 38 and 39; B′ is negligible) we estimate a dihedral angle for the β1-proton between 12° and 16°. This result is close to the value of 17° that we obtained from the high-resolution structure for the reduced tyrosine residue Y122-OH, and indicates that a possible reorientation of the radical should not significantly affect this dihedral angle.
is the spin density at the adjacent π-carbon atom, B′ and B" are empirical constants, and θ is the dihedral angle of the β-proton. From the measured value Aiso(Hβ1) ≈ 2.0 mT, and by using a spin density at C1 of 0.38–0.39 (38, 39) and B" values between 5.24 and 5.7 mT (refs. 38 and 39; B′ is negligible) we estimate a dihedral angle for the β1-proton between 12° and 16°. This result is close to the value of 17° that we obtained from the high-resolution structure for the reduced tyrosine residue Y122-OH, and indicates that a possible reorientation of the radical should not significantly affect this dihedral angle.
The key result of this study is, however, the orientation of the radical Y122• with respect to the x-ray structure of Y122-OH, which deviates by 10°, 8°, and 5° (errors ± 1°; see Table 3) for the respective x, y, and z axes. To estimate the location of the radical, in particular the position of the tyrosyl oxygen, which is proposed to be involved in a transient hydrogen abstraction reaction (9), we assume that the origin of the measured rotation is in the flexible tyrosine side chain, because the headgroup is a rigid structure. We further assume that no larger translational motion (which would not affect the EPR spectra) occurs, because this would require movement of the helix backbone, which is quite unlikely. The energetically most favorable way to obtain the observed radical orientation is by dihedral angle rotations about the Cα–Cβ and Cβ–C1 bonds of the side chain. A rotation about Cα–Cβ by 11° and about Cβ–C1 by −3° results in the observed g-tensor orientation with remaining deviations of <2° for each axis. Fig. 5 Upper shows the obtained location of the radical Y122• (green) for these rotations. This clearly differs from the electron density for Y122-OH. It is interesting to note that the larger rotation of 11° about Cα–Cβ does not affect the dihedral angles of the β-protons. The rotation about Cβ–C1 (Fig. 2), which changes the dihedral angles, is only −3°, which is too small for a detectable change of Aiso(Hβ1). This finding is in line with the earlier conclusion that the hf coupling of the β-proton agrees with the x-ray structure of the reduced tyrosine Y122-OH (see above).
Compared with the reduced tyrosine, the radical is rotated away from the diiron center. In the met form the tyrosine oxygen is at a distance of ≈3.2 Å from one of the Asp-84 oxygens, probably forming a weak hydrogen bond. The rotation of the radical increases this distance to >4 Å, which is clearly beyond hydrogen-bonding distance (Fig. 5 Lower). This result is consistent with its large gx-component (gx = 2.00912) observed both in frozen solution and in the crystal, indicative of a nonpolar and not-hydrogen-bonding environment (11–15). The above-mentioned remaining deviation of <2° for the gx axis and the rotated Y122 x axis moves the tyrosine oxygen in a perpendicular plane, and therefore further changes its distance from Asp-84 by only <0.05Å.
Until now the extent of conformational changes induced upon tyrosyl radical formation in a protein have not been described. Previously, a single-crystal EPR study has been performed on Y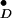 in photosystem II (PSII), which allowed the radical orientation to be related to the orientation of the membrane (32). However, when no high-resolution crystal structure is available of PSII, the orientation of the radical cannot be compared with its molecular structure and the structure of its protein environment.
in photosystem II (PSII), which allowed the radical orientation to be related to the orientation of the membrane (32). However, when no high-resolution crystal structure is available of PSII, the orientation of the radical cannot be compared with its molecular structure and the structure of its protein environment.
The conformational changes now observed in the R2 system upon oxidation of Tyr-122 are significant, although they are fairly small. The conformational motion of the tyrosine headgroup is, according to our model, assuming a rigid main-chain structure, mainly a rotation along the Cα–Cβ bond. The implication is that a relatively small energetic cost has to be paid for the conformational change on redox changes. Albeit small, the rotation of ≈10° leads to a significant displacement of the tyrosine oxygen. This oxygen is a key group for the function of the radical Tyr-122 when it can harbor a proton in a radical-mediated electron-transfer event. The reduced tyrosine Y122-OH is connected by a weak hydrogen bond to the diiron site, whereas the radical Y122• is disconnected from the diiron site and the hydrogen-bond network. The lack of a proton between Asp-84 and Y122• leads to a repulsion between these two residues, which might in fact be the major driving force for the observed conformational change. As a consequence, Y122• is “disconnected” from the iron coordination sphere, something that likely contributes to its unusual stability.
The radical transfer path between Tyr-122 on R2 and the active site on R1 has been suggested to involve several conserved residues (2, 40, 41). A tryptophan (W48) and tyrosine (Y356) on R2, and two tyrosines (Y730 and Y731), and an active site Cys (C439) on R1, could potentially serve as “high-potential” redox intermediates in this electron transfer. It was noted that most of these residues do in fact form conserved hydrogen-bonding paths. In R2, Trp-48 is connected to the metal-coordinating His-118 through the conserved Asp-237 (Fig. 5 Lower), and the two tyrosine residues on R1 form a hydrogen-bonded path with the active site Cys-439. These observations have served as a basis for a range of more or less detailed proposals for how a coupled proton/electron transfer could be in effect along this chain (3, 4–9, 42). However, a hydrogen-bonded path, able to transport protons, does not appear to extend all of the way between Tyr-122 and the active site Cys-439, and some steps along the proposed chain might solely involve electron transfer. One such example is between Asp-84 and His-118 of the R2 protein, and this may preclude any net proton transfer to the diiron site during the reaction. The major function for the conserved hydrogen-bonded networks is thus probably local rather than global, where they can serve in proton handling, redox tuning, and structural stabilization of intermediate states, such as a Trp-48 carbocation radical, which was proposed and reported to occur during the reconstitution reaction (2, 42).
However, local coupling of proton/electron transfer most likely occurs during the reaction cycle. It has been proposed in an earlier theoretical study that the first step of the long-range radical-transfer reaction is a transient hydrogen atom abstraction from the water coordinated to Fe1 (Fig. 5 Lower) by the radical Y122•, and subsequent formation of a radical character on Trp-48 in one step (9). In this case, the x-ray structure for Y122-OH shown in Fig. 5 Lower may well be similar to that of the transient state, which also exhibits a protonated Tyr-122; the H-bond connection between Y122 and the terminal water has been shown to be structurally possible (43). If no proton is transferred into the diiron-Tyr 122 site at this point, the result would be a “one-proton-deficient” site, which could in fact be an advantage, as this could provide the reversibility of the reaction. The resulting iron-coordinated hydroxide can then serve as a sufficiently strong and stable base to drive the proton abstraction, when Tyr-122 is reoxidized, with only a net transfer of an electron into this region. The observed conformational change, coupling or uncoupling Tyr-122 from the metal site, may assist in controlling this reversible protonation/deprotonation reaction.
Acknowledgments
We thank Prof. R. Bittl for helpful discussions; Prof. W. Lubitz for continuous support; and Dr. K. Knudsen and Prof. K. K. Andersson for help at the Swiss–Norwegian beam line at the European Synchrotron Radiation Facility. This work was supported by Deutsche Forschungsgemeinschaft Grant Le 812/1-2 (to F.L. and G.L.), the Swedish Research Council (to P.N.), and the European Community (to P.N.).
Abbreviations
RNR, ribonucleotide reductase
hf, hyperfine Y122•, functional tyrosyl radical in WT RNR subunit R2 from E. coli
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code ).
References
- 1.Logan D. T., Su, X.-D., Aberg, A., Regnström, K., Hajdu, J., Eklund, H. & Nordlund, P. (1996) Structure (London) 4, 1053-1064. [DOI] [PubMed] [Google Scholar]
- 2.Nordlund P. & Eklund, H. (1993) J. Mol. Biol. 232, 123-164. [DOI] [PubMed] [Google Scholar]
- 3.Eklund H., Uhlin, U., Färnegardh, M., Logan, D. T. & Nordlund, P. (2001) Prog. Biophys. Mol. Biol. 77, 177-268. [DOI] [PubMed] [Google Scholar]
- 4.Stubbe J. & Riggs-Gelasco, P. (1998) Trends Biochem. Sci. 23, 438-443. [DOI] [PubMed] [Google Scholar]
- 5.Stubbe J. & van der Donk, W. A. (1998) Chem. Rev. (Washington, D.C.) 98, 705-762. [DOI] [PubMed] [Google Scholar]
- 6.Siegbahn P. E. M. (1998) J. Am. Chem. Soc. 120, 8417-8429. [Google Scholar]
- 7.Mao S. S., Holler, T. P., Yu, G. X., Bollinger, J. M., Jr., Booker, S., Johnston, M. I. & Stubbe, J. (1992) Biochemistry 31, 9733-9743. [DOI] [PubMed] [Google Scholar]
- 8.Sjöberg B.-M. (1997) Struct. Bonding (Berlin) 88, 139-173. [Google Scholar]
- 9.Siegbahn P. E. M., Eriksson, L., Himo, F. & Pavlov, M. (1998) J. Phys. Chem. B 102, 10622-10629. [Google Scholar]
- 10.Andersson K. K. & Gräslund, A. (1995) Adv. Inorg. Chem. 43, 359-408. [Google Scholar]
- 11.Gerfen G. S., Bellew, B. F., Un, S., Bollinger, J. M., Jr., Stubbe, J., Griffin, R. G. & Singel, D. J. (1993) J. Am. Chem. Soc. 115, 6420-6421. [Google Scholar]
- 12.van Dam P. J., Willems, J.-P., Schmidt, P. P., Pötsch, S., Barra, A.-L., Hagen, W. R., Hoffman, B. M., Andersson, K. K. & Gräslund, A. (1998) J. Am. Chem. Soc. 120, 5080-5085. [Google Scholar]
- 13.Allard P., Barra, A.-L., Andersson, K. K., Schmidt, P. P., Atta, M. & Gräslund, A. (1996) J. Am. Chem. Soc. 118, 895-896. [Google Scholar]
- 14.Bleifuss G., Kolberg, M., Pötsch, S., Hofbauer, W., Bittl, R., Lubitz, W., Gräslund, A., Lassmann, G. & Lendzian, F. (2001) Biochemistry 40, 15362-15368. [DOI] [PubMed] [Google Scholar]
- 15.Un S., Gerez, C., Elleingand, E. & Fontecave, M. (2001) J. Am. Chem. Soc. 123, 3048-3054. [DOI] [PubMed] [Google Scholar]
- 16.Atkin C. L., Thelander, L., Reichard, P. & Lang, G. (1973) J. Biol. Chem. 248, 7464-7472. [PubMed] [Google Scholar]
- 17.Sahlin M., Sjöberg, B.-M., Backers, G., Loehr, T. & Sanders-Loehr, J. (1990) Biochem. Biophys. Res. Commun. 167, 813-818. [DOI] [PubMed] [Google Scholar]
- 18.Sjöberg B.-M., Hahne, S., Karlsson, M., Jörnvall, H., Göransson, M. & Uhlin, B. E. (1986) J. Biol. Chem. 261, 5658-5662. [PubMed] [Google Scholar]
- 19.Sjöberg B. M., Karlsson, M. & Jörnvall, H. (1987) J. Biol. Chem. 262, 9736-9743. [PubMed] [Google Scholar]
- 20.Nordlund P., Uhlin, U., Westergren, C., Joelsen, T., Sjöberg, B.-M. & Eklund, H. (1989) FEBS Lett. 258, 251-254. [DOI] [PubMed] [Google Scholar]
- 21.Otwinowski Z. (1993) in Proceedings of the CCP4 Study Weekend: Data Collection and Processing, eds. Sawyer, L., Isaacs, N. & Bailey, S.[Science and Engineering Council (SERC) Daresbury Laboratory, Daresbury, U.K.].
- 22.Brünger A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D. 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 23.Perrakis A., Sixma, T. K., Wilson, K. S. & Limzin, V. S. (1997) Acta Crystallogr. D. 53, 448-455. [DOI] [PubMed] [Google Scholar]
- 24.Stesmans A. & VanGorp, G. (1989) Rev. Sci. Instrum. 60, 2949-2952. [Google Scholar]
- 25.Lendzian F., Sahlin, M., MacMillan, F., Bittl, R., Fiege, R., Pötsch, S., Sjöberg, B.-M., Gräslund, A., Lubitz, W. & Lassmann, G. (1996) J. Am. Chem. Soc. 118, 8111-8120. [Google Scholar]
- 26.Press W. H., Teukolsky, S. A., Vetterling, W. T. & Flannery, B. P., (1992) Numerical Recipes in C (Cambridge Univ. Press, Cambridge, U.K.).
- 27.Nordlund P., Sjöberg, B.-M. & Eklund, H. (1990) Nature 345, 593-598. [DOI] [PubMed] [Google Scholar]
- 28.Nordlund P., (1990) Ph.D. thesis (Swedish Univ. of Agricultural Sciences, Uppsala).
- 29.Ivancich A., Mazza, G. & Desbois, A. (2001) Biochemistry 40, 6860-6866. [DOI] [PubMed] [Google Scholar]
- 30.Bennati M., Farrer, C. T., Bryant, J. A., Inati, S. J., Weis, V., Gerfen, G. J., Riggs-Gelasco, P., Stubbe, J. & Griffin, R. G. (1999) J. Magn. Reson. 138, 232-243. [DOI] [PubMed] [Google Scholar]
- 31.Gurbiel R. J., Bolin, J. T., Ronco, A. E., Mortenson, L. & Hoffman, B. M. (1991) J. Magn. Reson. 91, 227-240. [Google Scholar]
- 32.Hofbauer W., Zouni, A., Bittl, R., Kern, J., Orth, P., Lendzian, F., Fromme, P., Witt, H. T. & Lubitz, W. (2001) Proc. Natl. Acad. Sci. USA 98, 6623-6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fasanella E. L. & Gordy, W. (1969) Proc. Natl. Acad. Sci. USA 62, 299-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mezzetti A., Maniero, A. L., Brustolon, M., Giacometti, G. & Brunel, L. C. (1999) J. Phys. Chem. A 103, 9636-9643. [Google Scholar]
- 35.Andersson M. E., Högbom, M., Rinaldo-Matthis, A., Andersson, K. K., Sjöberg, B.-M. & Nordlund, P. (1999) J. Am. Chem. Soc. 121, 2346-2352. [Google Scholar]
- 36.Miller M. A., Gobena, F. T., Kauffmann, K., Münck, E., Que, L., Jr. & Stankovich, M. T. (1999) J. Am. Chem. Soc. 121, 1096-1097. [Google Scholar]
- 37.Engström M., Himo, F., Gräslund, A., Minaev, B., Vahtras, O. & Agren, H. (2000) J. Phys. Chem. A 104, 5149-5153. [Google Scholar]
- 38.Hoganson C. W., Sahlin, M., Sjöberg, B.-M. & Babcock, G. T. (1996) J. Am. Chem. Soc. 118, 4672-4679. [Google Scholar]
- 39.Himo F., Gräslund, A. & Eriksson, L. A. (1997) Biophys. J. 72, 1556-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uhlin U. & Eklund, H. (1994) Nature 370, 533-539. [DOI] [PubMed] [Google Scholar]
- 41.Ekberg M., Sahlin, M., Eriksson, M. & Sjöberg, B.-M. (1996) J. Biol. Chem. 271, 20655-20659. [DOI] [PubMed] [Google Scholar]
- 42.Baldwin J., Krebs, K., Ley, B. A., Edmondson, D. E., Huynh, B. H. & Bollinger, J. M., Jr. (2000) J. Am. Chem. Soc. 122, 12195-12206. [Google Scholar]
- 43.Högbom M., Andersson, M. E. & Nordlund, P. (2001) J. Biol. Inorg. Chem. 6, 315-323. [DOI] [PubMed] [Google Scholar]